
High-resolution physical maps of vertebrate species’ chromosomes empower comparative genomics discovery and are indispensable for sequence assembly precision. Beginning in 2003, the NIH–NHGRI launched an initiative that designated 24 species of mammals for low-coverage whole-genome sequencing in order to provide evolutionary context to human genome annotation (Green 2007) (http://www.genome.gov/25521745). Four principal goals were anticipated for the bold sequencing initiative: (1) to discover evolutionarily conserved sequence motifs, particularly outside of protein-coding genes, which are responsible for regulatory and other critical genomic functions; (2) to provide a framework for reconstruction of genome organization, content, and dynamics that have occurred during the mammalian radiations; (3) to empower new models of human disease and heritable phenotypes; and (4) to provide a starting point for assessment of the expansion, contraction, and adaptation of gene families in different evolutionary lineages.
Although the new mammal sequences have been eagerly anticipated, it is now becoming evident that draft and even “finished” genome sequence of evolutionarily divergent species by themselves can fail to provide sufficient granularity for confident comparison of genome organization and structure to fulfill goals 2–4 listed above. We suggest here to look “back to the future” in developing high-resolution chromosome-based physical maps as an essential and cost-effective framework for the annotation and evolutionary analysis of mammalian and other vertebrate genomes. Independent physical maps in concert with draft or complete sequence assemblies will greatly empower the precise view of comparative genome organization by facilitating the correct ordering of genic and nongenic DNA segments on chromosomes for whole-genome alignments. Accurate comparative physical maps enable discovery of conserved chromosome segments and evolutionary breakpoint regions (EBRs) that are useful for reconstructing the origins of mammalian genomes and the evolutionary forces that molded them (Murphy et al. 2005a; Larkin et al. 2009).
Genetic maps have formed the bedrock of genetic analysis since Sturtevant, Bridges, and their peers conjured the first gene maps nearly a century ago. More recently, linkage, radiation hybrid (RH), bacterial artificial chromosome (BAC), and/or ZOOFISH maps of human, macaque, mouse, rat, dog, cat, horse, opossum, and cattle anchored the genome sequence assemblies for those species (Table 1). Because current methods of DNA sequence assembly produce “contigs” and scaffolds of limited length, leaving scores, or in some cases, thousands of sequence gaps, a high-quality map of densely spaced markers is invaluable for affirming the correct placement of scaffolds on the chromosomes and for “proofing” the order of markers within the assemblies. Unplaced or misplaced scaffolds, plus improperly ordered markers within scaffolds, result in errors in comparative genome analysis by introducing heretical evolutionary breakages where none exist.
Coverage of available whole-genome sequences and physical/linkage maps used as anchors for mammalian genome assemblies
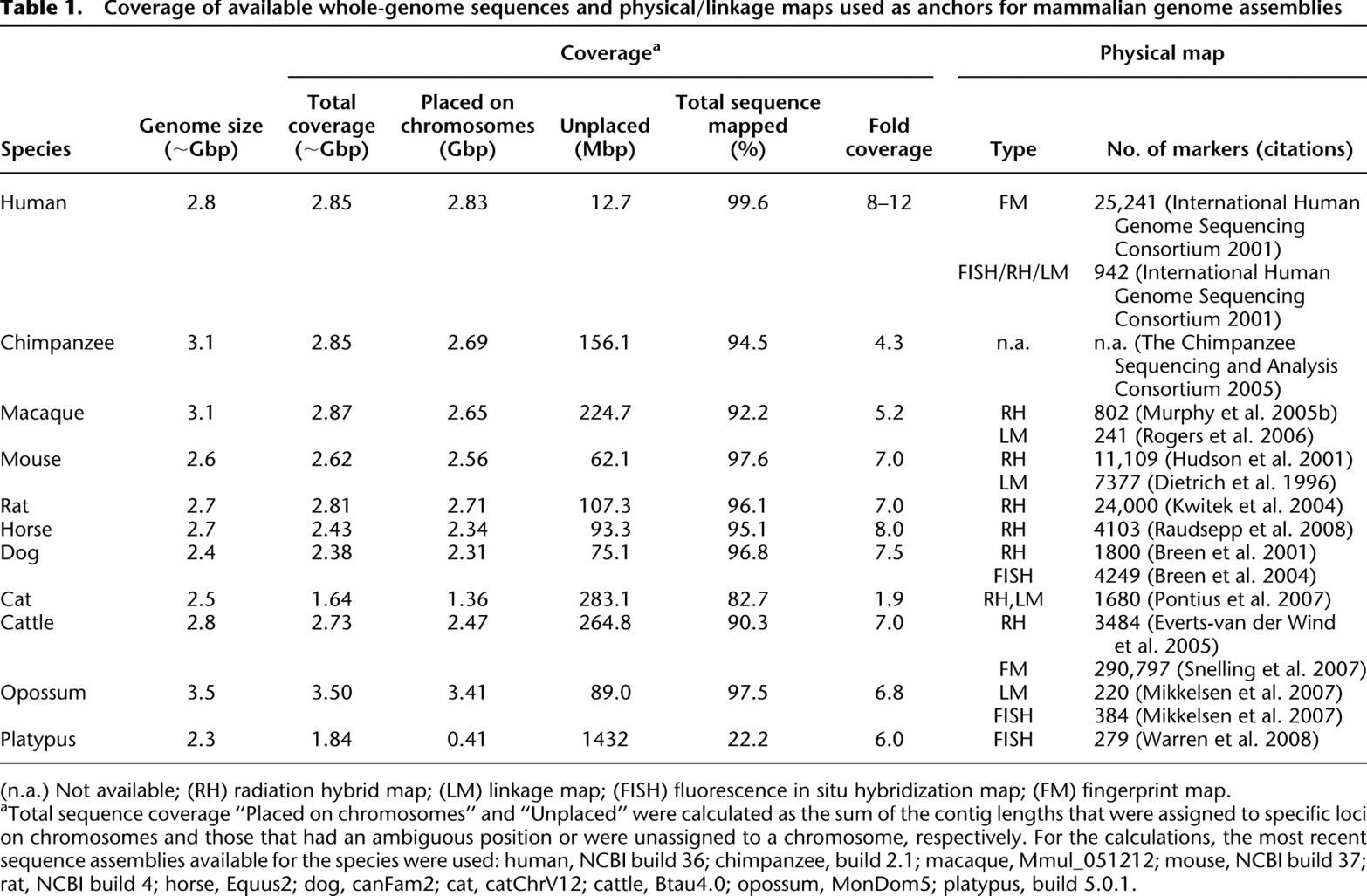
[i] (n.a.) Not available; (RH) radiation hybrid map; (LM) linkage map; (FISH) fluorescence in situ hybridization map; (FM) fingerprint map.
[ii] aTotal sequence coverage “Placed on chromosomes” and “Unplaced” were calculated as the sum of the contig lengths that were assigned to specific loci on chromosomes and those that had an ambiguous position or were unassigned to a chromosome, respectively. For the calculations, the most recent sequence assemblies available for the species were used: human, NCBI build 36; chimpanzee, build 2.1; macaque, Mmul_051212; mouse, NCBI build 37; rat, NCBI build 4; horse, Equus2; dog, canFam2; cat, catChrV12; cattle, Btau4.0; opossum, MonDom5; platypus, build 5.0.1.
The conundrum is illustrated in the assembly of the platypus genome, which was compiled with no map or related species’ reference sequence (O'Brien 2008; Warren et al. 2008). The platypus is a fascinating species for comparative evolutionary inference because its monotreme order serves as a “missing link” between two of the major dominating groups in the history of life on earth—the reptiles (345–363 million years ago [Mya]) and mammals (230 Mya–present). Nonetheless, and in part due to its distant evolutionary roots, the sequence assembly and comparative analysis of the platypus genome presented a huge challenge. Although sequenced to approximately sixfold coverage using a combination of whole-genome shotgun (WGS) and BAC sequencing, only 409.4 Mbp (22.2%) out of 1.84 Gbp of the sequence assembly (or 17.4% of 2.3 Gbp whole genome) could be ordered on the 20 platypus chromosomes, and then only by the assistance of 279 markers mapped by fluorescence in situ hybridization. Thus, while producing valuable new information and insights into early mammalian evolution, a detailed genomic architecture of the platypus genome suitable for comparison to other mammalian genomes remains hidden.
Recently, three species whose genome assemblies benefited appreciably from a framework RH map are the dog (Lindblad-Toh et al. 2005), cat (Pontius et al. 2007), and cattle (The Bovine Genome Sequencing and Analysis Consortium 2009). The cat genome, sequenced 1.9-fold, initially covered 65% of the total genome and contained 217,790 unconnected scaffolds and as many sequence gaps (Pontius et al. 2007). A multistep “assisted” assembly first mapped 1680 ordered RH markers to scaffolds and then placed “reciprocal best match” sequences between RH markers in the order their homologous counterparts occurred in human and dog genome maps. The strategy “humanized” or “canineized” the intervals between the cat RH markers for certain; however, the intervals between RH markers were likely accurate over 90% of the time, yielding useful insight from ∼2× sequence as determined by comparison to “finished” cat sequence of MHC, ENCODE, and selected gene regions (Yuhki et al. 2003; The ENCODE Project Consortium 2004; Fyfe et al. 2006; Menotti-Raymond et al. 2007; Murphy et al. 2007; Pontius et al. 2007). The combined 2× cat draft sequence, RH framework map, and the assisted assembly strategy were sufficient to allow informative, if preliminary, whole-genome assessment of chromosome breakpoint exchanges between cat and six other “finished” mammal genome sequences, leading to important insights on the rates of inter- and intrachromosomal rearrangements among the different mammalian lineages.
The cattle genome assembly (Btau4.0) utilized high-quality integrated RH and BAC physical maps consisting of >3000 markers, 2759 BAC-end sequences, and >240,000 fingerprinted BAC clones (Everts-van der Wind et al. 2005; Snelling et al. 2007; The Bovine Genome Sequencing and Analysis Consortium 2009). The final assembly covered 90.3% of the genome with a >95% consistency of marker order among all data sources. The remaining 11% of the genome not assigned to any chromosome consists mostly of difficult to assemble repetitive sequences and segmental duplications. The high-quality physical map of the cattle genome enabled detailed multispecies comparisons of chromosome organization that led to novel discoveries of ancient chromosome rearrangements and the sequence features of EBRs (The Bovine Genome Sequencing and Analysis Consortium 2009). For example, the four ferungulate-specific EBRs identified on BTA16 occurred before the divergence of Artiodactyla and Carnivora, but the human genome retains the Boreoeutherian ancestral form. The analysis of cattle-, artiodactyl-, and ferungulate-specific EBRs revealed significantly higher densities of LINE-L1 and other repeat families than in other parts of the genome, but lower densities of SINE-BovA repeats, providing support for the hypothesis that repeat elements promote chromosome rearrangements, and that older repeats (such as SINE-BovA) are disrupted by the insertion of more recent repetitive elements. In addition, segmental duplications larger than 10 kb were found at >10-fold higher density in cattle-specific EBRs than in other regions of the genome, consistent with previous findings for segmental duplications in the human genome (Murphy et al. 2005a). Finally, the cattle-specific EBRs were found to contain genes (often duplicated) with functions related to ruminant-specific and adaptive phenotypes, providing further support for the hypothesis that mammalian chromosome rearrangements may be adaptive (Larkin et al. 2009).
The value of high-resolution (≤1 Mbp) physical maps for studies of chromosome evolution has been illustrated in the above and other multispecies map-anchored whole-genome sequence comparisons (Table 2). These studies identified and compared the orders of thousands of syntenic segments using computational routines that define and visualize precise chromosome coordinates of EBRs and homologous synteny blocks (HSBs). Multispecies genome comparisons have already produced provocative observations about evolutionary genome dynamics (Pevzner and Tesler 2003; Bailey et al. 2004; Everts-van der Wind et al. 2005; Murphy et al. 2005a; Ma et al. 2006; The Bovine Genome Sequencing and Analysis Consortium 2009; Larkin et al. 2009) including: (1) frequent reuse of certain EBRs in different mammal lineages; (2) concordance of EBR and diagnostic chromosome breakpoints in human cancers; (3) selection signatures for large HSBs shared among multiple mammals and the chicken (some as great as 23 Mbp in length); (4) gene density enrichment within EBRs; (5) clustering of segmental duplication around EBRs in primates and artiodactyls, raising the prospect that copy number variation actually precipitates chromosome exchange; (6) 20-fold difference in rates of chromosome rearrangements in different epochs and mammal lineages; and (7) a remarkable balance between intrachromosomal and interchromosomal exchanges in different lineages, as if species with few translocations relative to ancestral mammals display more inversions and vice versa (see Table 2). These studies clearly demonstrate that contiguous and accurate chromosome sequence assemblies are necessary to gain a more complete understanding of the mechanisms of genome evolution. Without map-assisted chromosome assemblies, the revealing sentinels and consequences of chromosome rearrangements, such as those described above, will remain undiscovered.
Breakthrough discoveries derived from precision physical mapping of vertebrate genomes

Comparative genomics is now being enhanced by direct comparison of sequences for defining coordinates of chromosome exchange, offering considerable promise for an even more detailed understanding of the forces driving chromosome evolution (Ma et al. 2006). However, sequence-only approaches are restricted to mammalian species with very deep or map-assisted coverage, because the average size of the contigs is relatively short and insufficient to span the vast majority of EBRs, typically complex regions containing segmental duplications, deletions, large gene families, and complex repeats (Murphy et al. 2005a; The Bovine Genome Sequence and Anlaysis Consortium 2009; Larkin et al. 2009). The precise definition of EBR coordinates obtained from marker-dense comparative maps and high-coverage genome sequence is thus essential for a detailed view of the role of chromosome rearrangements in evolutionary processes of divergence, adaptation, and speciation (Larkin et al. 2009).
The current “next generation” sequencing technologies (454 Life Sciences [Roche]; Illumina Genome Analyzer, Applied Biosystems SOLiD System, and others) hold much promise in both sequence throughput and in cost-effectiveness, and are already being applied by sequencing centers to human resequencing projects as well as certain NHGRI-sponsored mammal species sequences “top-up” (http://www.genome.gov/). The current generation of short-read sequencing technologies will not solve the problem of obtaining reliable ordering of scaffolds on chromosomes or complete chromosome assemblies necessary for detailed comparative evolutionary studies of genome organization. This is because of the abundance of repetitive sequences, large gene families, and extensive segmental duplications that greatly complicate assemblies, but are tractable problems for physical mapping methods. For de novo assemblies without a guiding physical map it will take a read length >300 bp and more than 22-fold genome coverage to close enough assembly gaps to obtain a reliable global ordering of sequence scaffolds on the chromosomes (Sundquist et al. 2007). The sequence platform dynamic re-emphasizes the requirement for parallel physical maps for genome assembly and analyses, particularly in species that are evolutionarily divergent from those that have been assembled and have good physical maps. The transition to even higher throughput sequencing technologies with longer reads may solve the problem in the not-too-distant future, but in the interim, physical maps offer a well-tested resource for a deep analysis of genome architecture and evolutionary history.
New genomic technologies have also made the prospect of physical map development, particularly RH maps, faster, denser, and cheaper (McKay et al. 2007). For the 24 NHGRI-nominated species, the draft WGS itself is invaluable for selecting evenly spaced framework markers that are distinct from the RH partner species (typically Chinese hamster ovary cells). We estimate that it is now possible to produce the data for a 1-Mbp resolution RH map for less than $100,000US plus labor in under 6 mo using a custom 3000–5000 marker genotyping array. Building a physical map for a representative of each mammalian order (or even for each family) would be a critical and cost-effective approach for advancing comparative and evolutionary biology and maximizing the investments in whole-genome sequencing. Thus, a timely investment in methods for even more rapid and inexpensive physical map development may have an enormous impact on comparative genomics.
With less expensive and faster sequencing technologies on the horizon, the original goals of large-scale DNA sequencing programs will soon enjoy thousands of species’ genomes sequenced to draft coverage. The existing and envisioned programs all emphasize the importance of understanding how genomes evolve and how changes in genome organization may lead to the phenotypic and adaptive changes that permitted complex cellular life to emerge and flourish. As global explorers of the world's continents were the first to draw maps that guided human understanding of geography, genome sequencing of new species that is complemented by dense physical maps will chart the course of genomic interpretation.
Acknowledgments
This project has been funded in whole or in part with federal funds from the USDA-CSREES under contract 538 AG 2008-34480-19328 (H.A.L.) and the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400 (J.P.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- ↵JA BaileyR BaertschWJ KentD HausslerEE Eichler(2004) Hotspots of mammalian chromosomal evolution. Genome Biol 5:R23, http://genomebiology.com/2004/5/4/R23..
- ↵The Bovine Genome Sequencing and Analysis ConsortiumCG ElsikRL TellamKC Worley(2009) The genome sequence of Taurine cattle: A window to ruminant biology and evolution. Science 324:522–528.
- M BreenS JouquandC RenierCS MellershC HitteNG HolmesA ChéronN SuterF VignauxAE Bristow(2001) Chromosome-specific single-locus FISH probes allow anchorage of an 1800-marker integrated radiation-hybrid/linkage map of the domestic dog genome to all chromosomes. Genome Res 11:1784–1795.
- M BreenC HitteTD LorentzenR ThomasE CadieuL SabacanA ScottG EvannoHG ParkerEF Kirkness(2004) An integrated 4249 marker FISH/RH map of the canine genome. BMC Genomics 5:65, 10.1186/1471-2164-5-65.
- KV BulazelGC FerreriMD EldridgeRJ O'Neill(2007) Species-specific shifts in centromere sequence composition are coincident with breakpoint reuse in karyotypically divergent lineages. Genome Biol 8:R170, 10.1186/gb-2007-8-8-r170.
- The Chimpanzee Sequencing and Analysis Consortium (2005) Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87.
- E Darai-RamqvistA SandlundS MüllerG KleinS ImrehM Kost-Alimova(2008) Segmental duplications and evolutionary plasticity at tumor chromosome break-prone regions. Genome Res 18:370–379.
- WF DietrichJ MillerR SteenMA MerchantD Damron-BolesZ HusainR DredgeMJ DalyKA IngallsTJ O'Connor(1996) A comprehensive genetic map of the mouse genome. Nature 380:149–152.
- ↵The ENCODE Project Consortium (2004) The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306:636–640.
- A Everts-van der WindSR KataMR BandM RebeizDM LarkinRE EvertsCA GreenL LiuS NatarajanT Goldammer(2004) A 1463 gene cattle-human comparative map with anchor points defined by human genome sequence coordinates. Genome Res 14:1424–1437.
- ↵A Everts-van der WindDM LarkinCA GreenJS ElliottCA OlmsteadR ChiuJE ScheinMA MarraJE WomackHA Lewin(2005) A high-resolution whole-genome cattle-human comparative map reveals details of mammalian chromosome evolution. Proc Natl Acad Sci 102:18526–18531.
- ↵JC FyfeM Menotti-RaymondVA DavidL BrichtaAA SchäfferR AgarwalaWJ MurphyWJ WedemeyerBL GregoryBG Buzzell(2006) An ∼140-kb deletion associated with feline spinal muscular atrophy implies an essential LIX1 function for motor neuron survival. Genome Res 16:1084–1090.
- ↵P Green(2007) 2× genomes–does depth matter? Genome Res 17:1547–1549.
- TJ HudsonDM ChurchS GreenawayH NguyenA CookRG SteenWJ Van EttenAB CastleMA StrivensP Trickett(2001) A radiation hybrid map of mouse genes. Nat Genet 29:201–205.
- International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921.
- C KemkemerM KohnDN CooperL FroenickeJ HögelH HameisterH Kehrer-Sawatzki(2009) Gene synteny comparisons between different vertebrates provide new insights into breakage and fusion events during mammalian karyotype evolution. BMC Evol Biol 9:84, 10.1186/1471-2148-9-84.
- AE KwitekJ Gullings-HandleyJ YuDC CarlosK OrlebekeJ NieJ EckertA LemkeJW AndraeS Bromberg(2004) High-density rat radiation hybrid maps containing over 24,000 SSLPs, genes, and ESTs provide a direct link to the rat genome sequence. Genome Res 14:750–757.
- DM LarkinA Everts-van der WindM RebeizPA SchweitzerS BachmanC GreenCL WrightEJ CamposLD BensonJ Edwards(2003) A cattle-human comparative map built with cattle BAC-ends and human genome sequence. Genome Res 13:1966–1972.
- ↵DM LarkinG PapeR DonthuL AuvilM WelgeHA Lewin(2009) Breakpoint regions and homologous synteny blocks in chromosomes have different evolutionary histories. Genome Res 19:770–777.
- ↵K Lindblad-TohCM WadeTS MikkelsenEK KarlssonDB JaffeM KamalM ClampJL ChangEJ KulbokasMC Zody(2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438:803–819.
- ↵J MaL ZhangBB SuhBJ RaneyRC BurhansWJ KentM BlanchetteD HausslerW Miller(2006) Reconstructing contiguous regions of an ancestral genome. Genome Res 16:1557–1565.
- ↵SD McKayRD SchnabelBM MurdochJ AertsCA GillC GaoC LiLK MatukumalliP StothardZ Wang(2007) Construction of bovine whole-genome radiation hybrid and linkage maps using high-throughput genotyping. Anim Genet 38:120–125.
- ↵M Menotti-RaymondVA DavidAA SchäfferR StephensD WellsR Kumar-SinghSJ O'BrienK Narfström(2007) Mutation in CEP290 discovered for cat model of human retinal degeneration. J Hered 98:211–220.
- TS MikkelsenMJ WakefieldB AkenCT AmemiyaJL ChangS DukeM GarberAJ GentlesL GoodstadtA Heger(2007) Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 447:167–177.
- ↵WJ MurphyDM LarkinA Everts-van der WindG BourqueG TeslerL AuvilJE BeeverBP ChowdharyF GalibertL Gatzke(2005a) Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science 309:613–617.
- WJ MurphyR AgarwalaAA SchäfferR StephensC Smith JrNJ CrumplerVA DavidSJ O'Brien(2005b) A rhesus macaque radiation hybrid map and comparative analysis with the human genome. Genomics 86:383–395.
- ↵WJ MurphyB DavisVA DavidR AgarwalaAA SchäfferAJ Pearks WilkersonB NeelamSJ O'BrienM Menotti-Raymond(2007) A 1.5-Mb-resolution radiation hybrid map of the cat genome and comparative analysis with the canine and human genomes. Genomics 89:189–196.
- ↵SJ O'Brien(2008) The platypus genome unraveled. Cell 133:953–955.
- ↵P PevznerG Tesler(2003) Human and mouse genomic sequences reveal extensive breakpoint reuse in mammalian evolution. Proc Natl Acad Sci 100:7672–7677.
- ↵JU PontiusJC MullikinDR SmithAgencourt Sequencing TeamK Lindblad-TohS GnerreM ClampJ ChangR StephensB NeelamN Volfovsky(2007) Initial sequence and comparative analysis of the cat genome. Genome Res 17:1675–1689.
- T RaudseppA Gustafson-SeaburyK DurkinML WagnerG GohCM SeaburyC Brinkmeyer-LangfordEJ LeeR AgarwalaE Stallknecht-Rice(2008) A 4103 marker integrated physical and comparative map of the horse genome. Cytogenet Genome Res 122:28–36.
- J RogersR GarciaW ShelledyJ KaplanA AryaZ JohnsonM BergstromL NovakowskiP NairA Vinson(2006) An initial genetic linkage map of the rhesus macaque (Macaca mulatta) genome using human microsatellite loci. Genomics 87:30–38.
- L SchiblerA RoigMF MaheP LaurentH HayesF RodolpheEP Cribiu(2006) High-resolution comparative mapping among man, cattle and mouse suggests a role for repeat sequences in mammalian genome evolution. BMC Genomics 7:194, 10.1186/1471-2164-7-194.
- ↵WM SnellingR ChiuJE ScheinM HobbsCA AbbeyDL AdelsonJ AertsGL BennettIE BosdetM Boussaha(2007) A physical map of the bovine genome. Genome Biol 8:R165, 10.1186/gb-2007-8-8-r165.
- ↵A SundquistM RonaghiH TangP PevznerS Batzoglou(2007) Whole-genome sequencing and assembly with high-throughput, short-read technologies. PLoS One 2:e484, 10.1371/journal.pone.0000484.
- ↵WC WarrenLW HillierJA Marshall GravesE BirneyCP PontingF GrütznerK BelovW MillerL ClarkeAT Chinwalla(2008) Genome analysis of the platypus reveals unique signatures of evolution. Nature 453:175–183.
- ↵N YuhkiT BeckRM StephensY NishigakiK NewmannSJ O'Brien(2003) Comparative genome organization of human, murine, and feline MHC class II region. Genome Res 13:1169–1179.
Notes
[3] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.094557.109.