
Embracing the complexity of genomic data for personalized medicine
- 1 Duke Institute for Genome Sciences & Policy,
- 2 Department of Molecular Genetics and Microbiology
- 3 Department of Medicine Duke University Medical Center, Durham, North Carolina 27710, USA;
- 4 Koo Foundation Sun Yat Sen Cancer Center Taipei, 112 Taiwan;
- 5 Institute of Statistics and Decision Sciences, Duke University, Durham, North Carolina 27708, USA
Abstract
Numerous recent studies have demonstrated the use of genomic data, particularly gene expression signatures, as clinical prognostic factors in cancer and other complex diseases. Such studies herald the future of genomic medicine and the opportunity for personalized prognosis in a variety of clinical contexts that utilizes genome-scale molecular information. The scale, complexity, and information content of high-throughput gene expression data, as one example of complex genomic information, is often under-appreciated as many analyses continue to focus on defining individual rather than multiplex biomarkers for patient stratification. Indeed, this complexity of genomic data is often—rather paradoxically—viewed as a barrier to its utility. To the contrary, the complexity and scale of global genomic data, as representing the many dimensions of biology, must be embraced for the development of more precise clinical prognostics. The need is for integrated analyses—approaches that embrace the complexity of genomic data, including multiple forms of genomic data, and aim to explore and understand multiple, interacting, and potentially conflicting predictors of risk, rather than continuing on the current and traditional path that oversimplifies and ignores the information content in the complexity. All forms of potentially relevant data should be examined, with particular emphasis on understanding the interactions, complementarities, and possible conflicts among gene expression, genetic, and clinical markers of risk.
Clinical disease states represent exceedingly complex biological phenotypes reflecting the interaction of a myriad of genetic and environmental contributions. A case in point is cancer, which represents a hugely heterogeneous disease. The characteristics of an individual tumor and its life course results from multiple mutations acquired over time (e.g., RAS, RTK) and continual evolution of the responses to environment (e.g., estrogen or tobacco exposure), overlaying inherent germline variations (e.g., BRCA1/2). Multiple oncogenes affecting critical pathways lead to gene expression data that reflect very many and diverse aspects of the oncologic state. While the effect of some oncogenes may be quite subtle, their combined effects—together with and in the context of environmental, lifestyle, and other factors—can make an important contribution to tumor aggressiveness. It is the aggregate of these effects that places the individual on a complex, high-dimensional risk “spectrum.” The complexity of the disease process leads to immense natural heterogeneity in tumor phenotypes, disease outcomes, and response to therapies. A major challenge is to develop information that can describe this complexity so as to facilitate an understanding of the disease mechanisms as well as to guide the development and application of therapies. Unfortunately, the available array of clinical and biochemical markers fall well short of being capable of describing the disease complexity. The challenge, as well as the opportunity, of personalized medicine lies in the capacity to develop quantitative data that can match the complexity of the disease.
The advent of genomic technologies has now offered the potential to develop data that do provide this complexity, identifying discrete subsets of disease that have not been recognized prior to the use of genomic data. Clear examples can be seen for both lymphoma (Golub et al. 1999) and breast cancer (Perou et al. 2000). High-density DNA microarray technologies for genome-scale measures of gene expression are uniquely adaptable to a broad array of biological and medical questions. Not too far behind, but lacking the throughput of RNA expression analyses, are whole-proteome analyses of tissue homogenates or serum using surface enhanced laser desorption ionization-time of flight (SELDI-TOF) or liquid chromatograph/mass spectrometry/mass spectrometer (LC/MS/MS) technologies (Calvo et al. 2005). The ability to find structure in the data—in the form of patterns of gene or protein expression that provide snapshots of gene activity in a cell or tissue sample at a given instant of time, and that can be used to describe a phenotype—is transforming biology from an observational molecular science to a data-intensive quantitative genomic science. The dimension and complexity of such data provide opportunity to uncover patterns and trends that can distinguish subtle phenotypes in ways that traditional methods cannot.
Among the most visible applications have been studies in human cancer where gene expression patterns can be identified that provide phenotypic detail not previously obtainable by traditional methods of analysis: profiles and patterns that identify new subclasses of tumors, such as the distinction between acute myeloid leukemia and acute lymphoblastic leukemia, without prior knowledge of the classes (Golub et al. 1999; Alizadeh et al. 2000). Over the past 5 yr, a very large collection of studies has detailed the power in the use of gene expression data to characterize, classify, and predict outcomes in cancer (Ramaswamy and Golub 2002; Staudt 2003). Other areas of biology and medicine have also benefited from the technology; examples in cardiovascular medicine, neurosciences, and others illustrate the opportunities for uncovering subtle distinctions in biological states (Tudor et al. 2002; Sarwal et al. 2003; Seo et al. 2004). Such studies are opening the way to an improved understanding of the complexities of biology and disease and, hence, the development of novel diagnostics and therapeutics. In the study of cancer, the use of such information in predicting the status of future cases represents the start toward moving modern genomics into clinical medicine by providing an opportunity to match an individual patient with the most appropriate therapeutics. Equally important is the opportunity for using gene expression profiles, or other forms of molecular profiles, in the process of new drug development, increasing the likelihood of success by again matching the therapeutic with the population of patients most likely to benefit from the drug, as well as improving prediction of adverse events (Stoughton and Friend 2005).
While much of our discussion focuses on the use of gene expression data given the wealth of experience now showing the value of this information, we also recognize the importance of other sources of data in the integrated view of personalized medicine. The Human Genome Project has provided genomic sequence and information on sequence variation that distinguish individuals and their susceptibility to disease or prospects for health. The transcripts from the 22,000 genes, their translated protein products and post-translational modifications and splice variants, and small molecule metabolites—the physiologic workhorses of the genomic program and its interplay with the environment—are all now available to measure disease states and potentially to correlate with clinical outcomes and drug response. Genomic complexity contained in DNA based information, combined with RNA/protein/metabolite profiles and clinical data, offers the opportunity to define multidimensional risk stratifiers with fidelity and precision that have never been possible (Fig. 1).
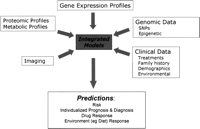
An integrated use of genomic, clinical, and other data to predict clinical and biological phenotypes.
Pharmacogenetic/genomic data have already provided examples where individual differences in drug response can be identified on the basis of variation in drug metabolizing enzymes or variants in the drug target itself (Tate and Goldstein 2004). Likewise, proteomic assays of both tumor material and serum samples from cancer patients have identified patterns reflecting outcomes (Yanagisawa et al. 2003; Calvo et al. 2005). In short, multiple forms of data will ultimately contribute to the goal of developing individualized risk predictors near and long-term and the development of the enabling paradigms for personalized medicine.
Integrated analysis: Utilizing the complexity of genomic and clinical data
The development of integrated analyses, making use of multiple forms of complex data, is an issue of critical relevance to clinical medicine. The benefit of data integration can be illustrated by a consideration of the Framingham Heart Study, the landmark longitudinal analysis of risk determinants for coronary artery disease (Dawber 1980). As a result of the massive and comprehensive collection of clinical and biological data in relation to coronary disease, the Framingham predictive models were developed that combine a wide variety of variables including age, gender, tobacco use, diabetes, hypertension, body mass index (BMI), low density lipoprotein/high density lipoprotein (LDL/HDL) cholesterol, and family history (Wilson et al. 1998); the resulting Framingham risk score predicts the relative likelihood for risk of developing coronary artery disease. It is clear that this prediction could be greatly enhanced by the inclusion of data that address the subtle distinctions in individuals that will be revealed through genomic analysis. The opportunity for impact on clinical decision making offered by genome technologies lies in increased resolution: the potential to better place a patient on the complex, multidimensional risk spectrum based on detailed, individual molecular characteristics on a genomic scale. The Framingham heart study example emphasizes the value of making use of the full spectrum of available clinical and demographic data; the genomic era simply expands this view toward integrated approaches that embrace and exploit genomic data in conjunction with other data.
To fully realize the clinical potential of genome-scale information requires a paradigm shift in the way complex, large-scale data are viewed, analyzed, and utilized. For example, the tradition of identifying one or a small number of biomarkers continues in the context of cancer genomics with the identification of a single gene expression signature from tumor-derived DNA microarray data being the goal, evaluated for its prognostic significance by association with disease outcomes but without regard to the myriad other dimensions of cancer biology reflected in the expression data. Typically, with patient samples stratified by such a simplified signature, the study will then define a predictor of risk. However, it is frequently found that, in follow-on studies with expanded and new patient samples, the potency of the signature as a “risk predictor” is diminished, as is also often the case with traditional clinical and genetic markers. The issue here is not the failure of analytical methods or genomic technologies; rather, it is the focus. The prognostic role of any one or a small number of molecular markers must generally be much more broadly evaluated in conjunction with multiple other factors, including biologically meaningful pathway data and clinical data. Cancer biology and the disease process are hugely complex. Individual risk factors, be they genetic, clinical genomic, or other, represent only single or low-dimensional snapshots of the disease process and state. What is needed is the integrative view that takes all forms of data into consideration and aims to identify an individual patient on a complex spectrum of risk that is measured by multiple factors, while addressing issues of interaction, complementarity, redundancy, and—critically—conflict among the risk factors at the individual patient level (Nevins et al. 2003). For example, a biologically inspired gene expression predictor of cancer recurrence that reflects subtle aspects of deregulation of a relevant signaling pathway or a biological process may well stratify a given patient sample according to high versus low risk of recurrence. But, what of other, interacting, and potentially conflicting expression signatures of other pathways, hormonal responses, hypoxia, and so forth? The need for comprehensive, integrative analysis that presents and evaluates these multiple factors, and that fairly assesses the combined prognostic implications with due regard for the uncertainty that arises when two or more biomarkers conflict, is simply paramount.
Breast cancer prognosis: Highlighting the importance of complexity
To illustrate the complexity of the oncogenic process and the need to employ equally complex data to predict clinical outcomes, we focus on four recent studies of breast cancer. A pivotal study was published in 2002 in the emerging field of genomic medicine describing the use of a DNA microarray-based 70-gene expression profile as a prognostic factor in breast cancer (van’t Veer et al. 2002). That this genomic marker has predictive value was demonstrated in the improved stratification of stage I and II breast cancer patients into two broad subgroups, relatively high- versus low-risk groups with respect to the long-term risk (over 10 yr) of cancer recurrence. The 70-gene predictor has now become the basis for an expanded European study that aims to measure its performance in a more diverse set of patients. A parallel prospective clinical trial aims to measure the effectiveness of the predictor in guiding adjuvant chemotherapy compared with predictions based solely on the traditional clinical parameters for prognoses.
A second recent study (Ein-Dor et al. 2005) describes further analysis of the data presented in the 2002 New England Journal of Medicine (NEJM) study. The investigators demonstrate one key point: The original 70-gene predictor is not unique and that multiple such predictors derived from the same data set can offer comparable predictive performance. Ein-Dor and colleagues identified at least eight separate gene sets whose aggregate expression patterns can stratify the same patient sample into comparable high-risk and low-risk categories, suggesting that multiple forms of information derived from this data set can equally well stratify patients according to recurrence risk. While this may surprise some readers, the result is perfectly consistent with the experience of multiple studies in breast cancer and other diseases. Heterogeneity in patient populations, methods of assay, varieties of array technologies, and other factors certainly underlie some of the differences. The fundamental reason is, however, simply the diversity, scale, and complexity of genetic and genomic factors defining this disease. This study highlights the many dimensions of genomic information that have a bearing on clinical outcomes, and the consequent need to define analytical approaches that interpret, combine, contrast, and evaluate multiple such aspects. Simply choosing one “best” gene expression signature, but ignoring multiple other choices that reflect other relevant aspects of cancer biology, is an oversimplification and a potentially dangerous, misleading strategy. In short, all relevant data, whether genomic or otherwise, should be taken together to achieve the maximal power in predicting the phenotype.
A third recent study (Chang et al. 2005) demonstrates the value of combining a novel, previously defined signature reflecting a wound healing response (Chang et al. 2004) with the original 70-gene risk predictor described by van de Vijver et al. (2002). This approach can be seen as clear evidence and support for the thesis that integrative analysis is needed. This work compares the prognostic value of the analysis with traditional methods based on nongenomic clinical variables (St. Gallen and the National Institutes of Health), and demonstrates that analysis combining two gene expression signatures improves outcome predictions. This conclusion highlights the importance and value of using additional signatures to dissect the evident heterogeneity left by stratification on a single gene expression pattern. In addition, this study takes the observation of Ein-Dor and colleagues (2005) one step further by demonstrating not only that there are multiple signatures that carry information regarding outcome but also that they can be combined to more effectively predict risk.
Our own work, published in 2004, provided a comprehensive demonstration of the value of combining multiple gene expression signatures together with clinical data in breast cancer prognosis (Pittman et al. 2004). This work evaluated the contribution of hundreds of gene expression signatures, assembling the multiple patterns in a combined model to predict patient survival. Consonant with the work of Ein-Dor et al. many of the signatures utilized in the predictive models were capable of stratifying patients into clinically relevant, distinct risk groups. That is, a number of individual signatures could perform in a manner not dissimilar from the 70-gene predictor in terms of stratifying patients into higher- versus lower-risk groups with respect to recurrence. It was an integrated analysis that generated the most relevant and robust predictive models. The logic in the approach is conceptually simple, recognizing the limitation of any one profile to go beyond a broad categorization into low risk versus high risk, and thus making use of multiple profiles to further dissect out subgroups based on prediction of risk (Fig. 2A). The benefit is clearly seen in survival analysis where subpopulations of higher- or lower-risk patients are identified from the original high-risk or low-risk groups (Fig. 2B). The generation of such models, which make use of many gene expression patterns to address the true complexity of the disease, point the way toward predictions for individual patients (Fig. 2C). The analysis went further in demonstrating mechanisms to effectively assemble the multiple profiles using statistical classification and regression tree analysis to weigh the relative importance of each pattern, providing a mechanism to incorporate multiple biomarkers such as expression signatures and clinical risk factors (Fig. 3A).
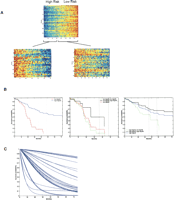
Utilizing multiple gene expression profiles to dissect cancer heterogeneity. (A) An example of the use of multiple profiles to predict recurrence of breast cancer. An initial gene expression profile (top pattern) is shown to split the patient population into high-risk and low-risk groups. The image shows the expression pattern of the genes in an aggregate signature or metagene labeled Mg440 (ordered vertically by correlation with Mg440) on the entire group of 158 patients. Samples are ordered (horizontally) by the value of Mg440, and the vertical black line indicates the split of patients into two subgroups underlying the empirical survival curves shown in the left panel of Figure 2B. The two subgroups of patients defined by this initial split are then further split with two additional metagenes. The “low Mg440” subgroup is further split based on Mg408, and the “high Mg440” group is split on Mg109. The two subsequent images show the patterns of genes within each of Mg408 and Mg109 for the corresponding two subgroups of patients, arranged similarly within each group and also indicating the second level splits. Red color in the plot defines high expression, and blue is low expression. Modified with permission from the National Academy of Sciences, U.S.A. © 2004, Pittman et al. 2004. (B) Sample survival characteristics based on a single gene expression profile versus multiple profiles. Empirical survival estimates based on a partition into two groups via a threshold on the gene expression pattern of Mg440 (left panel), and then the subgroups identified by splits with Mg408 (middle panel) and Mg109 (right panel). Modified with permission from the National Academy of Sciences, U.S.A. © 2004, Pittman et al. 2004. (C) Survival predictions for the entire population of 158 breast cancer patients using a full, integrated clinico-genomic model that combines multiple gene expression patterns with clinical information.

Combining multiple data to build predictive models. (A) Breast cancer recurrence. (Upper panel) An example of a single statistical classification tree model illustrating the utilization of gene expression profiles together with clinical variables to determine successive partitions of the patient sample with associated predictions. The boxes at each node of the tree identify the number of patients, and the number under each box is the corresponding model-based point estimate of the 4-yr recurrence-free probability (given as a percentage) based on the tree model predictions for that group. Overall practical predictions of survival risk are based on statistical aggregation of predictions across multiple such models; this multiplex prediction reflects the inherent complexity of the disease process and its bearing on survival risks. (Lower panel) The figure summarizes the level of the tree model in which each variable appears and defines a node split. The numbers on the left simply index tree models, and the probabilities in parentheses on the left indicate the relative weights of tree models based on fit to the data. The probabilities associated with clinical or metagene predictor variables (in parentheses on horizontal axis) are sums of the probabilities of trees in which each occurs, and so define overall weights indicating the relative importance of each variable to the overall model fit and consequent recurrence predictions. Clinical predictors, including lymph node status, tumor size and ER status, maintain predictive roles in some of the combined clinico-genomic tree models, whereas metagene predictors replace them in others. Adapted with permission from the National Academy of Sciences, U.S.A. © 2004, Pittman et al. 2004. (B) Coronary artery disease. Score sheet for estimating the risk of coronary heart disease over a period of 10 yr based on data from the Framingham Risk Study. Reprinted with permission from Lippincott, Williams and Wilkins © 1998, Wilson et al. 1998.
In the Pittman et al. (2004) study, the most predictive models for recurrence were defined via the synthesis of multiple gene expression profiles and clinical data (estrogen receptor (ER) and lymph node), combined together in integrative clinico-genomic models (Fig. 2A). In principle, this concept is similar in nature to the creation of models that predict risk of cardiovascular events based on the Framingham data (Fig. 2B), making use of various relevant clinical and biological data in a combined fashion. The goal in the two examples is the same—to define integrative models that, by building on multiple aspects of information that reflect the individual circumstances, lead to personalized predictions rather than stratification into broader patient subgroups. The distinction lies in the use of multiple aspects of the genome-scale molecular profiles for the prediction of cancer outcomes—profiles that have the capacity to add substantially increased resolution to tailor models toward this goal.
Adding further complexity
The example given for breast cancer prognosis, which makes use of multiple gene expression profiles together with clinical information to generate the most robust outcome predictor, should be viewed as just the initial step toward the final goal of a truly integrated predictive model that assesses all forms of useful data. Ultimately, a completely integrated set of data will be required for individualized prognosis inclusive of genomic variation of germline DNA, expression signatures from the tumor, serum protein markers, and clinical data. Where this exploration of relevant data eventually stops will be determined by the complexity of the phenotypes under study and recognized by the fact that the complexity of the data has matched the complexity of the biology. At a practical level, this is a question balancing statistical and technological considerations: Broader evaluation of the prognostic accuracy of predictive models across larger and diverse patient samples must be stressed in order to define improved understanding of the robustness and practical accuracy, while advances in genomic and other technologies will generate increasingly rich and precise determinations of molecular states that need to be contrasted and evaluation in expanded analyses.
While the most significant advances have been made by using expression profiling from disease tissue, it is also clear that other forms of data have the potential to contribute valuable information. As an example, proteomic profiling of tumors has identified patterns of protein peaks from mass spectrometry analyses that have the capacity to accurately predict clinical outcome in lung cancer (Yanagisawa et al. 2003). As illustrated in Figure 4A, protein profiles can be obtained that clearly distinguish normal from tumors; more importantly, a pattern of 15 distinct mass spectrometric peaks can be selected that distinguishes good and poor outcomes (Fig. 4B). Thus, similar to the utilization of gene expression data, protein profiles have the capacity to dissect the complexity of clinical phenotypes. Although the information contained in gene expression data and protein profile data are likely overlapping, it is also likely that there are distinctions between the two that will be synergistic in building integrated outcome models.
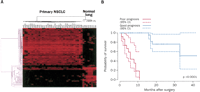
Protein patterns that classify and predict outcome in lung cancer. (A) A profile of protein abundance measured by mass spectrometry that was selected to distinguish samples of primary non-small-cell lung carcinoma (NSCLC) from normal lung. Reprinted with permission from Elsevier © 2003, Yanagisawa et al. 2003. (B) Kaplan Meier survival analysis of NSCLC patients stratified based on proteomic profiles. Reprinted with permission from Elsevier © 2003, Yanagisawa et al. 2003.
A clinical phenotype, whether this is disease outcome, response to therapy, or some other measure of the disease process, reflects events in the disease tissue (such as a tumor) as well as the inherited genetic constitution of the patient. The latter defines the potential response to drugs, the effectiveness of immune interactions, and more. As such, measures of this germline variation will also contribute to the overall goal of developing the most effective predictor of outcome. The concept is straightforward—variations in key genes that encode drug metabolizing enzymes or immune system activities can influence the onset and course of disease. Nevertheless, compared with the use of gene expression profiling of tumor tissue, the development of pharmaco-genomic markers that predict outcomes has been slow, in part reflecting the difficulty in identifying the relevant genes and gene variants but also the challenge of adoption in clinical practice.
Other forms of data may potentially act as surrogates for the impact of the germline genetic variation. For instance, serum proteomic profiles may reflect unique patterns that predict susceptibility. Likewise, expression profiles from peripheral lymphocytes may serve as indicators of events ongoing elsewhere in the body with the lymphocyte serving as a “sensor” of the host environment.
Next steps
Three key areas represent logical and critical next steps in the use of complex genomic profiling data toward the goal of personalized medicine. First, analyses that have developed profiles that predict future events—such as an adverse event or the response to a particular therapy—must now move into actual clinical practice by forming the basis for the next generation of clinical trials that will employ these methodologies to stratify patients. No longer should drug treatment studies be performed without a component that attempts to identify those patients most likely to respond to a particular therapeutic regimen. Although the ability to make this transition clearly depends on the strength of validation of genomic/integrated predictions, this transition must be a clear goal of the ongoing work. A distinction should be made between research discovery and eventual application where the latter might demand a more simplified approach for practical reasons. The continued emphasis toward the goal of a single, “silver bullet” gene expression index, or other form of genomic data, is clearly derived from the convention of using assays of single biomarkers or limited numbers of genes assayed by other means such as polymerase chain reaction (PCR) to reflect the state of the disease. It is important to recognize that while this may be worthwhile in a practical sense for an eventual clinical assay, it should not influence the research discovery of the most valuable information to describe a complex phenotype. Even in the setting of practical application, one must be cautious about reducing the information content and thus the ability to recognize the underlying complexity of the biological state. Multiple expression patterns can define concurrent risk stratifications of broad patient groups; such collections of markers will almost surely also generate conflicting information for individual patients as they bear on genes and pathways related to linked, although differing, aspects of the tumor. Indeed, this issue was described in discussions of several individual patients in Pittman et al. (2004), highlighting the need for detailed evaluation and understanding of the nature of interactions among expression signatures as representing multiple aspects of the complex cancerous state.
Second, the availability of multiple sources of genomic-scale data relevant to clinical phenotypes must be integrated to develop more precise descriptions of clinical phenotype. We have already discussed the opportunities for merging multiple gene expression patterns to develop more powerful predictive models. The availability of other forms of data, including proteomic, metabolomic, and DNA structure profiles now provides opportunities for integrating these additional forms of data into comprehensive models of disease outcome. Moreover, the analysis of disease tissue, whether tumors or other, represents only half of the story, with germline gene variation information representing yet another opportunity to add richness to the genomic-based predictors and classifiers.
The multidimensional nature of these assays (DNA, RNA, protein-based) along with the clinical data for an individual highlights several challenges that must be addressed for this paradigm to become a reality. One challenge is the nature of the testing platform and the development of performance standards for multicomponent biomaker assays. A second challenge is the delivery of information robustly that allows health care providers to incorporate it into risk assessment seamlessly. The Framingham nomogram (Fig. 3B) (Wilson et al. 1998) is one example of this, and software tools such as Adjuvant (www. adjuvantonline.com) are another example of technologies that must be developed to assist physicians in complex decision-making. A third and important challenge is the need for health and economic outcomes as an objective of the clinical research studies and trails employing the predictor. These will facilitate the approval of the diagnostic by the Food and Drug Administration and provide the basis for reimbursement by third party payers such as Centers for Medicare and Medicaid Services (CMS).
Lastly, while there is immediate utility in the application of genomic profiles as prognostic and predictive tools to guide therapy decisions, these profiles also hold information that distinguishes the underlying biological processes that define these differences in risk or response. Clearly, this is a major issue facing the opportunities for stratification since it is of little value to identify patients at increased risk for recurrence, or likely to be resistant to a given therapy, if there are no options for what to then offer such patients. Now, the challenge of extracting an understanding of the underlying biology from the gene expression profiles that define resistant or high-risk patients, which might represent new opportunities for therapeutic intervention, is daunting. While one can often identify function associated with some of the genes underlying a component signature, the challenge is to put this in perspective of the entire genomic profile—what is the underlying unifying aspect of the profile that distinguishes the two classes of patients? The gene-by-gene approach is limited by the lack of information that can easily connect these genes in a functional sense.
An alternative approach is to look for higher-order structure in the gene expression profiles that might offer clues to the underlying biology (Fig. 5). A recently described application known as gene set enrichment analysis (GSEA) (Mootha et al. 2003; Monti et al. 2005; Sweet-Cordero et al. 2005) provides one strategy to this problem. GSEA aims to compare the list of genes within a selected profile against large collections of pathway and other categorizations to determine if there is enrichment for one of these functional groupings. These functional groups (gene sets) are assembled from known pathway databases (i.e., KEGG, GenMAPP, etc.), cytogenetic loci, units of chromosome amplification, as well as any other relevant functional groupings such as previously defined gene signatures of clinical phenotypes. The power of this method is the ability to provide a quantitative enrichment score that views the gene expression profiles in a functional context as defined by pathway and other functional group organization, going beyond the process of single gene analysis. A distinct but related method of analysis makes use of gene expression signatures generated to reflect pathway activation (Black et al. 2003; Huang et al. 2003; Bild et al. 2006). The logic is to predefine relevant biology, in the form of gene expression profiles characterizing relative activity of components of known pathways, as well as subpathways, that can then be used to evaluate the extent to which any of these signatures are represented in a biological sample such as a tumor. Other work has made use of a defined mouse tumor model to develop a gene expression signature that could be applied to the analysis of human cancers (Sweet-Cordero et al. 2005).
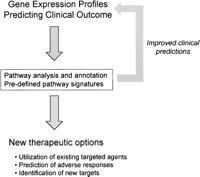
Extracting and using the biological meaning from complex genomic profiles. Outline of methods for identifying relevant pathways and therapeutic options in gene expression profile data.
In each of these examples, the goal is to convert the profile into an enhanced biological understanding with the goal of placing the genes in a functional context. This provides an opportunity to identify new therapeutic targets that might be suggested based on an understanding of the pathway that is affected. For instance, while no single gene within a given gene expression profile may represent an attractive therapeutic target, the pathway that is identified could be rich in potential targets. Further, it is also very possible that several therapeutics are already available that target components of the pathway. This approach anticipates the opportunity to generate improved biological understanding into predictive models that classify or predict clinical outcomes (Fig. 5). As already emphasized, it is paramount that all available data should be utilized in the development of models that can most effectively predict important clinical outcomes. Moreover, the realization that biologically defined signatures can add value to the development of outcome predictions clearly suggests that additional signatures tailored more closely to the biological context should be defined and evaluated. The relevance of a wound-healing profile in breast cancer (Chang et al. 2005) and of multiple specific oncogenic pathway signatures across multiple cancers (Bild et al. 2005) are examples that illuminate this potential for biologically defined signatures to aid in the improvement of disease course and outcome prediction for the individual patient. The need here is then for iteration between improved biological understanding and clinical prognostic models’ development and evaluation with an integrative focus.
Footnotes
- 6
↵6 Corresponding author.
↵6 E-mail j.nevins{at}duke.edu; fax (919) 681-8973.
-
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.3851306
- Cold Spring Harbor Laboratory Press








