
Localization of a long-range cis-regulatory element of IL13 by allelic transcript ratio mapping
Abstract
It appears that, for many genes, the two alleles possessed by an individual may produce different amounts of transcript. When such allelic differences in transcription are observed for some individuals but not others, a plausible explanation is genetic variation in the cis-acting elements that regulate the gene in question. Here we describe a novel analytical approach that uses such observations, combined with genotyping data from the HapMap project, to define the genomic location of cis-acting regulatory elements. When applied to the human 5q31 chromosomal region, where complex regulatory mechanisms are known to exist, we demonstrate the sensitivity of this approach by locating a highly significant cis-regulatory element operating on IL13 at long range from a position 250 kb upstream from the gene (P = 2 × 10−6). As this method is unaffected by other sources of variation, such as environmental and trans-acting genetic factors, it provides a tractable approach for dissecting the complexities of genetic variation in gene regulation.
It is now possible to identify regions of the human genome that determine individual variation in gene expression, by combining the powerful techniques of genome-wide expression profiling, genome-wide SNP genotyping, and genetic association analysis (Cheung et al. 2005; Stranger et al. 2005). The next step is to use such findings to identify functional elements that regulate gene expression. Here we explore a way of mapping genetic determinants of gene expression that is complementary to genetic association analysis and may in some circumstances be more robust.
Imagine a functional polymorphism (F) that affects the expression of gene X. The aim of genetic association analysis is to detect a significant difference in gene X expression between individuals of different F genotypes. However, if the expression of gene X is affected by multiple genetic or environmental factors, these are potential confounders that might obscure or distort the association of gene X expression with F genotypes.
The number of potential confounders can be reduced by using allelic transcript quantification to focus on cis-regulatory effects, where F acts only on the copy of gene X that lies on the same chromosome (Goldsborough and Kornberg 1994; Knight et al. 2003; Buckland 2004; Pastinen and Hudson 2004; Pastinen et al. 2004, 2005). This approach can be applied if there exists some polymorphism (typically a SNP) on the transcript of gene X (here called transcript marker T) that allows us to compare the abundance of transcripts carrying different T alleles within heterozygous individuals. For each heterozygous individual, we calculate the ratio of the different T alleles, here called the allelic transcript ratio (ATR). We would expect ATR measurements to be largely unaffected by trans-acting genetic factors and to be relatively robust against environmental confounders, because allelic comparisons are always made within individuals, not between individuals. The purpose of the present study was to develop a simple way of using ATR measurements to map the genomic location of F.
Results
We use as an example chromosome 5q31, a region that is rich in immune genes and has been implicated in several common diseases (Marquet et al. 1996; Rihet et al. 1998; Rioux et al. 2001). Patterns of gene expression in this region are determined by complex long-range regulatory mechanisms, making it an interesting test case (Ansel et al. 2003; Fields et al. 2004; Spilianakis and Flavell 2004). We performed allelic transcript quantification on the genes CSF2, SLC22A4, RIL (currently known as PDLIM4), IRF1, IL13, and IL4, using immortalized lymphoblastoid B-cell lines from 16 unrelated individuals. The most striking deviation from an ATR of 1 was observed for rs20541, a transcript marker of IL13. A strong effect was observed in two out of eight individuals who were heterozygous for rs20541, and this was confirmed by further experiments using three other transcript markers of IL13 that were in phase with rs20541 (Fig. 1A). This observation raised the possibility that some but not all individuals who are heterozygous for the transcript marker rs20541 are also heterozygous for a functional polymorphism F with cis-regulatory effects on IL13. In an individual who is homozygous for cis-regulatory polymorphism F, we expect an ATR of 1, while in an individual who is heterozygous for F, we expect an ATR that can be either above or below 1, depending on the phase relationship of F with the transcript marker T (Fig. 2A).
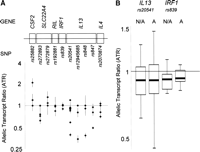
(A) Allelic transcript ratios (±SEM) observed in 16 CEPH cell lines, for 10 transcript SNPs covering six genes across the 5q31 cytokine cluster. Four transcript SNPs in complete linkage disequilibrium across IL13 demonstrate consistent ATRs in all cell lines tested, suggesting that these results are real and not a product of asymmetrical primer affinity or extension reaction bias. (B) Allelic transcript ratios observed in 22 CEPH cell lines used in the HapMap resource show more variation in ATR for IL13 (using rs20541 as transcript SNP) than for IRF1 (using rs839), in both non-activated (N/A) and activated (A) cell aliquots. In each plot, the median allelic transcript ratio, the interquartile range, and the full range are shown for the group of cells assayed.
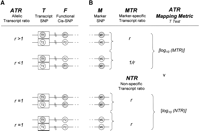
(A) If a cis-acting polymorphism F is heterozygous, the observed ATR at transcript Marker T will be expected to deviate from 1. The direction of ATR deviation in a given cell line will depend on the phase relationship between F and T. In the example shown in A, allele F1 up-regulates gene expression. The observed ATR, T1:T2, is therefore >1 when it represents F1:F2 and <1 when it represents F2:F1. (B) In individuals who are heterozygous for M, the ATR is phase-corrected with respect to M so that in all cases it represents the relative abundance of transcripts derived from the M1 chromosome compared to the M2 chromosome. We call the phase-corrected ATR the marker-specific transcript ratio (MTR) and for a marker SNP M in high LD with the functional SNP F, but in low LD with the transcript SNP T, MTR values will cluster where ATR values may not. In individuals who are homozygous for M, ATR values provide a measure of experimental variation that cannot be attributed to the allele-specific effects of M. We call this the non-specific transcript ratio (NTR). The ATR Mapping Metric is a two-tailed t-test with unequal variance for the comparison ATR values in the two groups MTR and NTR. The Log10s of all ATR values are used in the t-test. A significant P-value implies that the variation seen in the observed ATR is attributable to the allele-specific effects at M.
The above findings are typical of allelic transcript quantification studies that have been reported for a growing number of genes (Pastinen et al. 2004). Cis-acting regulation can act over hundreds of kilobases (Lettice et al. 2002; West and Fraser 2005), and this adds to the difficulty of identifying the functional element precisely. Conventional linkage disequilibrium mapping approaches cannot be applied to allelic transcript ratio data, but by inspecting haplotypes across the genomic region surrounding T, we can identify SNP markers that are correlated with allele-specific expression (Pastinen et al. 2005). The problem we address in this study is how to most effectively screen across a wide genomic region to determine the location of F.
We propose a simple method of allelic transcript ratio mapping. For any SNP marker M, with alleles M1 and M2, in the genomic region of T we can measure ATR in a group of individuals who are heterozygous for T, and determine pairwise haplotypes between M and T in each individual. In individuals who are heterozygous for M, this haplotypic information allows us to establish the phase of the ATR with respect to M, and to derive a marker-specific transcript ratio (MTR), that is, the relative abundance of transcripts derived from the M1 chromosome compared to the M2 chromosome (Fig. 2B).
In individuals who are homozygous for M, the distribution of ATR values provides a measure of experimental variation that cannot be attributed to the allele-specific effects of M. We call this the nonspecific transcript ratio (NTR). We then derive some measure of the statistical significance of the MTR taking the NTR into account—we call this the ATR mapping metric. The P-value of a t-test comparison of the MTR and NTR distributions is used here as a simple example of a mapping metric. By charting the ATR mapping metric for a set of SNP markers across the genomic region surrounding T, we can build up a picture of the location of F.
To test this method on the IL13 locus in a model system, we used immortalized lymphoblastoid B-cell lines derived from 90 individuals of European ancestry that form part of the HapMap project (Altshuler et al. 2005). Allelic transcript quantification of the IL13 transcript polymorphism rs20541 was carried out in 22 unrelated individuals who were heterozygous for this SNP, both in non-activated cells and after activation with PMA/ionomycin. Individuals with extreme ATR values were found to give consistent results when the test was repeated on different cell aliquots (Supplemental Fig. 1; Supplemental Table 1). As an experimental control, we studied the IRF1 transcript polymorphism rs839, and observed greater variation in the ATR for IL13 than for IRF1 (Fig. 1B), consistent with our earlier data (Fig. 1A). Genotyping data and pedigree information obtained from the HapMap database (http://www.hapmap.org) allowed us to derive pairwise haplotypes for SNP markers in the region surrounding the IL13 transcript polymorphism. Using this haplotypic information, we calculated MTR and NTR values for each SNP marker, and from a comparison of these values by t-test, we derived a log probability to use as the ATR mapping metric.
We examined 300 SNPs across an ∼1200-kb region centered on IL13 (Fig. 3). This revealed a region located 200–300 kb upstream of IL13 containing nine SNP markers with marked differences between MTR and NTR in non-activated cells (t-test, P = 2 × 10−6; permutation analysis, P < 1 × 10−5; Mann Whitney, P = 4 × 10−4) (Fig. 3A) and somewhat less marked differences in activated cells (t-test, P = 2 × 10−4; permutation analysis, P < 1 × 10−3; Mann Whitney, P = 7 × 10−3) (Fig. 3B).

Allelic transcript ratio mapping for 300 SNPs across ∼1200 kb centered on IL13 revealed a region located 200–300 kb upstream of IL13 containing nine SNP markers with marked differences between MTR and NTR in both (A) non-activated cells and (B) activated cells. These nine SNPs fall within a genomic block of high linkage disequilibrium spanning APXL2 (SHROOM1), GDF1, QP-C (UQCRQ), LEAP2, and AFF4 and are all unique to (C) haplotype A4, which has an average haplotype-specific transcript ratio (HTR) of 1.28 (95% CI, 1.19–1.37). (D) Haplotype analysis of the proximal 100-kb region containing IL13, IL4, and RAD50 highlighted haplotype B4 with an HTR much more apparent in activated cells (HTR = 0.78, 95% CI, 0.65–0.91) than in non-activated cells (HTR = 0.93, 95% CI, 0.76–1.10).
These nine SNPs fall within a genomic block of high linkage disequilibrium (Fig. 3B), and their minor alleles appear to be specific for a long-range haplotype spanning the genes APXL2 (currently known as SHROOM1), GDF1, QP-C (currently known as UQCRQ), LEAP2, and AFF4 (haplotype A4 in Fig. 3C). By analogy with MTR, we can use individuals who are heterozygous for a given haplotype to estimate the allelic expression ratio between that haplotype and all other haplotypes, which we call the haplotype-specific transcript ratio (HTR). Haplotype A4 has an average HTR of 1.28 (95% CI, 1.19–1.37) in non-activated cells and 1.26 (95% CI, 1.15–1.37) in activated cells. The t-test for average HTR in A4 haplotype carriers versus non-A4-haplotype carriers was significant in both resting cells (P = 2 × 10−5), and in activated cells (P = 8 × 10−4).
These IL13 data highlight a crucial feature of ATR mapping, namely, that this method does not require F to be in linkage disequilibrium with T. If F is in strong linkage disequilibrium with T, then ATR values observed in different individuals will tend to be consistently different from 1. If F is not in linkage disequilibrium with T, then ATR values observed in different individuals will range above and below 1, depending on the haplotypic relationship of F and T in the individual being tested, and there may be many ATR values close to 1 arising from individuals who are heterozygous for T but homozygous for F. This has interesting implications for large-scale efforts to use allelic transcript quantification to screen for cis-regulatory factors across the whole genome (Pastinen et al. 2004, 2005). By focusing on loci where the ATR is consistently different from 1, we will detect functional polymorphisms that are in strong linkage disequilibrium with the transcript marker, and this tends to bias the experimental design toward the detection of cis-regulatory factors that operate over a short range. It is important not to reject loci where the ATR ranges widely above and below 1 because, as we have observed for IL13 (Figs. 1B and 3), this may indicate a functional polymorphism that acts on but is not in linkage disequilibrium with the transcript marker, and by investigating such loci we may discover cis-regulatory factors that operate at long range.
Since ATR data are determined by cis-acting regulation and are potentially refractory to trans-acting effects (which will affect absolute expression but not the allelic transcript ratio), genetic diversity on a different chromosome should not influence the variation observed in ATR data for IL13. We therefore applied the ATR mapping metric for the IL13 data (chromosome 5) to 10,000 consecutive SNPs on chromosome 20 (available for the same cell lines from the HapMap resource), to gain further insight into the potential false discovery rate of the test. Because the ATR mapping metric relies on phasing of the transcript SNP with each marker SNP being tested, we “transferred” the IL13 transcript SNP to chromosome 20 “in silico” for this simulation, so that it could be phased with each SNP in the analysis. Of the 10,000 SNPs on chromosome 20, only eight independent loci reached significance at or above the level of that seen for the nine SNPs identified 250 kb upstream of IL13 (Fig. 4), giving a false discovery rate for the ATR mapping metric, at a designated significance level of P < 2 × 10−6, of 1 in 1250 SNPs.
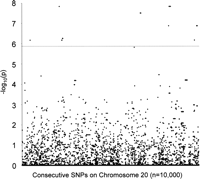
Simulation of the ATR mapping metric for IL13 data on 10,000 consecutive SNPs on chromosome 20 identified only eight independent loci with a significance at or above the level of that seen for the nine SNPs identified 250 kb upstream of IL13, giving a false discovery rate for the ATR mapping metric, at a designated significance level of P < 2 × 10−6, of 1 in 1250 SNPs.
To corroborate the finding of a potential long-range cis-regulator for IL13, we interrogated publicly available human cell line expression data at Gene Expression Omnibus, the NCBI gene expression and hybridization array data repository (Edgar et al. 2002). We sourced quantitative expression data for IL13 from expression profiles performed on 15 CEPH families using a 25K human gene oligonucleotide microarray (Monks et al. 2004). Using only the unrelated individuals in this cohort, we sourced genotype status at rs11739417 for 30 individuals using publicly available genotype data from the HapMap and Perlegen resources. Expression levels are correlated with genotype at rs11739417 in Figure 5, and demonstrate a reduction in mean IL13 production for AG heterozygotes compared to AA homozygotes (t-test, P = 0.016). These independent expression data support the conclusions from ATR mapping, namely, that allele G at rs11739417 (and haplotype A4 to which allele G is unique) is associated with a reduction in IL13 expression.
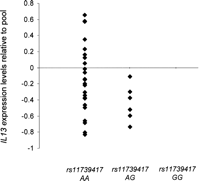
Quantitative expression data for IL13 from expression profiles performed on 15 CEPH families using a 25K human gene oligonucleotide microarray (Monks et al. 2004) are correlated with genotype at rs11739417 and demonstrate a reduction in mean IL13 production for AG heterozygotes compared to AA homozygotes (t-test, P = 0.016). These independent expression data support the conclusions from ATR mapping, namely, that allele G at rs11739417 (and haplotype A4 to which allele G is unique) is associated with a reduction in IL13 expression.
In addition to the 200–300-kb upstream region, there may exist other cis-regulatory polymorphisms that determine IL13 expression. For example, the region ∼400–500 kb downstream from IL13 shows several potentially interesting differences between MTR and NTR (Fig. 3A).
When we focused on the proximal 100-kb region containing IL13 and its flanking genes RAD50 and IL4, single-locus analysis revealed no striking effects, but there were potentially interesting haplotypic effects (Fig. 3D). We analyzed common haplotypic groups across this region in 22 unrelated individuals, after removing the potential confounding effect of the distal haplotype A4 (Fig. 3C) by excluding four individuals known to be A4 heterozygotes. One haplotype (B6) (see Fig. 3D) emerged from this analysis as having a possible effect on gene expression that was much more apparent in activated cells (HTR = 0.78, 95% CI, 0.65–0.91) than in non-activated cells (HTR = 0.93, 95% CI, 0.76–1.10). The t-test for average HTR in B6 haplotype carriers versus non-B6-haplotype carriers was significant in activated cells (P = 9 × 10−3) but not in resting cells. The direction of HTR distortion in B6 haplotype carriers upon activation was consistent in all lines (paired t-test, P < 5 × 10−3).
Discussion
We demonstrate a method of ATR mapping that is capable of detecting functional polymorphism acting on a gene, irrespective of whether it lies proximally to and in high LD with the gene, or operates at long range and is in low LD with the gene. A pattern of unidirectional ATR distortion in the majority of the individuals assayed would be consistent with the presence of cis-regulatory polymorphism in high LD with the gene, whereas bidirectional ATR distortion in a subgroup of the individuals assayed would be consistent with cis-regulatory polymorphism in low LD with the gene.
For IL13 we identify distal cis-regulatory polymorphism highlighted by nine SNPs all common to a single haplotype spanning the genes from APLX2 to AFF4. Complex mechanisms of gene regulation have been described in the 5q31 gene region. A three-dimensional chromatin configuration that approximates regulatory elements and target genes has been proposed at 5q31 (Ansel et al. 2003; Fields et al. 2004; Spilianakis and Flavell 2004), and it is conceivable that the distal cis-regulatory element described here may also integrate into such a multiloop chromatin structure, or may yet be part of a higher-order configuration. Replicating these findings for IL13 in other populations with less extensive LD may localize the effect to a smaller region and help in the focused application of reporter assays and EMSAs in the assessment of transcription factor dynamics.
Single-locus analysis revealed no effects in the region proximal to IL13 despite the presence of a positive haplotype effect. The ATR mapping metric proposed here has the greatest power to detect a cis-regulatory effect in a Marker SNP (M) where there are equal numbers of heterozygotes and homozygotes. Although as a consequence, this metric is less well powered for the detection of proximal cis-polymorphism existing in high LD with the transcript marker (T), it is likely that single-locus analysis would have been successful in identifying the proximal effect seen for IL13 with a greater depth of SNP ascertainment.
In the context of complex gene regulation, it is possible that those cell lines with observed ATR distortion may carry one of a number of independent or interdependent cis-acting components. The two regulatory elements identified with these data appear to be independent effects that show different characteristics, with the distal haplotype affording a constitutive down-regulating effect and the proximal haplotype affording an inducible up-regulating effect. The context specificity of these findings will need to be addressed in primary T-cell experiments.
If the complexity of cis-regulatory mechanisms is high, the observed ATR for an individual may be a composite reflection of multiple cis-acting elements. In this situation, the observed ATRs for a group of cell lines are likely to cover a broad spectrum of values, and the data may prove more difficult to resolve.
For all modes of quantifying cell line expression (Cheung et al. 2003; Pastinen and Hudson 2004), the success of locating an existing cis-regulator will be dependant on many factors, including the complexity of the regulatory mechanisms that exist, the context specificity of those regulatory processes in the cell lines or tissues analyzed, on the stochastic sampling of individuals, on cohort size, and on the haplotypic structure of the population studied (Forton and Kwiatkowski 2006).
Nevertheless, the complexity of regulatory mechanisms that are assayed using the ATR approach is significantly less than that using traditional expression profiling as trans-acting regulation and environmental confounders will not influence ATR measurements. By focusing only on the cis-acting framework of gene regulation, ATR measurement and mapping (Knight et al. 2003; Pastinen et al. 2004, 2005) may provide a tractable approach for dissecting the complexities of variation in gene expression.
Methods
Cell lines
Lymphoblastoid B-cell lines from the CEPH collection (Center d’Etude de Polymorphisme Humaine) were sourced from the Coriell Repository. All 30 CEPH HapMap families and a further eight CEPH family trios were used in analysis. Unrelated individuals only were used in expression assays.
Cell culture and activation
Immortalized lymphoblastoid B-cells were cultured at 37°C in a humidified, 5% CO2 environment in RPMI 1640 with 10% fetal calf serum, 200 mM L-glutamine, penicillin, and streptomycin. Cell density was maintained between 200,000 and 800,000 cells/mL. DNA was extracted from 20 million cell aliquots. For RNA extraction and cDNA synthesis, cell cultures were set up in aliquots of 15 ml in T25 flasks at a cell density of 200,000 cells/mL. Cells were activated at a cell concentration between 600,000 and 800,000 cells/mL (∼10 million cells) using PMA (200 mM) and ionomycin (200 mM). Cells were harvested at 6 h post-activation. Equivalent non-activated aliquots were prepared in parallel.
cDNA preparation
Cell aliquots (15 mL) for RNA were centrifuged, the media was decanted, and the cell pellet was lysed in TRIREAGENT. Cell lysates were stored at −80°C until RNA extraction. RNA was extracted as per the TRIREAGENT protocol, using chloroform, isopropanol, and ethanol precipitation. Total RNA was quantified using UV spectrophotometry (Nanodrop). Samples with an A260/ A280 ratio of 2.0 ± 0.2 were accepted. mRNA was extracted from 20-μg total RNA aliquots using the Dynabeads mRNA purification kit, as per protocol, and cDNA was synthesized using Stratascript reverse transcriptase with oligo(dT) primers. One microliter of cDNA was derived from 100 ng of total RNA. Parallel RT negative controls were generated in all cDNA syntheses.
Allele-specific transcript quantification
The Allelotype platform from Massarray (Sequenom) was used for accurate relative quantification of allele-specific cDNA species (Hacking et al. 2004; Elvidge et al. 2005). Primers were designed using the dedicated software Spectrodesigner (Sequenom).
For a single cDNA assay, we performed four PCR replicates. We performed four extension reaction replicates on each PCR product, therefore producing 16 nested technical replicates per assay. Each replicate was run using a 2-μL aliquot of cDNA (derived from 200 ng of total RNA). Genomic DNA in 16 equivalent nested technical replicates (4 μg per replicate) was assayed as a control for each assay, using the same allelotype chip and primer mix. RT negative cDNA controls for all assays were run in parallel.
For a given cDNA assay, the allelic transcript ratio was calculated on each of the 16 technical replicates from the relative quantity of the two allele-specific cDNA species. The mean allelic transcript ratio for the whole assay was then calculated and normalized to the mean allelic transcript ratio for the genomic controls. An assay was accepted for further analysis if the standard error of the mean for the 16 technical replicates was <10%.
SNP and haplotype analysis
HapMap release 16 was accessed at http://www.hapmap.org. All other genotyping was performed using MassArray (Sequenom). All haplotypes were generated with the software PHAMILY and PHASE2.1.1 (Stephens et al. 2001) using family trios.
Statistical analysis
The ATR Mapping metric applied to the data in this study is a simple two-tailed t-test with unequal variance for the comparison of ATR values in the two groups bracketed as MTR and as NTR for a given SNP M. The Log10s of all ATR values were used in the t-test.
To generate a corrected P-value for multiple testing, permutation analysis for SNPs of interest was performed by randomizing observed ATRs with individuals. For the SNP of interest, a two-tailed t-test with unequal variance for the comparison of groups bracketed as MTR and as NTR was performed for each of 10,000 permutations to generate a distribution of P-values. (The Log10s of all ATR values were used in the t-test.) The corrected P-value was assigned from the position of the observed P-value within this distribution of random P-values.
All statistics were programmed in visual Basic (Microsoft Excel) or obtained using SSPS.
Acknowledgments
We thank Evelyn Harvey for her contribution to initial ATR experiments in the 5q31 region. This work was funded by a Wellcome Trust Clinical Research Training Fellowship (to J.T.F.) and a MRC Program Grant (to D.P.K.).
Footnotes
-
↵3 Present address: Kennedy Institute of Rheumatology, Imperial College, London, UK.
-
↵4 Corresponding author.
↵4 E-mail Julian.forton{at}paediatrics.ox.ac.uk; fax 44-1865-220479.
-
[Supplemental material is available online at www.genome.org.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.5663007
-
- Received June 19, 2006.
- Accepted October 18, 2006.
- Copyright © 2007, Cold Spring Harbor Laboratory Press








