
Genome-wide analyses of alternative splicing in plants: Opportunities and challenges
Abstract
Alternative splicing (AS) creates multiple mRNA transcripts from a single gene. While AS is known to contribute to gene regulation and proteome diversity in animals, the study of its importance in plants is in its early stages. However, recently available plant genome and transcript sequence data sets are enabling a global analysis of AS in many plant species. Results of genome analysis have revealed differences between animals and plants in the frequency of alternative splicing. The proportion of plant genes that have one or more alternative transcript isoforms is ∼20%, indicating that AS in plants is not rare, although this rate is approximately one-third of that observed in human. The majority of plant AS events have not been functionally characterized, but evidence suggests that AS participates in important plant functions, including stress response, and may impact domestication and trait selection. The increasing availability of plant genome sequence data will enable larger comparative analyses that will identify functionally important plant AS events based on their evolutionary conservation, determine the influence of genome duplication on the evolution of AS, and discover plant-specific cis-elements that regulate AS. This review summarizes recent analyses of AS in plants, discusses the importance of further analysis, and suggests directions for future efforts.
An introduction to pre-mRNA processing and alternative splicing
The discovery that gene sequences are interrupted by noncoding segments (introns) that are removed during message processing (Berget et al. 1977) was initially surprising, but mRNA processing is now known to be common in eukaryotic genes. Most intron splicing is carried out by the spliceosome, a large macromolecular machine composed of five small nuclear riboproteins (snRNPs) and numerous accessory proteins (Staley and Guthrie 1998; Zhou et al. 2002). Spliceosome biochemistry and intron processing have been reviewed substantially (Staley and Guthrie 1998; Ast 2004). In metazoans, intron removal and the joining of flanking exons is directed by four sequence signals: the exon–intron junctions at the 5′ end and 3′ end that are the splice donor and acceptor sites, respectively, and two sites within the introns—the branch site sequence located upstream of the 3′ splice site, and the polypyrimidine tract located between the 3′ splice site and the branch site. Interestingly, in plants the pyrimidine tracts are mostly uridine, and the branch point sequences are not obvious (Reddy 2007). Although plant genomes are known to encode homologs of many proteins that are included in animal spliceosomes, plant spliceosomes have never been isolated, and their exact protein composition is yet unverified
Alternative splicing
Alternative splicing (AS) creates multiple mRNA transcripts, or isoforms, from a single gene. While AS had been observed in several genes by the early 1980s (Early et al. 1980; Rosenfeld et al. 1982), it was characterized at the single gene level and thought to occur in <5% of human genes (Sharp 1994). However, analysis of genome sequence data has demonstrated that AS is widespread in metazoans (Ast 2004; Sorek et al. 2004; Blencowe 2006; Kim et al. 2007). AS can affect message stability and translation efficiency as well as influencing and increasing protein diversity (Stamm et al. 2005). Indeed, the human genome is predicted to contain ∼32,000 genes (Lander et al. 2001; Venter et al. 2001), while the proteome is defined by ∼90,000 proteins. The observation that upward of 80% of human genes have been demonstrated to undergo AS supports its use as a mechanism for resolving this discrepancy (Modrek and Lee 2002; Leipzig et al. 2004).
AS is well studied in humans where altered expression of splicing variants has been correlated with numerous diseases (Soleymanlou et al. 2005; Tan et al. 2005; Ule et al. 2005; Agrawal and Eng 2006; Speek et al. 2006; Venables 2006; Zhong et al. 2006). Alternatively spliced isoforms result from the use of alternate splice sites during mRNA processing (Fig. 1). A potentially large number of alternatively spliced mRNAs can be created by these mechanisms both singularly or in combination (Black 2003; Sorek et al. 2004). Changes that affect the coding regions may change protein structure, while changes in the 3′ or 5′ UTRs may affect message stability. Approximately 60%–75% of AS events occur within the translated regions of mRNAs (Gupta et al. 2004; Stamm et al. 2005), and this can have dramatic effects on binding properties, intracellular localization, protein stability, enzymatic, and signaling activities (for review, see Stamm et al. 2005). Some splice isoforms contain a premature stop codon (PTC). These are often not translated, but are targeted for nonsense-mediated decay (NMD) (Belgrader et al. 1994). NMD is an RNA surveillance system that recognizes mRNAs containing premature termination codons (PTCs) and targets them for degradation (Maquat 2004). Lewis et al. (2003) determined that 35% of a set of >3000 alternatively spliced human genes have predicted isoforms that result in PTCs, concluded that ∼75% of these are apparent targets for NMD, and proposed that coupled AS and NMD plays a functional role in regulating protein expression levels. This process, termed RUST (regulated unproductive splicing and translation) may function to regulate protein expression by generating NMD-targeted isoforms. Evidence of such a process has been demonstrated in Caenorhabditis elegans (Morrison et al. 1997; Mitrovich and Anderson 2000). Recently, ultraconserved elements in mammals associated with some serine/arginine-rich splicing activator proteins (see below) have been discovered (Lareau et al. 2007; Ni et al. 2007). The transcripts of SR (serine-arginine-rich protein) genes that include these conserved sequences contain PTCs and are subject to NMD. The conserved nature of these elements argues for their functional significance, and their apparent role in message degradation illustrates the role of NMD in the autoregulation or homeostatic control of these splicing regulators (Lareau et al. 2007; Ni et al. 2007).

Common types of alternative splice events. The total numbers of EST/cDNAs with validated alignments (similarity ≥ 97% and EST/cDNA coverage ≥ 75%) included in this analysis are: Arabidopsis, 541,594; Rice, 903,022; Maize, 914,822. MAGI3.1 genome data (Fu et al. 2005) was used for maize AS analysis, Transcript isoform determination for all three plant species was carried out with the PASA spliced alignment software described by Haas et al. (2003). The data for human AS event types was reported by Kim et al. (2007)
Tools for identification and analysis of alternative splicing
Global identification of AS can be examined by stringently aligning ESTs/cDNA to genomic regions, ensuring fidelity of the gene model by assessing splice-site consensus sequences, and comparing transcripts originating from the same genomic location to identify alternative isoforms. The availability of sequenced genomes and large collections of transcript sequences provide a rich source for identifying AS events by computational methods (Modrek and Lee 2003), and tools have been recently developed that can automate much of this process (Haas et al. 2003). Limitations of transcript sequence analysis for AS detection include coverage biased toward transcript ends, insufficient numbers of transcript sequences yielding poor gene coverage and under-representation of AS isoforms, expression biases that affect abundance, and the inability to properly sample transcripts that are tissue specific, temporal, or treatment (stress, etc.) responsive. In addition, it is difficult to distinguish biologically relevant intron retention from transcriptome artifacts originating from genomic DNA contamination or incompletely processed transcripts. Oligonucleotide microarrays based on exon arrays composed of probes that hybridize to constitutive or alternative exons (Hu et al. 2001), or junction arrays composed of probes that hybridize to exon–exon junction regions (Johnson et al. 2003), or a combination of the two (Le et al. 2004; Pan et al. 2004; Sugnet et al. 2006) can sidestep the limitations inherent in EST analysis. While these provide a more sensitive detection platform and are well suited to identifying and comparing tissue-specific or treatment-specific AS, a complete and accurate description of the exon–intron structure of each gene included on the array is a prerequisite to their construction. An alternative array is composed of oligonucleotide probes distributed uniformly across the length of chromosomes or genomic regions. These tiling arrays (Yazaki et al. 2007) can identify and characterize regions of the genome that are transcriptionally active (Schadt et al. 2004) and have succeeded in identifying new transcript isoforms (Kapranov et al. 2002; Kampa et al. 2004). Combining microarrays with protein–RNA cross-linking and immunoprecipitation (CLIP) provides the opportunity to discover those mRNAs associated with specific SR proteins or other trans-acting protein factors involved in splice-site recognition. Genome-wide RNA-binding analysis of Nova, a neuron-specific RNA-binding protein that functions as a splicing regulator, has revealed the rules of Nova-dependent splicing regulation in vivo (Ule et al. 2006).
There are few bioinformatics tools for de novo prediction of AS. AUGUSTUS is an ab initio gene prediction tool based on a generalized hidden Markov model that identifies the most probable gene structure for each predicted gene based on its training parameters (Stanke et al. 2006b). Recently, this algorithm has been extended to predict multiple gene structures for each gene, some of which may represent biologically relevant alternative transcripts (Stanke et al. 2006a). However, the accuracy of an ab initio gene finder depends on the comprehensiveness of its training set and only a fraction of predictions represent the true gene structure. Therefore, it’s likely that many predicted genes and their predicted isoforms will be incorrect, and bench validation will be required to substantiate these.
Alternative splicing in plants
The characterization of spinach and Arabidopsis ribulosebisphosphate carboxylase/oxygenase (rubisco) activase provided one of the first demonstrations of AS in plants (Werneke et al. 1989). Subsequent characterization of plant genes that are alternatively spliced included RNA polymerase II (Dietrich et al. 1990), chorismate synthase (Gorlach et al. 1995), H protein (Kopriva et al. 1995), Arabidopsis U1 snRNP 70K (Golovkin and Reddy 1996), the maize regulatory MuDR transposable element (Hershberger et al. 1995), the maize loci for glutathione S-transferase (bronze2) (Marrs and Walbot 1997), and wx (Marillonnet and Wessler 1997). While AS in humans is known to be common, AS in plants was not extensively observed and previously thought to be rare (Brett et al. 2002). Recent estimates of AS in plants based on genome data sets (Table 1) suggest that it occurs more frequently than originally expected. However, the abundance of AS in plants may be underestimated, because these analyses were based only on EST data that may contain artifacts such as genomic contamination and single aberrant events, and are generally biased (Xing and Lee 2006). For instance, EST coverage in humans is far more extensive than in Arabidopsis or rice, and it covers numerous tissue and cell types. Completion of the Arabidopsis and rice genome sequences and the availability of EST/cDNA sequences enabled initial genome-wide examination of AS events in these plants (Table 1).
Summary of recent genome-wide analyses of AS in plants
The abundance of Arabidopsis and rice AS revealed by these studies varies with the size of the EST collection analyzed. This confirms that the depth of the sequence data sets impact the discovery of AS, which implies that the relatively poorly characterized plant EST sets are likely underestimating the importance of AS in the plant kingdom. Two recent studies predict that over 20% of Arabidopsis and rice genes with EST/cDNA evidence undergo AS (Campbell et al. 2006; Wang and Brendel 2006a). While substantially less than that observed for human genes, the observation that 1/5 of plant genes have EST evidence predictive of AS argues that AS in plants is not rare. Furthermore, this predicted rate (∼20%) is consistent between rice (a monocot) and Arabidopsis (a dicot) that diverged from a common ancestor 140–200 million years ago (MYA) (Wolfe et al. 1989; Sanderson 1997; Chaw et al. 2004), and have similarly sized EST collections (∼300,000) (Wang and Brendel 2006a), suggesting that AS is likely common in most plant species. Recent analysis of genome data from maize (Table 1), moss (Rensing et al. 2008), and three legume species (Wang et al. 2008) further support this view.
Factors influencing the predicted rates of AS in plants
It is likely that the number of alternatively spliced genes identified in plants will increase with larger and more comprehensively sampled tissue-specific transcriptome sequence collections. In addition, plants respond to the environment in more diverse and complex ways than do animals, and only a small proportion of these conditions have been addressed in EST sequencing projects. It is worthy to note that the rice orthologs of 40% of the Arabidopsis genes demonstrated to encode multiple splice isoforms are themselves alternatively spliced (Wang and Brendel 2006a). Although the specific AS events are not necessarily conserved, this supports some splice variants having a biological role, rather than simply reflecting RNA processing errors. While conserved AS is observed in both the plant and animal kingdoms, and conserved AS has been used to imply functional significance (Kalyna et al. 2006; Lareau et al. 2007; Irimia et al. 2008; Wang et al. 2008), some cases of conserved AS may reflect conserved sequence features that form mRNA structures that hinder mRNA processing. This may increase the possibility of recovering an incompletely processed message that may be mistaken for AS.
The proclivity for genome duplication and/or polyploidization observed in plants may have had an influence on AS abundance. Many eukaryotic genomes have undergone whole or partial duplication events in their evolutionary history, and like AS, gene duplication followed by divergence is another potential source of proteomic functional diversity. Genome duplication seems to be commonly involved in promoting functional diversity within plant genomes (Moore and Purugganan 2005; Ober 2005). Su et al. (2006) observed a negative correlation between gene family size and AS in several model animal genomes and suggested that a transition from AS diversity to functional divergence of duplicated genes occurs early after duplication, resulting in isoform loss. Most plant genomes have undergone multiple genome duplication events during their evolutionary history (Paterson 2005), and some (e.g., soybean) have experienced very recent duplication events (Blanc and Wolfe 2004). It is supposed that AS arose early in eukaryotic evolution and that at least a simple form of AS was present in the unicellular ancestor of plants, animals, and fungi (Irimia et al. 2007). Therefore, the prevalence of duplication in the evolutionary history of plant genomes may have contributed to the lower abundance of AS in plants relative to animals. At least two examples of AS loss following gene duplication have been found in plants (Cusack and Wolfe 2007; Rosti and Denyer 2007). Within the common ancestor of mangrove and poplar, the gene encoding chloroplast ribosomal protein RPL32 was transferred to the nuclear genome and inserted into the last exon of a Cu-Zn superoxide dismutase (SOD). The chimeric gene can be processed by AS to produce either a transcript identical in structure to the original Cu-Zn superoxide dismutase mRNA, or one in which exons 1–7 of SOD were spliced onto a novel exon corresponding almost exactly to the whole RPL32 coding region. After its divergence from mangrove, the chimeric gene was duplicated, resulting in loss of AS and subfunctionalization: the daughter genes encode either RPL32 or SOD (Cusack and Wolfe 2007). The genes that encode small subunits (SSU) of ADP-glucose pyrophosphorylase in grasses provide a second example. There are two types of gene. One type encodes two SSU proteins through AS and is found widely in grasses, with the exception of maize. In maize, two separate genes, bt2 and L2 (also known as agpsl1), are known to have the same role as the alternatively spliced type. Rosti and Denyer (2007) demonstrated that bt2 and L2 are paralog genes that arose as a result of the allo-tetraploidization of the maize genome. bt2 and L2 derive from an ancestral alternatively spliced gene orthologous to that found in other grasses. After duplication, the bt2 and L2 genes diverged in function, and each took one of the two functions of the ancestral gene.
Plants and animals differ in their preferred AS types
In addition to fewer genes exhibiting AS than in animals, plants preferentially utilize different AS mechanisms. The most abundant human AS event is exon-skipping (42%) (Fig. 1). The second most abundant AS events in human are alternative donor/acceptor, while intron retention is the least common (Kim et al. 2007). In contrast, ∼40% of the AS events observed in Arabidopsis and rice are intron retention (IR) isoforms (Fig. 1; Ner-Gaon et al. 2004), while only 9% of human AS events are this type (Fig. 1; Kim et al. 2007) and exon-skipping is relatively rare in plants. These differences suggest that the mechanism of splice site recognition may differ between plants and animals (intron definition vs. exon definition, see below). Evidence suggests that splice mechanisms are closely tied to gene structure (McGuire et al. 2008), and perhaps differences between plant and animal splice regulatory components also influence the splicing mechanisms.
Plants and animals differ in their gene structure and may differ in their splice regulatory components
Global analysis of Arabidopsis and rice gene sets (B. Wang, unpubl.) and Arabidopsis and rice gene sets (Korf 2004) reveals C(A)AG/GTAA and TGCAG/G as the consensus sequences for donor and acceptor splice sites, respectively. Aside from some slight position-specific differences in nucleotide frequency, these are noticeably consistent with animal splice sites (Korf 2004; Reddy 2007). Plants and animals show dramatic differences in their gene sizes and structures (Table 2). Human genes show enormous variation in gene size, have large introns (5500 bp average) (Sakharkar et al. 2006), and like other vertebrates, relatively short exons (170 bp average) (Sakharkar et al. 2006). In contrast, plant genes are generally smaller. Analyses of genes with known structures from rice and Arabidopsis indicate that plant exons are slightly larger than those in human, while their intron lengths, similar to those in yeast, Drosophila, and C. elegans, are substantially shorter (Deutsch and Long 1999); approximately 50%∼70% introns are ≤150 bp in plant genes (Wang and Brendel 2006a). In addition, the branchpoint site located upstream of the 3′ splice site is only loosely conserved in plants, and the polypyrimidine tract located between the 3′ splice site and the branch site is absent, being replaced by a U-rich sequence (Reddy 2001; Jurica and Moore 2002).
Summary of average gene size, average exon size, and average intron size across several species
The spliceosome is well understood in metazoans (Staley and Guthrie 1998; Zhou et al. 2002; Ast 2004; Lev-Maor et al. 2007), but plant spliceosomes have not been isolated, and thus, their exact composition is not known (Reddy 2007). However, most animal spliceosome components appear to be well conserved within plants. A total of 74 snRNAs and 395 spliceosome and spliceosome-associated protein-encoding genes are predicted by sequence similarity to reside within the Arabidopsis genome (Wang and Brendel 2004, 2006b) suggesting that plant and animal spliceosomes are likely similar. The mechanism of splicing is thought to be well conserved between plants and animals (Lorkovic et al. 2000; Reddy 2001; Jurica and Moore 2002), but the known splicing regulatory sequences (5′ and 3′ splices sites, branch points, and intron–exon sequence elements, see above) lack sufficient information on their own to direct the splicing machinery to the correct splice sites (Lim and Burge 2001).
Specificity is conferred through the interaction between splicing regulatory proteins and additional cis-sequence elements. These elements, referred to as exonic or intronic splicing enhancers (ESEs or ISEs) and exonic or intronic splicing silencers (ESSs or ISSs), are located within the exon or adjacent introns at variable distances from the splice site, and have been identified in several mammalian genes, and direct splice-site choice (Matlin et al. 2005). In addition, a collection of potential Arabidopsis ESE sequences have been recently described (Pertea et al. 2007).
In human, the best-characterized splicing cis-regulatory elements are ESEs and ESSs. In addition to their roles in constitutive splicing, ESEs and ESSs play roles in regulating AS, which is regulated in different developmental stages and tissues (Black 2003). The selection of correct splicing variants is believed to be coordinated by multiple and potentially overlapping exonic and/or intronic splicing enhancers and suppressors (Cartegni et al. 2002; Ladd and Cooper 2002). These elements act by recruiting protein factors that interact with components of the core splicing machinery (Wu and Maniatis 1993; Kohtz et al. 1994) to affect the splicing process. There are two general classes of splicing regulatory proteins that have RNA-binding domains specific to splicing regulatory sequences: members of the hnRNP (heterogeneous ribonucleoprotein) protein family (Weighardt et al. 1996), and the serine–arginine-rich SR proteins (Schaal and Maniatis 1999). SR proteins are essential for splicing as well as spliceosome assembly (Bentley 2002) and have demonstrated tissue-specific patterns of expression and different sequence specificity (Liu et al. 1998). Some hnRNP proteins have been demonstrated to silence splicing (Hastings and Krainer 2001; Caceres and Kornblihtt 2002; Szeszel-Fedorowicz et al. 2006). ESEs function by recruiting members of the serine–arginine protein family (Wang et al. 2004), which interact with one another, pre-mRNA, or spliceosome components to enhance recognition of adjacent splice sites (Wang and Brendel 2004). In contrast, ESSs have been demonstrated to inhibit the use of adjacent splice sites, often interacting with members of the hnRNP family (Zheng et al. 1998; Zhu et al. 2001; Wang et al. 2004). Intriguingly, there are some human splicing factors for which homologs were not identified in the sequenced Arabidopsis genome, and there has been an apparent expansion of splicing regulators in Arabidopsis relative to humans (Wang and Brendel 2004).
Studies in metazoan splice-site selection demonstrate that a 5′ splice-site mutation commonly results in skipping of the preceding exon, suggesting that the exon is initially recognized by the interaction of the splicing machinery with the splice site (Berget 1995). In contrast, mutating splice sites in Schizosaccharomyces pombe leads to intron retention (Romfo et al. 2000). Additionally, Talerico and Berget (1994) point out that many Drosophila genes contain short introns that lack polypyrimidine tracts (similar to plant introns), and suggest that the intron, rather than the exon, serves as the initial unit of recognition during spliceosome assembly in flies. These analyses predict two models for spliceosome assembly: the intron-definition and the exon-definition model (Berget 1995), which splice short and long introns, respectively (Berget 1995; Lorkovic et al. 2000; Wang and Brendel 2006a). Intuitively, inaccurate splicing under the intron-definition or exon-definition model would result in intron retention or exon-skip events, respectively. Consistent with this is the observation that over half of the observed plant AS events are intron retention (Fig. 1; Ner-Gaon et al. 2004; Wang and Brendel 2006a) while exon-skipping predominates AS events in vertebrates (Gupta et al. 2004). The observed increase (∼5%) of exon-skip events in rice compared with Arabidopsis may reflect the presence of more long introns in rice than in Arabidopsis (Wang and Brendel 2006a).
As mentioned previously, some splice isoforms contain a premature termination codon (PTC) and many such transcripts are often not translated, but are targeted for nonsense-mediated decay (Maquat 2004). Analysis of rice and Arabidopsis AS events suggest that greater than one-third of all events may be coupled with NMD based on the presence of a PTC, and ∼50% of all intron retention events may be subject to NMD. Because NMD is a surveillance mechanism that targets and removes mRNA containing PTCs (Maquat 2004), and because it has been suggested that NMD can be used by the cell to regulate gene expression (Lejeune and Maquat 2005), intron retention and concomitant NMD may serve as an important regulatory mechanism in plants. Unfortunately, it is difficult to distinguish biologically relevant intron retention from transcriptome artifacts originating from genomic DNA contamination or incompletely processed transcripts. It is possible that some transcripts may process slowly, and isolating total rather than cytoplasmic RNA may increase the incidence of recovering an immature message that may be mistaken for a mature isoform.
Because intron retention occurs infrequently in mammals (Gupta et al. 2004), its functional significance is not well known and so remains an open question. However, it has been implicated in important processes in animals such as the autoregulation of SR gene expression in human and mouse (Lareau et al. 2007). Likewise, the low frequency of intron retention in animals cannot be taken to be a reliable predictor of its frequency in plants, or its relevance to plant growth and development. Ner-Gaon et al. (2004) demonstrated that some intron-retaining messages could be copurified with ribosomes, thus confirming nuclear export and supporting intron retention as a valid AS mechanism. Additionally, intron retention is involved in important plant processes, such as floral development, where AS of Arabidopsis FCA pre-mRNA regulates the switch from the vegetative to the reproductive phase (Quesada et al. 2003; Razem et al. 2006; Reddy 2007).
Evidence that plant AS has a biological role
The majority of known plant AS events have not been functionally characterized, but several lines of evidence suggest that AS has a biological role. As suggested by Reddy (2007), the majority of intron-containing genes should produce splice variants if most isoforms resulted from random splicing errors. However, AS is predominant in some gene families, while absent in others. Furthermore, several studies link the occurrence of AS to tissue-specific and/or developmental cues, and alternatively spliced isoforms (specifically intron retention, which is abundant in plants) have been associated with ribosomes (see above). Results of the few functional analyses that have been conducted indicate roles for AS in plant processes such as some metabolic pathways (Gorlach et al. 1995), catabolic pathways (Kopriva et al. 1995), and mRNA processing (Golovkin and Reddy 1996; Kalyna et al. 2006), and AS impacts many important plant process such as photosynthesis, defense response, flowering, and cereal grain quality (for review, see Reddy 2007).
Conservation of alternatively spliced genes between evolutionarily distant plant species is further evidence that AS products may play biologically significant roles in plants. Wang and Brendel (2006a) determined that 47% of Arabidopsis genes with at least one AS isoform (1988 of total genes exhibiting AS) were found to have potential orthologs in rice that were also alternatively spliced, and 58% of these orthologous gene pairs conserved the same AS type. Extending this type of analysis to identify specific AS events that are conserved among multiple species will identify those “ancestral” candidate AS events that are more likely to be functionally significant. For example, one of the FCA gene AS events involved in flowering control (see above) is conserved between Arabidopsis and rice (Lee et al. 2005). Computationally identifying conserved AS events requires the identification of robust cross-species orthologous gene sets. Additionally, the splice sites have to be similarly compared and unambiguously paired between orthologs (Fig. 2). Wang and Brendel (2006a) searched for conserved intron pairs that defined conserved AS events, and were able to identify only 41 conserved AS events that include just one exon-skip event. Given the assumption that Arabidopsis and rice diverged 200 MYA, the method of Wang and Brendel (2006a) is probably under-representing conserved events. Careful comparison of loci that have at least one defined exon-skip event in rice to Arabidopsis loci with similar events identifies five unambiguous conserved exon-skip events between rice and Arabidopsis (At1g72050, At3g55460, At4g25500, At4g35785, At5g56140).
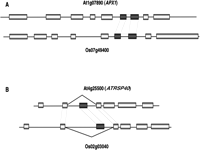
Two examples of AS events conserved between rice and Arabidopsis. (A) A conserved intron retention event associated with Arabidopsis APX1 (L-ascorbate peroxidase) gene and its rice orthlogous gene Os0749400; (B) A conserved exon-skipping event associated with At4g25500 (ATRSP40) gene and its rice orthologous gene Os07g38730. RSp40 is one of SR proteins for splicing regulation. The orthologous exon pairs are in black.
This approach would exclude functionally important AS events that are species specific or recent events that are conserved only among a small group of related species. Including multiple species in the analysis of conserved events might solve the latter problem. For example, AS events important for the development of cereal monocots may be conserved within maize, rice, and sorghum, but absent in dicotyledonous species. Indeed, this approach has successfully predicted conserved alternative splice events within related legume species (Wang et al. 2008). Unfortunately, resolving whether or not many AS events are important for proper gene functioning or simply artifacts requires detailed functional analysis; an overwhelming task.
Directions for future study
Investigate the increased levels of intron retention in plants
AS is common in plants and plays a critical role in many aspects of plant biology. In addition, there are differences between animals and plants in both gene structure and the frequency of AS (see previous) that may reflect important mechanistic differences or evolutionary pressures. Instances of intron retention have been found in 42 eukaryotic organisms studied (McGuire et al. 2008). Why intron retention is so prevalent in plants is not understood, but its abundance provides an opportunity to study its regulation and identify the underlying sequence signals responsible. Sakabe and de Souza (2007) sought to identify sequence features associated with intron retention in humans by comparing cDNAs that represented intron retention as the major splice isoform with cDNAs representing intron retention as a minor splice form. In general, intron retention is associated with weaker splice sites, short intron lengths and higher expression levels, and a reduction in ESS signature sequences. However, this study did not permit the discovery of novel regulatory sequences. Sequence features that regulate the inclusion/exclusion of human alternatively spliced exons have been identified by using an in vivo splicing reporter system (Wang et al. 2004) and by contrasting exons with strong vs. weak (nonconcensus) 5′ or 3′ splice signals. Another powerful method for identifying alternative splice regulatory sequences is to contrast alternatively spliced exons conserved in human and mouse with exons that are constitutively spliced (Sorek and Ast 2003; Yeo et al. 2005; Goren et al. 2006). Because selective pressure should cause nonfunctional sequences to evolve faster than functional sequences, splicing regulatory sequences should be conserved within human–mouse orthologous alternatively spliced exons, and these sequences should be enriched within these exons relative to constitutively spliced exons. Although the vast majority of alternatively spliced exons are species specific (Yeo et al. 2005), the prevalence of exon-skip events in vertebrates enables the identification of enough orthologous events to perform comparative analysis. The relatively low frequency of intron retention events in mammals precludes such an analysis. In contrast, while exon-skipping events in plants are rare, intron retention events are very common, suggesting that a similar computational analysis might be possible using plant intron retention AS events. If the majority of intron retention events serve a functional purpose, then several events should be conserved throughout multiple plant species.
Investigate the roles of plant splice regulators
Flowering plants have more SR proteins than other eukaryotes (Reddy 2007). SR proteins play roles in splice site choice and spliceosome assembly, they are differentially expressed, and most SR proteins display distinct as well as overlapping expression patterns (Wang and Brendel 2006a). Extensive phosphorylation of SR proteins has been documented, and perhaps this influences their RNA-binding activity, interaction with other protein components, and localization, which may in turn affect their splicing activity (Bourgeois et al. 2004; Shen and Green 2006; Reddy 2007). Given the complexity of animals and the abundance of AS events, it is likely that a given animal SR protein is active within more cell types than are plant SR proteins, or that animals have additional signals or regulative proteins that have yet to be discovered. Another role for an increase in SR protein diversity in plants over animals could relate to the fact that plants are required to respond and adapt to environmental changes in order to ensure survival; perhaps some SR proteins respond to environmental cues.
Determine the relationship between AS and plant stress response
Several reports demonstrate that AS can be influenced by abiotic stresses (i.e., temperature fluctuations) (Marrs and Walbot 1997; Palusa et al. 2007; Reddy 2007; Tanabe et al. 2007), and biotic stresses (i.e., pathogen infection) (Iida et al. 2004; Attallah et al. 2007; Reddy 2007). Zhang and Gassmann (2007) demonstrated that the Arabidopsis disease resistance gene, RPS4, produces multiple transcripts via alterative splicing. Regulation of RPS4 function was demonstrated to occur at multiple levels and included dynamic changes in AS that adjust the transcript isoform ratios during the resistance response. Regulation of alterative splicing in this case is thought to fine-tune resistance gene activity, and may limit damage inflicted by activated RPS4 protein. The wheat DREB2 homolog (Wdreb2) is a transcription factor that is activated by several abiotic stresses to produce three alternatively spliced transcripts that are differentially expressed. Under cold and drought/salt stress conditions, the amount of WDREB2 transcription factor is differentially controlled by the level of transcription and AS. Wdreb2 is regulated through two independent pathways, ABA (abscisic acid) dependent (drought and salt response) and ABA independent (cold response), suggesting that significant changes in splicing factors occur under abiotic stress conditions and that these affect AS patterns of the Wdreb2 transcripts (Egawa et al. 2006).
Further support for AS having a role in stress response comes from a study performed by Xiao et al. (2005) that tested the functionality of “hypothetical” genes in Arabidopsis that were computationally predicted during genome sequence annotation, but lacked support from Arabidopsis EST/cDNA or cross-species protein homologs. Attempting to amplify a selection of these genes from diverse cDNA collections that included several obtained from stressed (abiotic) tissues resulted in confirmation of over 50% of the genes tested. Interestingly, the rate of AS observed within these “hypothetical” genes was greater than the rate observed genome-wide. This result is likely due to the diverse pool of tissues and biological conditions used for cDNA, as well as deep sequence sampling of this group of transcripts. However, it is also possible that “hypothetical” genes, as a class in plants, exhibit a greater rate of AS. The predicted protein products of a subset (357) of the 399 chromosome 2 loci amplified and sequenced by Xiao et al. (2005) were aligned by BLAST to the GenBank nonredundant protein collection (release 164). A total of 35% (126) of the Arabidopsis hypothetical proteins were not found to have matches to non-Arabidopsis proteins within NRaa, suggesting that at least a portion of the hypothetical Arabidopsis genes analyzed by Xiao et al. (2005) may be Arabidopsis specific.
The absence of cross-species protein homologs implies that these are either species specific or evolving rapidly. It has been demonstrated that disease resistance loci in plants are highly divergent between closely related species, an indication that they may evolve rapidly to remain effective (Bishop et al. 2000). Therefore, genes required to ameliorate various stress responses or respond to changing environmental conditions may also evolve rapidly, and the acquisition of alternative splice isoforms may provide an additional mechanism to facilitate such behavior. The concept of a simple genetic change that changes protein sequence to result in a “molecular hopeful monster” was proposed by Kramer et al. (2006) to describe the events leading to evolution of the type II MADS box genes involved in flowering. Kramer et al. (2006) suggest that the evolution of the APETALA3 lineage was influenced by a single-base deletion event that produced novel protein sequence, which was conserved almost immediately on the basis of what appears to be a rapidly created new function. Similarly, the observation of increased AS under stressed conditions and the abundance of intron retention events may reflect an underlying mechanism to create novel function acting on many other genes in plants. Stress may regulate splicing by multiple mechanisms, including alteration of the population or distribution of splicing factors, or induction of changes in phosphorylation status or expression of SR proteins. While the actual mechanisms are not well understood, continued investigation will lead to a better understanding of how plants respond to stress and a changing environment, which will impact future improvement programs.
Investigate the relationship between transposable elements and AS
Many crop species exhibit vast molecular and phenotypic variation among landraces and cultivars. This variation drove ancient and modern plant breeding, which has placed intense selection pressure upon cultivated species. Interestingly, maize displays enormous genetic diversity, and this genetic diversity may influence, or be influenced by sequences involved in regulation of AS. Comparative analysis of sequences from homeologous regions of maize inbreds examined nonhomology at multiple loci in the maize genome and demonstrated significant differences in LTR-retroelement number and composition surrounding conserved genes (Brunner et al. 2005). These elements are usually inactive, but may act as enhancers effecting expression of neighboring genes under stress conditions, and different repetitive sequence environments may have profound effects on the temporal or spatial regulation of expression (Brunner et al. 2005). Transposable elements are associated with new exon creation (Lev-Maor et al. 2007; Sela et al. 2007) and transposable element-derived exons often are alternatively spliced (for review, see Xing and Lee 2006). In plants, transposable elements that carry host fragments such as the Pack-MULES (Jiang et al. 2004) and Helitrons (Gupta et al. 2005; Lai et al. 2005; Morgante et al. 2005) can create novel chimeric genes. Recently, a similarly behaving element in soybean, Tgm-express, has demonstrated the ability to insert gene fragments into downstream genes through complex AS (Zabala and Vodkin 2007).
Plant breeding serves to create lines that are better suited for agriculture, and crop species have been bred for multiple environments and agronomical characteristics. Given the prevalence of AS in plants, it is not unreasonable that there may be AS isoforms involved in conferring traits selected for during breeding programs. Interestingly, Ner-Gaon et al. (2007) demonstrated that cereals show ∼20% differences in their AS rates between cultivars, and concluded that these AS rate differences may be correlated with niche specialization resulting from domestication in different geographical regions. A potential limitation of this analysis is that all comparisons were made between EST collections, and none of these collections is likely to be complete. In addition, one has to use caution when comparing ESTs between cultivars, or aligning ESTs of one cultivar to a genomic reference from another, to ensure that transcript structural differences are true AS events rather than polymorphic differences between genotypes. At any rate, a global understanding of the AS potential between crop cultivars will complement ongoing molecular map construction and association studies in dissection and understanding complex agricultural traits such as yield.
Future analysis of alternative splicing in plants
The prevalence of AS in plants, differences in abundance and frequency of events between animals and plants, the role of AS in stress response, and the observation that cereal cultivars have vast differences in AS with implications for domestication and trait selection, justify further examination. Recent years have seen increases in DNA sequence data from plant species. Currently there are substantial EST collections for at least 34 plants, while full or draft genome sequences are available or underway for at least 29 plant species. The available sequences come from a diversity of plant species (Fig. 3), many of which are valuable crops for bioenergy and agriculture. This sequence resource provides an excellent platform to begin to identify important plant AS events based on their evolutionary conservation, to examine the influence of genome duplication on the evolution of AS, and to discover plant-specific cis elements that regulate AS, particularly intron retention. Of particular interest will be the genome sequence of Selaginella, a lower plant lacking true leaves and roots that is a clade intermediate between nonvascular and vascular plants, and that of Physcomitrella (moss), which diverged from vascular plants ∼450 MYA and is viewed as a key link to understanding plant genome evolution. Preliminary analysis suggests that ∼21% of Physcomitrella genes are alternatively spliced (Rensing et al. 2008), which is consistent with rice and Arabidopsis. As expected, the proportion of genes/events that undergo exon-skipping in moss is rare relative to human levels, but consistent with observed levels in Arabidopsis and rice. However, unlike other plants studied, the predominant type of AS event in moss is not intron retention, which accounts for only 25% of moss AS events, in contrast to ∼40% of events in Arabidopsis and rice (Fig. 1; Ner-Gaon et al. 2004).
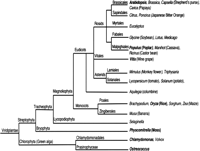
A dendogram approximating the phylogenetic relationships between plant species with completed genome sequencing projects (bold) or those currently ongoing.
Using EST collections to investigate AS is limited by the comprehensiveness of the EST collection, which is affected by library choice and depth of sequencing. Johnson et al. (2003) determined that 20% of human exons are not represented by any EST, and another 11% are represented by only a single EST, suggesting that the available EST data are insufficient for detection of AS events involving as many as 31% of the exons represented in the human RefSeq cDNA collection. This problem is magnified in plants, where the best-sampled transcriptome (Arabidopsis) is represented in an EST collection ∼1/7 the size of that for human. Furthermore, it is only possible to determine the tissue type of origin from the GenBank records for <10% of the Arabidopsis genes for which there is EST or cDNA-based evidence of AS, meaning that important information about the tissue or developmental specificity of a given splicing event may be lost for much of these data.
Exon junction (EJ) DNA microarrays are very sensitive indicators of AS (Johnson et al. 2003; Blanchette et al. 2005; Ule et al. 2005). Similar array-based assays will enable identification of many novel cases of AS in plant species, and provide a tool for assessing AS across many different genotypes, growth conditions, and tissue types. A potential weakness of exon-junction microarrays is their failure to detect all types of splicing events such as intron retention, which is common in plants; however, probes designed to detect unspliced intron sequence can be included in the probe design. Another weakness is that probes are designed to genes that have been defined by ESTs or identified during genome annotation efforts, thus assessing the AS potential of previously defined genes and exons. Whole-genome tiling arrays composed of probes designed to overlap across the genome (for review, see Yazaki et al. 2007) can define novel AS without having identified every gene up front. While these have been used in Arabidopsis (Ner-Gaon and Fluhr 2006), only Arabidopsis and rice of the 29 plant genome sequencing projects finished or underway (Fig. 3) have “complete and contiguous” coverage, while the remainder are highly gapped draft sequences. One solution is to construct exon-junction arrays modified to identify alternate donor/accepter sites and intron retention events. While draft genome sequences lack contiguity, much of the euchromatic regions are represented, and tiled oligo arrays could be constructed to represent these gene-rich regions. These arrays will not be completely comprehensive, but may be superior to exon-junction arrays that depend largely on the accuracy of the underlying gene models during construction.
Increasing plant transcriptome sequence collections will be vital to accurately define gene features during annotation, and new high-throughput genome sequencing tools, such as the 454 Life Sciences (Roche) pyrosequencer, have been applied to both animal (Bainbridge et al. 2006) and plant transcriptome sequencing (Cheung et al. 2006; Emrich et al. 2007). Plant transcriptome sequencing with 454 has successfully identified novel, tissue-specific, and/or rare plant transcripts (Emrich et al. 2007). A po-tentially promising application of high-throughput pyrosequencing will be to sequence plant tissue-specific cDNA pools that have been enriched for AS isoforms (Watahiki et al. 2004 Venables 2006). Coupling 454 sequencing with AS-enriched cDNAs isolated by laser capture microscopy will allow rapid identification of tissue-specific AS events within developmentally important cell populations such as the shoot apical meristem—a stemcell population from which all above ground plant tissue originates. Identification of additional AS isoforms by methods such as these will guide microarray probe design to investigate their spatial and temporal representation, and the effects of various environmental conditions upon them, such as abiotic and biotic stresses.
Concluding remarks
Studies of AS in plants are likely to benefit tremendously from previous and ongoing work in other systems. Additionally, studies of AS in plants will provide unique opportunities to investigate heretofore-unstudied questions, and are likely to contribute important and groundbreaking discoveries to the field. The availability of genome resources for ancient and higher plants will identify AS events that have been conserved through multiple species, permit investigation of the evolution of AS in plants and the impact that plant genome dynamics (duplication and polyploidization) have on AS, elucidate plant-specific regulation of AS, and enable comparative genomic analysis of intron retention to further identify cis regulatory elements involved in these events. Further exploration of AS in plants with microarray platforms will permit large-scale investigation of the effects of various environmental conditions on the regulation of AS, and reveal its role in stress response. Extending these analyses to important bioenergy and crop plants such as Zea mays will address the influence of AS on domestication and trait selection, which will be invaluable for future breeding programs.
Acknowledgments
We thank Richard Jorgensen (University of Arizona) and Ruth Davenport (Danforth Center) for productive discussions during the preparation of this manuscript, and Bing-Bing Wang (University of Minnesota) for communicating results of recent global analysis of alternative splicing in legumes. Research in the laboratory of W.B.B. is supported by grants from the National Science Foundation (DBI-0501758); The National Research Initiative (NRI) Plant Genome Program of the USDA Cooperative State Research, Education and Extension Service (CSREES); and the Donald Danforth Plant Science Center.
Footnotes
-
↵3 Present address: Department of Botany and the Genetics Institute, University of Florida, Gainesville, FL 32611, USA.
-
↵4 Corresponding author.
↵4 E-mail bbarbazuk{at}danforthcenter.org; fax (314) 587-1378.
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.053678.106.
- Copyright © 2008, Cold Spring Harbor Laboratory Press








