
Abstract
Recent reports indicate that copy number variations (CNVs) within the human genome contribute to nucleotide diversity to a larger extent than single nucleotide polymorphisms (SNPs). In addition, the contribution of CNVs to human disease susceptibility may be greater than previously expected, although a complete understanding of the phenotypic consequences of CNVs is incomplete. We have recently reported a comprehensive view of CNVs among 270 HapMap samples using high-density SNP genotyping arrays and BAC array CGH. In this report, we describe a novel algorithm using Affymetrix GeneChip Human Mapping 500K Early Access (500K EA) arrays that identified 1203 CNVs ranging in size from 960 bp to 3.4 Mb. The algorithm consists of three steps: (1) Intensity pre-processing to improve the resolution between pairwise comparisons by directly estimating the allele-specific affinity as well as to reduce signal noise by incorporating probe and target sequence characteristics via an improved version of the Genomic Imbalance Map (GIM) algorithm; (2) CNV extraction using an adapted SW-ARRAY procedure to automatically and robustly detect candidate CNV regions; and (3) copy number inference in which all pairwise comparisons are summarized to more precisely define CNV boundaries and accurately estimate CNV copy number. Independent testing of a subset of CNVs by quantitative PCR and mass spectrometry demonstrated a >90% verification rate. The use of high-resolution oligonucleotide arrays relative to other methods may allow more precise boundary information to be extracted, thereby enabling a more accurate analysis of the relationship between CNVs and other genomic features.
In the last several years following completion of the human genome sequence (International Human Genome Sequencing Consortium 2004), new progress in unraveling the complexities of the genome’s architecture has revealed a remarkable degree of structural variation present among normal individuals (Fredman et al. 2004; Iafrate et al. 2004; Sebat et al. 2004; Sharp et al. 2005; Tuzun et al. 2005; Conrad et al. 2006; Hinds et al. 2006; Locke et al. 2006; McCarrol et al. 2006). Structural variants include a variety of molecular alterations such as duplications, deletions, and inversions, and are distinct from the genetic sequence diversity represented by single nucleotide polymorphisms (SNPs) (Feuk et al. 2006a, b; Freeman et al. 2006). Just as the genome-wide haplotype map has now provided the framework to identify the genetic basis of complex diseases, pathogen susceptibility, and differential drug responses (International HapMap Consortium 2005), a thorough map that catalogs and indexes structural variants (and, in particular, copy number variants [CNVs]) in the human genome is a necessary prelude to understanding their role in the context of both the normal and disease state. Although there are increasingly clear examples of how CNVs can, for example, influence susceptibility to HIV infection (Gonzalez et al. 2005), modulate drug responses (Ouahchi et al. 2006), or contribute to genomic microdeletion and duplication syndromes (Inoue and Lupski 2002), a comprehensive biological understanding of the roles of CNVs is not yet currently available. While several different molecular techniques can be used for CNV detection, array-based experimental approaches still offer the most efficient and cost-effective method for global, high-resolution scans of structural features of the genome (Sharp et al. 2005; Speicher and Carter 2005; Hoheisel 2006; Urban et al. 2006).
High-density DNA oligonucleotide arrays allow unsurpassed levels of genetic information to be acquired in single experiments (Fodor et al. 1991, 1993; Pease et al. 1994). These arrays, coupled with a DNA target preparation method termed whole-genome sampling analysis (WGSA), which involves PCR-mediated complexity reduction, have successfully been used to simultaneously genotype >10,000 SNPs on a single array and 100,000 SNPs on a two-array set (Kennedy et al. 2003; Matsuzaki et al. 2004a, b). Recently, by changing the choice of restriction enzymes and by increasing the information capacity of the arrays, highly accurate genotyping of 500,000 (500K) SNPs has been enabled on a pair of arrays (http://www.affymetrix.com). In addition to multiplexed SNP genotyping, these arrays, in concert with the development of specialized algorithms, have been used to detect genome-wide DNA copy number changes that include loss of heterozygosity (LOH), deletions, and gene amplification events (Bignell et al. 2004; Huang et al. 2004, 2006; Zhao et al. 2004; Ishikawa et al. 2005; Laframboise et al. 2005; Nannya et al. 2005; Slater et al. 2005; Beroukhim et al. 2006; Komura et al. 2006).
We have recently used two complementary experimental approaches, namely, BAC-based array CGH and high-density SNP genotyping arrays, to produce a first-generation global CNV map of the human genome, based on analyses of the HapMap population (Redon et al. 2006). Here we describe in detail the algorithm that was developed for this study to assess CNVs using probe intensity information from the GeneChip Human Mapping 500K EA (Early Access) arrays. The algorithm is predicated on improved intensity normalization methods originally used in the Genomic Imbalance Map (GIM) (Ishikawa et al. 2005) coupled with an optimized SW-ARRAY algorithm (Price et al. 2005) and a graph-theory based extraction of CNV results from a large reference set. Using this approach, we have identified 1203 CNVs and obtained a high rate of verification for these calls using multiple independent methods. The high-density SNP genotyping arrays afford a level of resolution that allows CNV boundaries to be called with relatively high precision at a genome-wide level.
Results
The 500K EA arrays, a pre-commercial version of the GeneChip Human Mapping 500K Array Set, contain 534,500 SNPs on two genotyping arrays (see Methods for details of the assay). To minimize the impact of cross-hybridization, probes were removed whose central 21 bases perfectly matched additional locations in the genome, with the exception of segmental duplications, which are enriched for CNVs. Probes corresponding to NspI or StyI restriction fragments in which the enzyme recognition site contains a SNP were also removed. These steps trimmed the total probe content to 474,642 SNPs (88.8% of the original) with a relatively minor effect on genome coverage (Supplemental Fig. 1).
CNV calling algorithm
Overview
In contrast to the detection of copy number changes in tumor samples, where DNA from the same individual can be used as a reference, the use of matched samples is not possible for CNV detection in normal individuals. Similarly, the use of a single reference, as is often used in BAC-array CGH, is limited by the inability to determine whether a copy number change is from the test or the reference sample. Thus we have developed an algorithm that builds on GIM and SW-ARRAY, two methods based on pairwise comparisons of array data, with the goal of accurately defining CNV regions using a large set of reference samples. GIM, which has been used previously for identification of copy number changes in cancer cells, focuses predominantly on intensity processing and reduces noise due to probe and restriction fragment sequences using a polynomial regression (Ishikawa et al. 2005; Midorikawa et al. 2006). Subsequent to GIM, SW-ARRAY identifies copy number changes using an adapted Smith-Waterman algorithm by finding isolated islands of substantially higher (or lower) intensity ratios and assigns significance to each finding by a permutation test (Price et al. 2005).
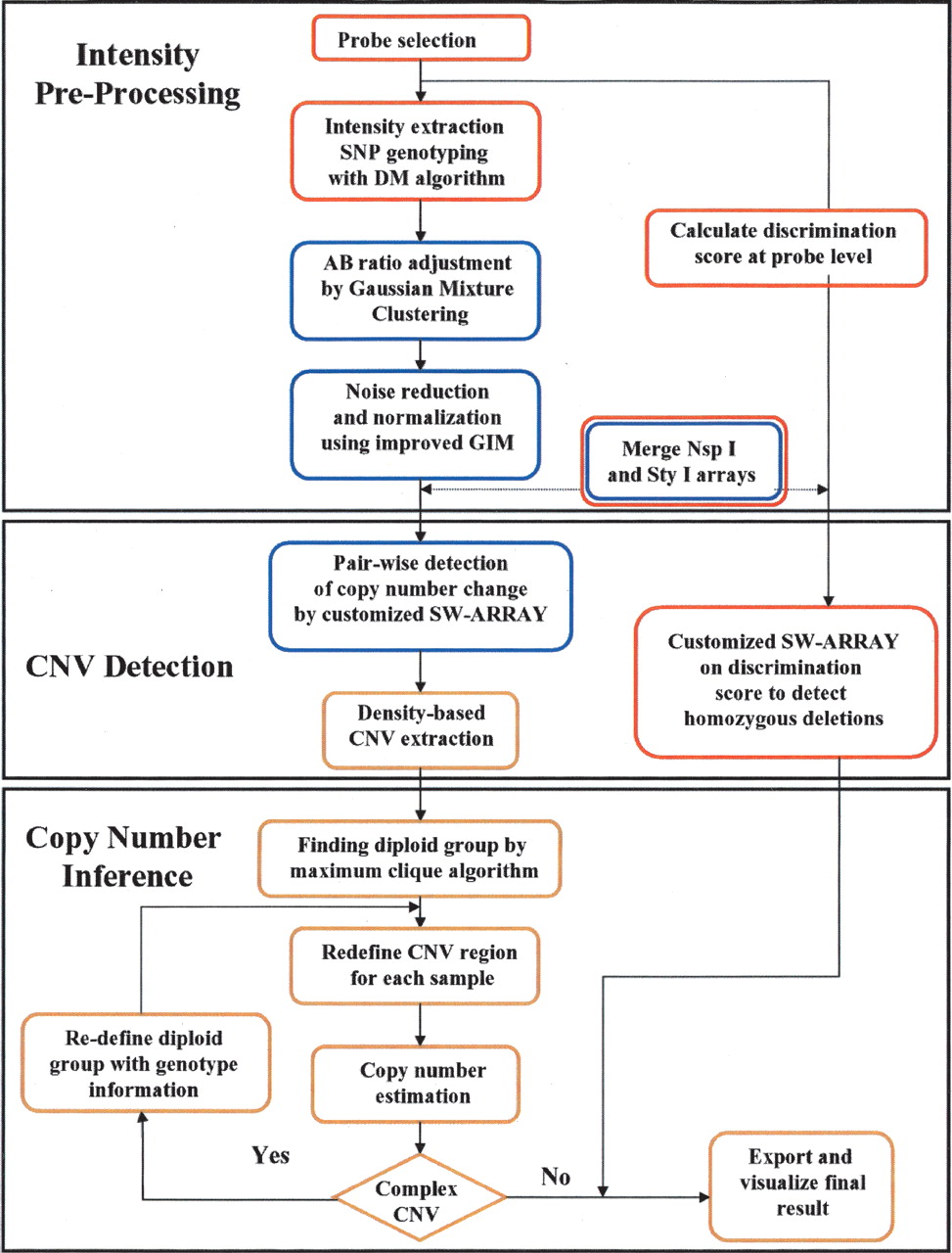
The algorithm described here contains three major parts as depicted in Figure 1. Intensity pre-processing includes probe selection, noise reduction, normalization, and merging signal ratios from the NspI and StyI arrays. CNV detection begins with pairwise comparisons of probe intensities for all possible pairs of samples (i.e., 269 comparisons for each HapMap sample), which are then merged to extract candidate CNV regions for each sample. Homozygous deletions are detected separately using an alternative approach that relies on the discrimination ratio between alternate SNP alleles in lieu of SNP genotypes (see Supplemental Methods for details). The copy number inference step uses signal ratios and SNP information to more precisely define CNV boundaries and the copy number within each region. The final step uses a maximum clique algorithm to define the diploid samples for any given region based on the results from the large reference data. Through a comparison of the test sample to the diploid subset, precise boundaries and accurate copy number inferences can be drawn (Fig. 2). Critical aspects of the CNV calling algorithm are highlighted in detail below.
Flowchart overview of the algorithm. Red, blue, and yellow boxes indicate that the process was carried out for each array, each sample pair, and each CNV region, respectively. GIM is used for intensity pre-processing, SW-ARRAY is used for pairwise CNV detection, and the maximum clique algorithm is used for CNV extraction.
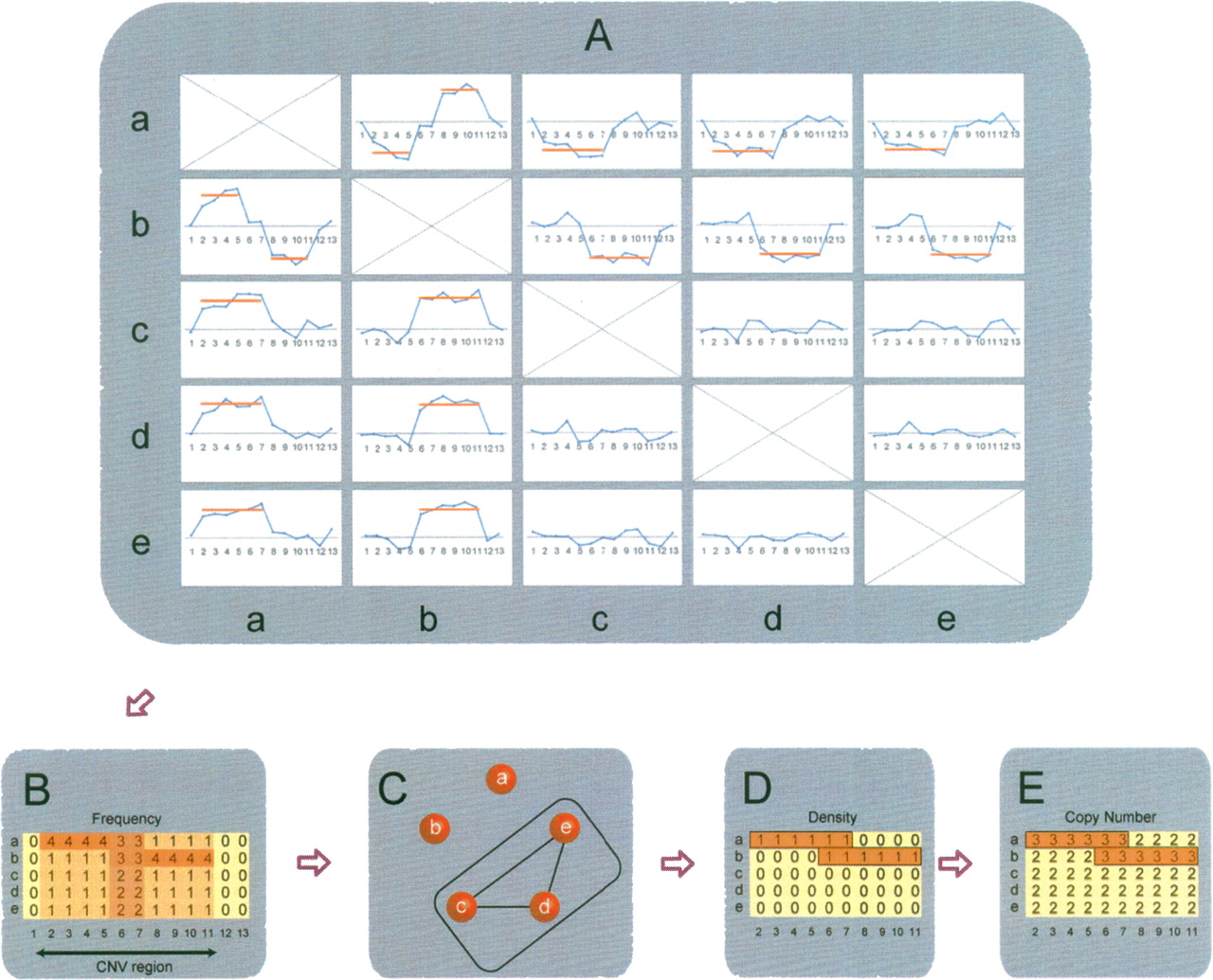
Overview of copy number inference. (A) Pairwise comparisons of five different DNA samples (a–e) in a given candidate CNV region. The x-axis represents the SNP positions, and the blue lines are log2 signal intensity ratios for any given pair. The red line indicates the significant CNVs detected by SW-ARRAY. (B) Summary of the comparisons of any given sample to the remaining four samples. Based on the physical location of copy number changes, the frequencies are calculated for each sample, and consecutive CNV regions are extracted. Each row represents a single sample, and each column represents the frequency of a given SNP. The frequency of a particular SNP is the number of times that it is called a CNV in all four pairwise comparisons. (C) Graph theory (the maximum clique algorithm) is applied to the frequency summarization results presented in B. In this example, samples c, d, and e, which have the lowest frequency and represent the maximum clique, are defined as the diploid group. (D) Density (the proportion of comparisons where a CNV is called) is calculated based on the diploid samples found by the maximum clique algorithm, and the boundary of the CNV region in each nondiploid sample is determined. (E) Copy number is determined based on the median ratio of each CNV region.
Intensity pre-processing
When intensity signals are compared from two individuals with different SNP genotypes, the signal ratio may be artificially skewed because of differences in the allele-specific probe affinities. Although the original version of GIM does not consider such comparisons, the current algorithm directly estimates the affinity differences using signal ratios between probe A and B in the AB genotype group and corrects the comparison accordingly. This increases the average number of SNPs used in any pairwise comparison from 256,257 to 429,104, resulting in a 67.5% improvement in resolution. GIM has also been improved through the use of robust BIC (Qian and Kunsch 1996; Komura et al. 2006) to remove signal fluctuations due to probe sequences, restriction fragments, and long-range genomic context surrounding each SNP (see Supplemental Methods for details). In addition, a new normalization step based on the recognition sites of the two restriction enzymes has been added to account for variation attributed to the differences in the intensity ratio distributions across restriction fragments with various recognition sites (Supplemental Fig. 2). To eliminate this difference, median scaling across recognition sites was applied to both enzymes, resulting in a 64% reduction in intensity ratio variation for a typical example (Supplemental Fig. 2C,D).
Modification of SW-ARRAY for CNV detection
To define CNVs, intensity values from two separately processed arrays must first be merged. Because each array contains unique outliers, the merged error distribution is non-Gaussian, making it difficult to define CNV regions based solely on the raw intensity-ratio distributions. For this reason, SW-ARRAY (Price et al. 2005), a nonparametric, dynamic programming algorithm, was adapted to identify copy number changes (Fig. 3A,B,C). In all cases, we used data generated from three replicates of NA15510, a DNA sample that has been extensively characterized by fosmid end-sequencing (Tuzun et al. 2005) and three replicates of a designated reference genome (NA10851) to define an optimized set of parameters that maximize reproducibility (percentage of CNVs called >50% of time in all pairwise comparisons) and minimize false-positive signals.
Parameter tuning. Several parameters were optimized for SW-ARRAY and CNV extraction including (A) intensity ratio threshold, (B) statistical significance, (C) number of SNPs and restriction fragments required for calling a CNV, and (D) density cutoff (the fraction of positive pairwise comparisons necessary for calling a CNV). For each parameter, CNVs were called from pairwise comparisons between NA15510 and NA10851 (A,B,C) or population-wide comparisons (D) for each sample. In A and B, the percentage of CNVs called more than half of the time (red line) is compared to the percentage that have been positively validated (blue line), or negatively validated, false positives (green line in B with the y-axis on the right-hand side). The final values chosen were 1.12 for the intensity ratio threshold, and 0.01 for the P-value (indicated by the vertical black lines). In C, the size distribution of CNVs detected using the 3 SNPs:2 fragments criterion (with a mean length of 300 kb) is compared to the 4 SNPs:3 fragments criterion (mean length 326 kb). In D, the number above each bar is the percentage of validated CNVs and validated diploid regions within certain density bins. A 10% density cutoff was chosen for further analyses. For any given cutoff, a false positive is defined as the percentage of all validated diploid regions that are incorrectly called as a CNV with a density that is greater than the cutoff; false negative is defined as the percentage of validated CNVs that are missed because their density is lower than the cutoff.
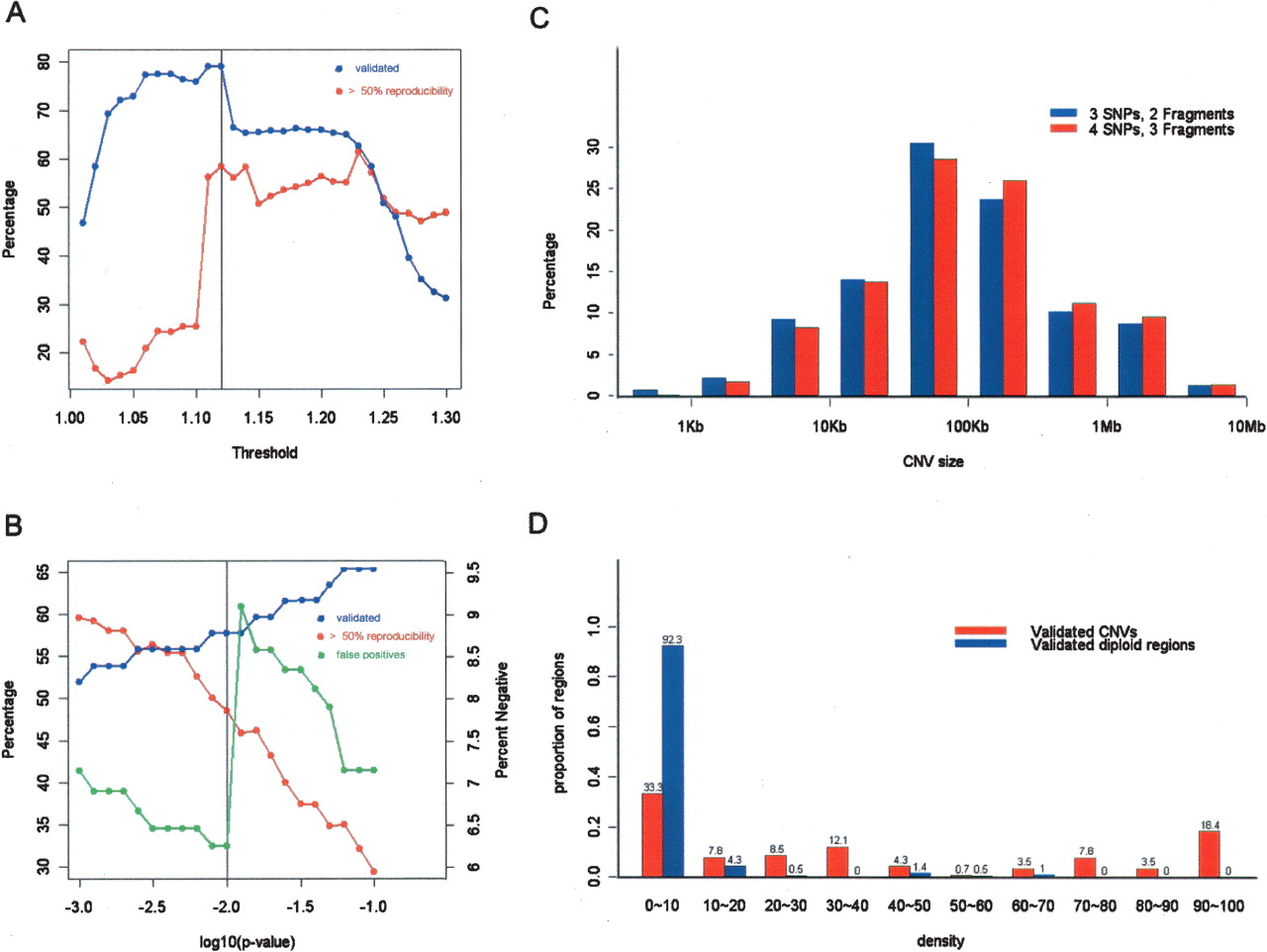
Four subsets of parameters were extensively studied including (1) intensity ratio threshold value, (2) significance cutoff, (3) constraints on number of SNPs and number of restriction fragments used to define a CNV, and (4) density optimization (Fig. 3). For the intensity ratio threshold, we first used samples with different numbers of X chromosomes, and observed an average intensity ratio of 1.3 between two versus three copies. The use of 1.3 as a stringent cutoff results in detection of 50% of the single-copy gains (based on the X chromosome data) with minimal false-positive signals. We then tested 31 threshold values evenly distributed between 1 and 1.3 using the three replicates of NA15510 and NA10851 and found the optimal threshold to be 1.12 (Fig. 3A). The significance value of 0.01 was derived using a similar approach (Fig. 3B). To increase the confidence of a CNV call, we introduced the new requirement that multiple probes on different fragments show consistent intensity change. Two combinations were examined, namely, four SNPs on three restriction fragments (4 SNPs:3 fragments) or three on two (3 SNPs:2 fragments). The former was chosen because the number and size of CNVs is similar using the two settings (Fig. 3C), while the reproducibility increased from 44% to 48% and the self–self false positives decreased 58% with the 4 SNPs:3 fragments setting.
CNV extraction based on a given density cutoff (the fraction of times that the region is called as a CNV when compared with reference samples) is a new parameter added during the summarization step to allow confident CNV regions to be extracted from a given test sample and a large reference set. To optimize this parameter, CNVs were called from the same triplicate experiments with NA15510 and NA10851 as described above, but this time they were compared to all 270 HapMap samples as a reference set. Independently verified regions that include both CNVs and diploid regions were placed into different density bins (Fig. 3D). The 10% cutoff gave the optimal result with 7.7% false positives and 33.3% false negatives.
Copy number inference: Identification of diploid samples
In order to accurately identify gains and losses in common CNV regions, each sample’s CNV copy number was calculated by comparison only with diploid samples, which were initially identified by a maximum clique algorithm as the largest copy number group. To confirm that the two-copy group was identified correctly, we used SNP genotypes to calculate the level of heterozygosity and the A/B ratio for each CNV region with the assumption that single-copy losses should be homozygous while three-copy number regions should show heterozygous A/B ratios significantly different from 1. For regions that did not satisfy these assumptions, the largest group remaining after removing the previously defined set was reselected as the diploid group. The majority of CNV regions did not deviate from expectations, and in the end only a small percentage (5.8%) of CNVs required a re-evaluation of the diploid set.
CNV boundary determination
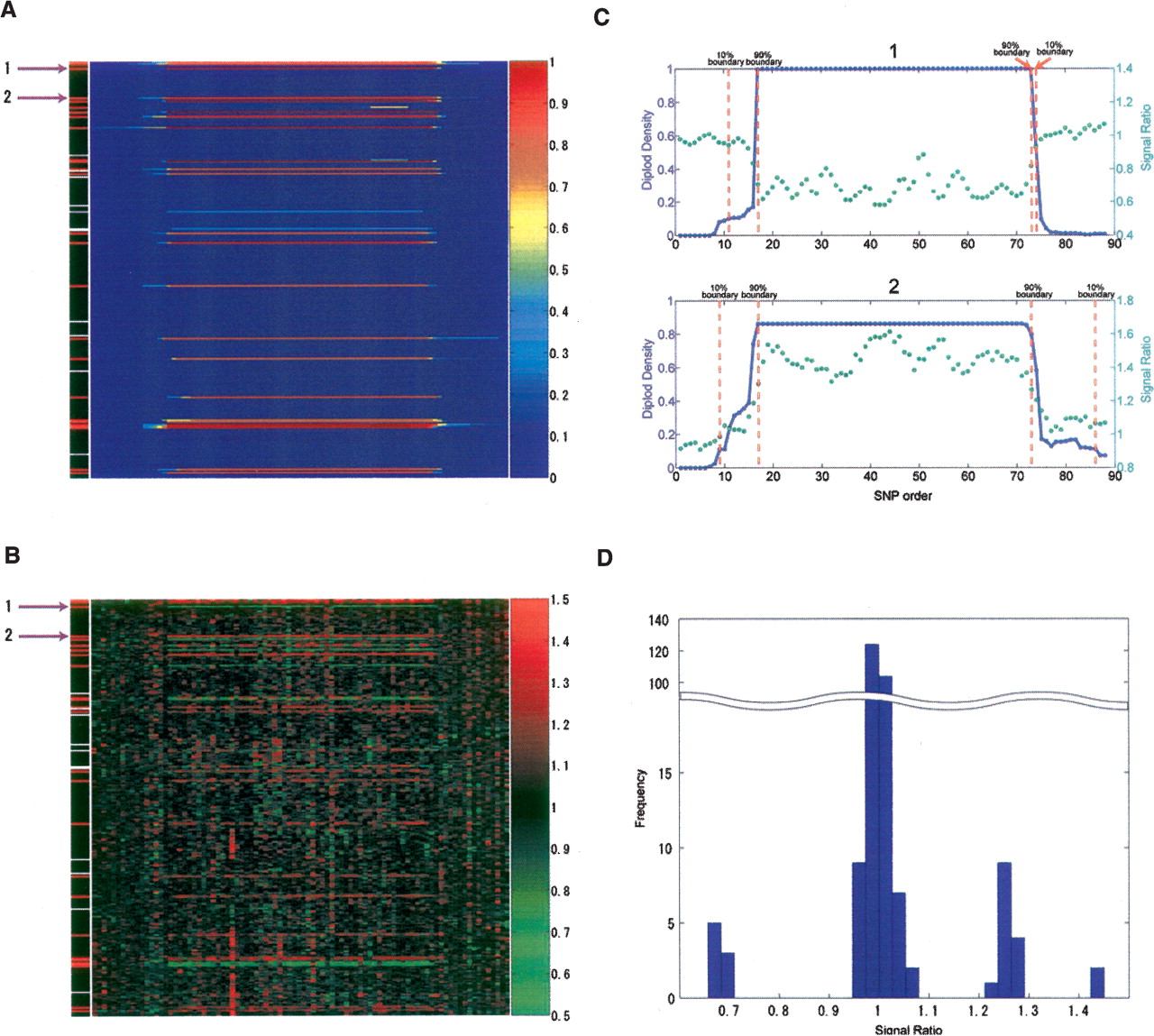
When individual CNVs are called for each sample, the borders are not always the same for the 269 comparisons (Figs. 2A, 4). Because of this variability, a density cutoff is assigned to each boundary as a measure of the confidence associated with the border position. In this case, the density cutoff is based on the maximum density, which is defined as the largest density value for any SNP in that CNV region after comparison with the diploid samples. Thus, the 10% and 90% boundaries are the outer SNP positions of the segments that maintain at least 10% or 90% of the maximum density, respectively (Fig. 4).
CNV boundary determination. In A,B,C, the x-axis is the sequential order of the SNPs both within and outside the CNV region; the y-axis represents individual samples. In D, the x-axis represents the intensity ratio, and the y-axis is the sample frequency. (A) Diploid density distribution for HapMap CNVID 1166 at chr22:23932716–24371067. In the left-most column, CNV samples detected by the algorithm are shown in red, the diploid samples selected by the maximum clique algorithm are shown in black, and samples that display the intensity trend but do not meet the CNV extraction criteria are shown in white. (B) Median ratio distribution in the same region as shown in A. The median ratios were calculated based on the diploid samples with the same genotype. The similar pattern with A indicates that the CNV regions were successfully detected by the algorithm. (C) The diploid density (blue solid line) and median ratio (green dotted line) smoothed with a 10 probe window of sample 1 (top graph) and sample 2 (bottom graph) indicated by the purple arrows in A and B. The 10% and 90% boundaries are shown as dashed red lines. (D) The intensity ratio histogram of all 270 samples in the same CNV region depicted in A, B, and C shows clear clusters that correspond to one, two, three, and four copies of the region. The histogram is compressed in the middle range of the y-axis as represented by the wavy double line.
HapMap CNVs
The CNV calling algorithm, with an optimized density cutoff of 10%, was applied to 270 HapMap samples, and 6469 sample-level CNVs in total were identified with an average of 24 CNVs per individual (Table 1; Redon et al. 2006). CNV calls were merged and summarized into 1203 CNV events (where CNVs are merged if they contain 30% SNP overlap) and 980 nonoverlapping CNV regions (Redon et al. 2006; Supplemental material). The size distribution of the 1203 CNV events ranges from 1 Kb to 3.6 Mb, with median size of 71 kb using the 10% boundaries, and the majority of CNV regions contain between five and 20 SNPs (Supplemental Table 1).
CNV coverage by chromosome
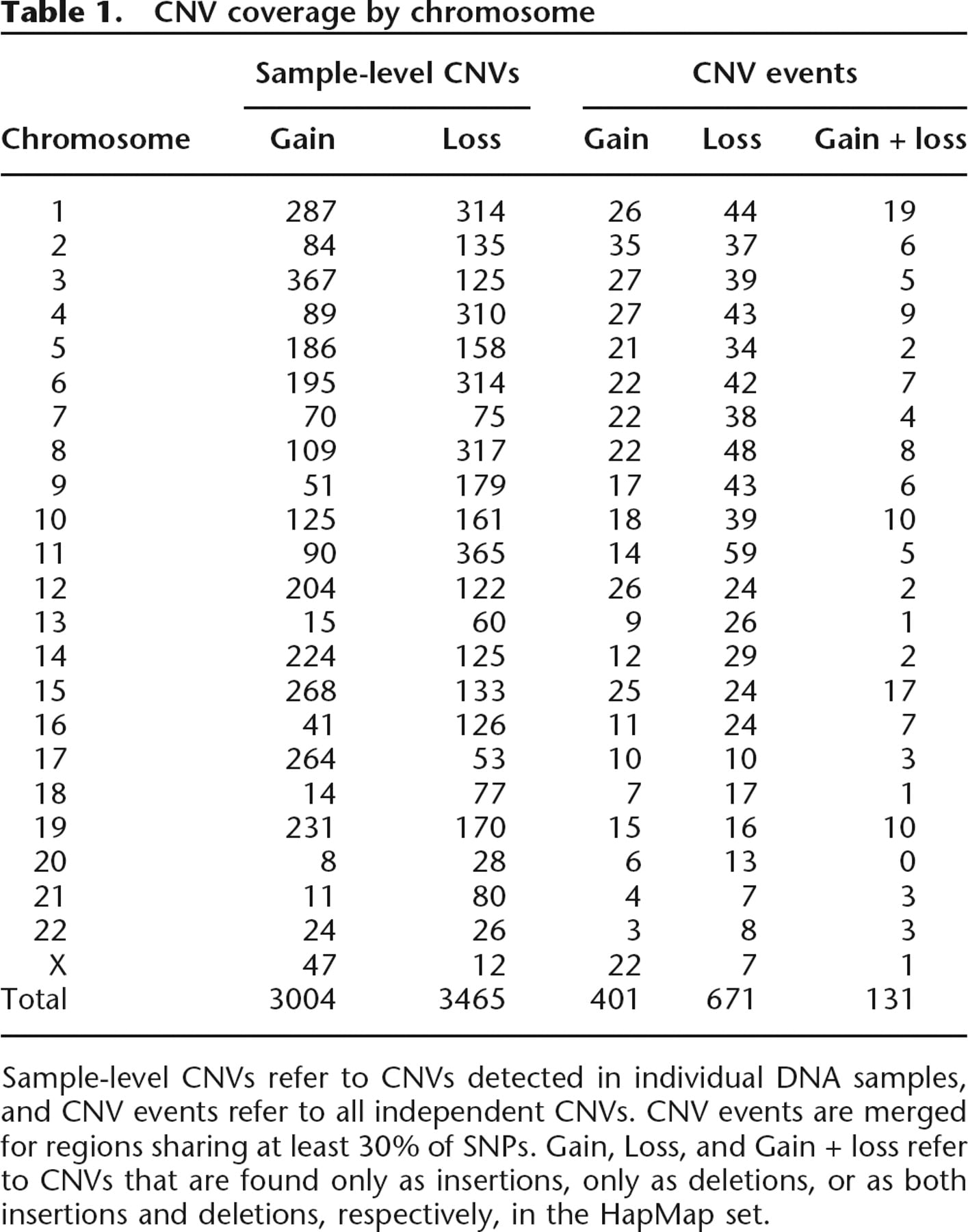
Sample-level CNVs refer to CNVs detected in individual DNA samples, and CNV events refer to all independent CNVs. CNV events are merged for regions sharing at least 30% of SNPs. Gain, Loss, and Gain + loss refer to CNVs that are found only as insertions, only as deletions, or as both insertions and deletions, respectively, in the HapMap set.
Mendelian inheritance
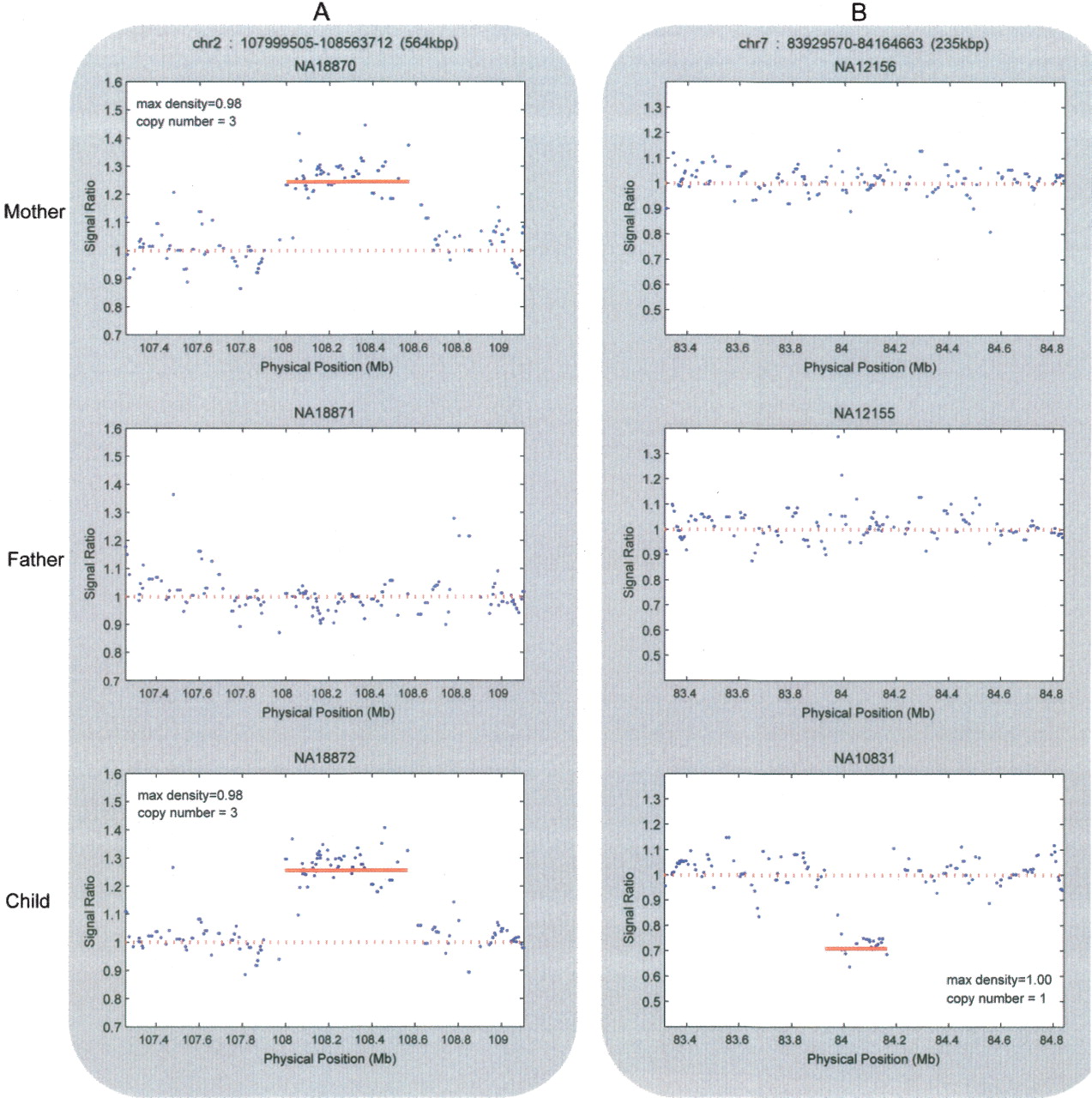
CNVs that were detected from 60 trios from the CEU and YRI populations were analyzed for Mendelian inheritance, and 1229 regions in 60 offspring were identified as CNVs with an inferred copy number of at least three for gains or at most one for losses. The signal intensities were evaluated in the parents for these regions, and 1185 (96.4%) of the CNVs were clearly inherited or displayed a signal intensity profile in one of the parents that is just below the threshold cutoff (Fig. 5A). In addition, 3.6% (44) of CNVs do not show any signal indicative of a possible copy number alteration in one of the parents (Fig. 5B). The latter category may represent de novo CNVs, CNVs present as gains and losses in both parents of the trio (i.e., both parents have one chromosome with two copies, and one chromosome with zero copies), or cell line artifacts. Taken together, these data suggest that at least 96.4% of CNVs display Mendelian inheritance, confirming previous conclusions that CNVs are highly heritable (Locke et al. 2006).
Mendelian and non-Mendelian CNV inheritance. (A) Transmission of a single copy gain transmitted from a YRI mother (NA18870), to the child (NA18872), and absent in the father (NA18871). These results were also confirmed by qPCR. (B) A single copy loss identified in a CEU child (NA10831) that is not present in either parent (NA12156 or NA12155). Each plot shows the smoothed (50-kb window) signal ratio intensity on the y-axis and the physical position of the probes on the x-axis.
Experimental validation and false-positive estimation of HapMap CNV calls
In order to estimate the percentage of HapMap CNV calls that are likely to be false positives, we used quantitative PCR (qPCR) and mass spectrometry for experimental validation and compared replicates of the same DNA sample (self–self comparisons). Experimental validation of CNVs called in three replicate experiments for DNA samples NA15510 and NA10851 (each compared to the HapMap reference set) indicated that the average percent of false positives was 2.5% (Table 2). Similarly, self–self comparisons of 10 HapMap samples, each done in triplicate, identified an average of 0.73 CNVs per experiment (Supplemental Table 3). In addition, when these 10 samples are compared to the HapMap reference set, 80% of the CNVs are called in all three replicates (see Redon et al. 2006). Taken together, the above data indicate that the false-positive signal due to intensity variability is <5%, and that the reproducibility is consistently high.
Independent verification of CNV calls
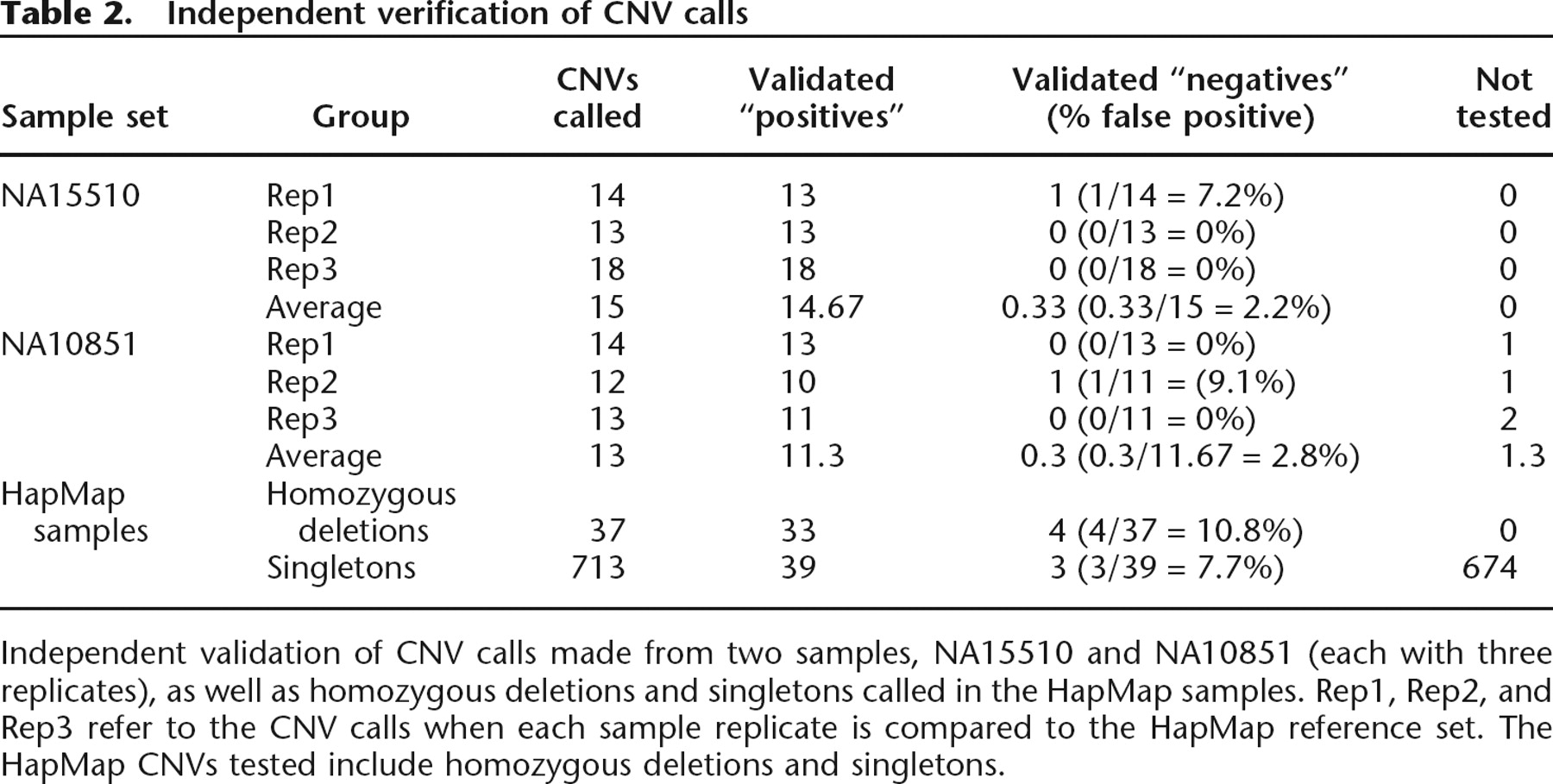
Independent validation of CNV calls made from two samples, NA15510 and NA10851 (each with three replicates), as well as homozygous deletions and singletons called in the HapMap samples. Rep1, Rep2, and Rep3 refer to the CNV calls when each sample replicate is compared to the HapMap reference set. The HapMap CNVs tested include homozygous deletions and singletons.
HapMap CNVs called in only one individual (singletons) represent a large percentage of the total CNVs found in the population (Redon et al. 2006), yet may include a higher number of false positives compared to CNVs called in multiple individuals. Nevertheless, 36 out of 39 singleton CNVs that were tested were experimentally validated, suggesting that singleton CNVs are correctly called >90% of the time (Table 2).
Homozygous deletions, which are called using a separate algorithm, were also tested and found to be correctly identified at least 89% of the time, with 11% of homozygous deletion regions giving rise to a PCR product using nonquantitative measurements (Table 2). The reproducibility of the homozygous deletion detection algorithm was assessed using the replicates of NA15510 and NA10851. In total, two homozygous deletion regions were identified and validated (Redon et al. 2006). In each case, the results were identical in all three replicates, and with the same boundaries, suggesting a high reproducibility for this calling algorithm.
As mentioned above, the precise delineation of CNV boundaries in each sample is challenging. Estimation of CNV ends can be complicated by chromosomal mosaicism in cell culture, where the CNV ends may exhibit differences from cell to cell, or, in the case of high-frequency CNVs, the edges may be sample-specific, making it difficult to define a single population consensus boundary. Another possibility is that the variability is simply a reflection of experimental noise. To test this idea, and to evaluate the accuracy of border estimations given by the algorithm, a representative sampling of CNVs was experimentally tested in the regions between the flanking SNP and the 10% borders, the 10% and the 90% borders, and within the 90% borders (Supplemental Table 4). In all six cases, the region between the highest confidence 90% borders was confirmed as a true region of copy number change. In four unique sequence CNVs (i.e., not in segmentally duplicated regions), all eight 10% borders were confirmed. For these same CNVs, the regions flanking the 10% borders were altered in four out of eight cases. In contrast, for CNV regions that contain segmental duplications, the border determinations were not as accurate, and in all four examples the 10% specific regions were not confirmed. This shows that borders of CNVs in unique sequence regions can be determined with high confidence, but less so for common CNVs, especially those associated with segmental duplications. Thus, the accuracy of border determination reflects the underlying genomic structure in regions of CNV.
Discussion
We have developed a multistep algorithm that allows accurate CNV calls to be derived from the GeneChip Human Mapping 500K EA arrays. The method described here has been developed to reduce systematic noise and precisely extract significant intensity information. It is substantially different from the previously developed GIM algorithm in several aspects including (1) an intensity pre-processing step, (2) an allele-specific ratio adjustment, (3) the incorporation of new variables (restriction enzyme recognition site normalization, signal ratio adjustment based on G:C content of SNP-surrounding sequence) to remove systematic noise, and (4) the use of a robust regression with Bayesian Information Criterion (BIC) selection (Qian 1996) to simplify the calculations without sacrificing the accuracy. SW-ARRAY, previously used only for large copy number changes associated with cancer and disease, has been optimized to call CNVs. Most importantly, the proposed algorithm enables comparisons of any DNA test sample against a large reference set, which allows precise assignment of CNVs to the test sample and derives more accurate estimates of the CNV boundaries. The CNV extraction step is completely novel, and uses a scoring system implemented following SW-ARRAY that summarizes all pairwise comparisons with the large reference set. Since the copy number inference step identifies diploid samples for any given region, there is no reduction in detection power of common CNVs.
When used with DNA samples from the HapMap population, the approach described here led to the identification of 1203 CNV events spanning a broad size range from <1 kb to >3 Mb (Redon et al. 2006). Although the 500K EA platform has good resolving power to identify CNVs <100 kb in size, CNVs spanning segmental duplications are underrepresented because of the difficulty in developing robust SNP assays in these regions (Fredman et al. 2004). Future generations of oligonucleotide-based copy number arrays can be designed to minimize this discrepancy and have appropriate representation for segmentally duplicated regions. For example, a new high-density array that contains multiple nonpolymorphic probes for every predicted NspI fragment and is used in conjunction with WGSA has been designed. This array covers >1.3 million fragments with a median intermarker distance of just less than 800 bp. Furthermore, >90% of genome-wide segmental duplications have at least one of these fragments within their boundaries. In addition to these higher-density arrays, an alternative approach might involve the use of molecular inversion probes (MIP), which have successfully been used for copy number analysis and can specifically target selected regions of the genome (Wang et al. 2005).
Efforts directed at a global characterization of CNVs are an important first step toward understanding the role of CNVs in the biology of the cell. The CNVs identified using this algorithm, combined with the complementary data derived from the WGTP platform, provide the framework for the first comprehensive global map of human CNVs (Redon et al. 2006). This information should also prove useful in better understanding the role of CNVs in disease pathology and will provide a more detailed baseline for discriminating DNA copy number changes in cancer cells. Lastly, the data described here have been used to study the genetic correlation between CNVs and SNPs (Redon et al. 2006). There is a decreased level of linkage disequilibrium between CNVs and SNPs, suggesting that SNPs are not an ideal surrogate for CNVs in association studies (Hinds et al. 2006; Locke et al. 2006; McCarrol et al. 2006; Newman et al. 2006; Redon et al. 2006). This implies that CNVs need to be assessed independently in whole-genome association studies. The algorithm described here, along with high-density DNA oligonucleotide arrays, offers an optimal solution by providing both SNP genotype information as well as CNV profiling in a single experiment.
Methods
Experiments
500K EA Arrays and the Whole-Genome Sampling Assay (WGSA)
The 500K EA arrays contain 534,500 SNPs on two enzyme-specific arrays and are used in conjunction with whole-genome sampling analysis (WGSA). Each array interrogates SNPs residing on NspI or StyI PCR amplicons that range in size from 200 bp to 1000 bp. The method described here should be directly applicable to the commercially available Affymetrix 500K array. As an example, 97.5% of the CNVs identified in this study contain at least four SNPs (the requirement for calling a CNV) that are present on the commercial array.
DNA from cell lines derived from the 270 HapMap individuals as well as NA15510 used for parameter tuning were purchased from NIGMS the Human Genetic Cell Repository, Coriell Institute for Medical Research (Camden, NJ). For preparation of the DNA prior to hybridization, we used the pre-commercial or early access version of WGSA, which is identical to the manufacturer’s commercial protocol (Affymetrix; http://www.affymetrix.com) with the following modifications. For PCR, 5 μL of diluted, adapter-ligated DNA and 3.5 μM primer were used in a total volume of 100 μL, and three reactions were prepared for each DNA sample per enzyme. Sixty micrograms of purified product were fragmented and end-labeled using 0.57 mM DLR (GeneChip DNA Labeling Reagent) and 105 U of TdT (Promega) for 2 h at 37°C. Hybridization onto the 250K Nsp and 250K Sty EA arrays and subsequent washing steps were done exactly as described by the manufacturer (Affymetrix).
For data quality assessment, genotype calls were generated using the DM (Dynamic Modeling) calling algorithm with a cutoff P-value of 0.17 (Di et al. 2005). Intensity information for each probe set was extracted using Affymetrix software (http://www.affymetrix.com). Any arrays giving rise to a call rate of <85% were redone. Approximately 10% of samples were reprocessed based on this criterion. In addition, 15 samples that showed high standard deviations of normalized intensity ratios were also reprocessed. Prior to GIM, genotype calls were generated using DM (Di et al. 2005). For an average experiment, the resulting genotyping quality was consistently high, with an average call rate of 96.8% and concordance of 99.5% with HapMap Phase I genotypes (Supplemental Table 3).
Validation
Quantitative PCR
Primer Express v3.0 (Applied Biosystems) was used for primer design, and, when possible, avoided segmental duplications, SNPs, or simple repeats. The UCSC In-Silico PCR tool (http://genome.ucsc.edu) was used to check for single amplicons. Multiplex real-time PCR reactions were performed using the ABI Prism 7700 Sequence Detection System (Applied Biosystems) and followed the manufacturer’s guidelines and cycling conditions. For normalization, a VIC-labeled TaqMan probe to the RPPH1 locus (RNA moiety of RNase P) was used (PE Applied Biosystems). At least three replicate reactions were run for each primer pair, and the comparative CT method (User Bulletin #2; Applied Biosystems) was used to calculate the fold change at each locus between the test and reference samples. In addition, a t-test based on the ΔCt values was used to determine the statistical significance of the result. All results that showed a fold change <0.9 or >1.10 as well as a P-value <0.05 were considered to be significant. Detailed QPCR results are presented as Supplemental material elsewhere (Redon et al. 2006).
Quantitative validation of CNVs using mass spectrometry
The determination of allele frequencies in test and reference samples was based on MALDI-TOF mass spectroscopy of allele-specific primer extension products (MassArray Sequenom Inc.) (Bansal et al. 2002; Mohlke et al. 2002; Downes et al. 2004). All assays for the PCR and associated extension reactions were performed as suggested by the manufacturer. At any given SNP, a CNV is considered validated in a heterozygous individual if the allele dosage ratios are statistically different from reference heterozygous individuals with no CNVs. DNA from individuals with homozygous genotypes in the CNV region are mixed with references homozygous for the alternate allele, and its allelic dosage ratio is compared with heterozygous references to calculate the significance of the deviations. The appropriate mixture procedure in these samples is verified by distinct CNV-free loci. In all cases, a P-value <0.05 (t-test) was considered significant. Detailed mass spectrometric results are presented as Supplemental material elsewhere (Redon et al. 2006).
PCR validation of homozygous deletions
All 37 homozygous deletion regions were tested using PCR. PCR consisted of 34 cycles of 94°C for 20 sec, 68° to 51°C for 20 sec (0.5°C decrease/cycle), and 72°C for 60 sec. One to three DNA samples that were called for the deletion were examined along with three diploid reference samples. Visual inspection of agarose gels stained with ethidium bromide was used to assess the presence or absence of the PCR product.
Summary of data analysis
The specific details of the CNV calling algorithm, including all mathematical formulas, can be found in the online Supplemental material. Prior to CNV calling in the HapMap samples, cell line artifacts were identified as any chromosomal segmental imbalance with a deviation of normalized intensity ratios >0.025 (or >0.05 for the X chromosome) for regions >5 Mb and that were observed in only one sample. Thirty-six such regions were identified and removed prior to analysis (Redon et al. 2006). An additional seven CNV events were removed based on loss of transmitted allele (LTA) analysis as described in Redon et al. (2006). (For a complete listing, see Supplemental material in Redon et al. 2006). In addition to cell line artifacts, immunoglobulin (Ig) genes were removed from the analysis. These regions include IgK at 2p11, IgL at 22q11, and IgH at 14q32 (Redon et al. 2006).
The parameters used for CNV calling required that four SNPs on three restriction fragments gave rise to a signal intensity ratio above 1.12 for insertions or 0.89 for deletions. CNVs were considered significant for P-values <0.01 using 5000 permutations of the data (see Results). For data integration, only CNVs called in at least 10% of the comparisons to the diploid samples were retained.
Data release
The raw data from this study are posted at the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE5013. The equivalent data for the commercially available Affymetrix GeneChip Human Mapping 500K arrays are also being released as part of this project (GEO accession no. GSE5173). Publicly available software called GEMCA (Genotyping Microarray based CNV Analysis), which implements this CNV calling algorithm, can be freely downloaded from http://www2.genome.rcast.u-tokyo.ac.jp/CNV/.
Acknowledgments
We thank Hiroko Meguro for technical assistance. This research was supported by the Core Research for Evolutional Science and Technology (CREST) from the Japan Science and Technology Agency (to H. Aburatani), Grants-in-Aid for Young Scientists, and Grant-in-Aid for Scientific Research on Priority Areas “Applied Genomics” (to S. Ishikawa) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and Grants-in-Aid from National Institute of Biomedical Innovation (to S. Ihara). S.W.S. is an Investigator of the CIHR and International Scholar of the Howard Hughes Medical Institute.
References
- ↵A. Bansal,D. van den Boom,S. Kammerer,C. Honisch,G. Adam,C.R. Cantor,P. Kleyn,A. Braun,(2002) Association testing by DNA pooling: An effective initial screen. Proc. Natl. Acad. Sci. 99:16871–16874.
- ↵R. Beroukhim,M. Lin,Y. Park,K. Hao,X. Zhao,L.A. Garraway,E.A. Fox,E.P. Hochberg,I.K. Mellinghoff,M.D. Hofer,(2006) Inferring loss-of-heterozygosity from unpaired tumors using high-density oligonucleotide SNP arrays. PLoS Comput. Biol. 2:e41.
- ↵G.R. Bignell,J. Huang,J. Greshock,S. Watt,A. Butler,S. West,M. Grigorova,K.W. Jones,W. Wei,M.R. Stratton,(2004) High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res. 14:287–295.
- ↵D.F. Conrad,T.D. Andrews,N.P. Carter,M.E. Hurles,J.K. Pritchard,(2006) A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 38:75–81.
- ↵X. Di,H. Matsuzaki,T.A. Webster,E. Hubbell,G. Liu,S. Dong,D. Bartell,J. Huang,R. Chiles,G. Yang,(2005) Dynamic model based algorithms for screening and genotyping over 100K SNPs on oligonucleotide microarrays. Bioinformatics 21:1958–1963.
- ↵K. Downes,B.J. Barratt,P. Akan,S.J. Bumpstead,S.D. Taylor,D.G. Clayton,P. Deloukas,(2004) SNP allele frequency estimation in DNA pools and variance components analysis. Biotechniques 36:840–845.
- ↵L. Feuk,A.R. Carson,S.W. Scherer,(2006a) Structural variation in the human genome. Nat. Rev. Genet. 7:85–97.
- ↵L. Feuk,C.R. Marshall,R.F. Wintle,S.W. Scherer,(2006b) Structural variants: Changing the landscape of chromosomes and design of disease studies. Hum. Mol. Genet. 15:R57–R66.
- ↵S.P. Fodor,J.L. Read,M.C. Pirrung,L. Stryer,A.T. Lu,D. Solas,(1991) Light-directed, spatially addressable parallel chemical synthesis. Science 251:767–773.
- ↵S.P. Fodor,R.P. Rava,X.C. Huang,A.C. Pease,C.P. Holmes,C.L. Adams,(1993) Multiplexed biochemical assays with biological chips. Nature 364:555–556.
- ↵D. Fredman,S.J. White,S. Potter,E.E. Eichler,J.T. Den Dunnen,A.J. Brookes,(2004) Complex SNP-related sequence variation in segmental genome duplications. Nat. Genet. 36:861–866.
- ↵J.L. Freeman,G.H. Perry,L. Feuk,R. Redon,S.A. McCarroll,D.M. Altshuler,H. Aburatani,K.W. Jones,C. Tyler-Smith,M.E. Hurles,(2006) Copy number variation: New insights in genome diversity. Genome Res. 16:949–961.
- ↵E. Gonzalez,H. Kulkarni,H. Bolivar,A. Mangano,R. Sanchez,G. Catano,R.J. Nibbs,B.I. Freedman,M.P. Quinones,M.J. Bamshad,(2005) The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 307:1434–1440.
- ↵D.A. Hinds,A.P. Kloek,M. Jen,X. Chen,K.A. Frazer,(2006) Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat. Genet. 38:82–85.
- ↵J.D. Hoheisel,(2006) Microarray technology: Beyond transcript profiling and genotype analysis. Nat. Rev. Genet. 7:200–210.
- ↵J. Huang,W. Wei,J. Zhang,G. Liu,G.R. Bignell,M.R. Stratton,P.A. Futreal,R. Wooster,K.W. Jones,M.H. Shapero,(2004) Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum. Genomics 1:287–299.
- ↵J. Huang,W. Wei,J. Chen,J. Zhang,G. Liu,X. Di,R. Mei,S. Ishikawa,H. Aburatani,K.W. Jones,(2006) CARAT: A novel method for allelic detection of DNA copy number changes using high density oligonucleotide arrays. BMC Bioinformatics 7:83.
- ↵A.J. Iafrate,L. Feuk,M.N. Rivera,M.L. Listewnik,P.K. Donahoe,Y. Qi,S.W. Scherer,C. Lee,(2004) Detection of large-scale variation in the human genome. Nat. Genet. 36:949–951.
- ↵K. Inoue,J.R. Lupski,(2002) Molecular mechanisms for genomic disorders. Annu. Rev. Genomics Hum. Genet. 3:199–242.
- ↵International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320.
- ↵International Human Genome Sequencing Consortium (2004) Finishing the euchromatic sequence of the human genome. Nature 431:931–945.
- ↵S. Ishikawa,D. Komura,S. Tsuji,K. Nishimura,S. Yamamoto,B. Panda,J. Huang,M. Fukayama,K.W. Jones,H. Aburatani,(2005) Allelic dosage analysis with genotyping microarrays. Biochem. Biophys. Res. Commun. 333:1309–1314.
- ↵G.C. Kennedy,H. Matsuzaki,S. Dong,W.M. Liu,J. Huang,G. Liu,X. Su,M. Cao,W. Chen,J. Zhang,(2003) Large-scale genotyping of complex DNA. Nat. Biotechnol. 21:1233–1237.
- ↵D. Komura,K. Nishimura,S. Ishikawa,B. Panda,J. Huang,H. Nakamura,S. Ihara,M. Hirose,K.W. Jones,H. Aburatani,(2006) Noise reduction from genotyping microarrays using probe level information. In Silico Biol. 6:9.
- ↵T. Laframboise,B.A. Weir,X. Zhao,R. Beroukhim,C. Li,D. Harrington,W.R. Sellers,M. Meyerson,(2005) Allele-specific amplification in cancer revealed by SNP array analysis. PLoS Comput. Biol. 1:e65.
- ↵D.P. Locke,A.J. Sharp,S.A. McCarroll,S.D. McGrath,T.L. Newman,Z. Cheng,S. Schwartz,D.G. Albertson,D. Pinkel,D.M. Altshuler,(2006) Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am. J. Hum. Genet. 79:275–290.
- ↵H. Matsuzaki,S. Dong,H. Loi,X. Di,G. Liu,E. Hubbell,J. Law,T. Berntsen,M. Chadha,H. Hui,(2004a) Genotyping over 100,000 SNPs on a pair of oligonucleotide arrays. Nat. Methods 1:109–111.
- ↵H. Matsuzaki,H. Loi,S. Dong,Y.-Y. Tsai,J. Fang,J. Law,X. Di,W.-M. Liu,G. Yang,G. Liu,(2004b) Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high density oligonucleotide array. Genome Res. 14:414–425.
- ↵S.A. McCarrol,T.N. Hadnott,G.H. Perry,P.C. Sabeti,M.C. Zody,J.C. Barrett,S. Dallaire,S.B. Gabriel,C. Lee,M.J. Daly,(2006) Common deletion polymorphisms in the human genome. Nat. Genet. 38:86–92.
- ↵Y. Midorikawa,S. Yamamoto,S. Ishikawa,N. Kamimura,H. Igarashi,H. Sugimura,M. Makuuchi,H. Aburatani,(2006) Molecular karyotyping of human hepatocellular carcinoma using single-nucleotide polymorphism arrays. Oncogene 25:5581–5590.
- ↵K.L. Mohlke,M.R. Erdos,L.J. Scott,T.E. Fingerlin,A.U. Jackson,K. Silander,P. Hollstein,M. Boehnke,F.S. Collins,(2002) High-throughput screening for evidence of association by using mass spectrometry genotyping on DNA pools. Proc. Natl. Acad. Sci. 99:16928–16933.
- ↵Y. Nannya,M. Sanada,K. Nakazaki,N. Hosoya,L. Wang,A. Hangaishi,M. Kurokawa,S. Chiba,D.K. Bailey,G.C. Kennedy,(2005) A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 65:6071–6079.
- ↵T.L. Newman,M.J. Rieder,V.A. Morrison,A.J. Sharp,J.D. Smith,L.J. Sprague,R. Kaul,C.S. Carlson,M.V. Olson,D.A. Nickerson,(2006) High-throughput genotyping of intermediate-size structural variation. Hum. Mol. Genet. 15:1159–1167.
- ↵K. Ouahchi,N. Lindeman,C. Lee,(2006) Copy number variants and pharmacogenomics. Pharmacogenomics 7:25–29.
- ↵A.C. Pease,D. Solas,E.J. Sullivan,M.T. Cronin,C.P. Holmes,S.P. Fodor,(1994) Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proc. Natl. Acad. Sci. 91:5022–5026.
- ↵T.S. Price,R. Regan,R. Mott,A. Hedman,B. Honey,R.J. Daniels,L. Smith,A. Greenfield,A. Tiganescu,V. Buckle,(2005) SW-ARRAY: A dynamic programming solution for the identification of copy-number changes in genomic DNA using array comparative genome hybridization data. Nucleic Acids Res. 33:3455–3464.
- ↵G. Qian,R.H. Kunsch,(1996) Technical Report 80, Seminar Fur Statistik, On model selection in robust linear regression (Eidgenossische Technische Hochschule (ETH) Zurich, Switzerland).
- ↵R. Redon,S. Ishikawa,K.R. Fitch,L. Feuk,G.H. Perry,T.D. Andrews,H. Fiegler,M.H. Shapero,W. Chen,(2006) Global variation in copy number in the human genome. Nature, (in press).
- ↵J. Sebat,B. Lakshmi,J. Troge,J. Alexander,J. Young,P. Lundin,S. Maner,H. Massa,M. Walker,M. Chi,(2004) Large-scale copy number polymorphism in the human genome. Science 305:525–528.
- ↵A.J. Sharp,D.P. Locke,S.D. McGrath,Z. Cheng,J.A. Bailey,R.U. Vallente,L.M. Pertz,R.A. Clark,S. Schwartz,R. Segraves,(2005) Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 77:78–88.
- ↵H.R. Slater,D.K. Bailey,H. Ren,M. Cao,K. Bell,S. Nasioulas,R. Henke,K.H. Choo,G.C. Kennedy,(2005) High-resolution identification of chromosomal abnormalities using oligonucleotide arrays containing 116,204 SNPs. Am. J. Hum. Genet. 77:709–726.
- ↵M.R. Speicher,N.P. Carter,(2005) The new cytogenetics: Blurring the boundaries with molecular biology. Nat. Rev. Genet. 6:782–792.
- ↵E. Tuzun,A.J. Sharp,J.A. Bailey,R. Kaul,V.A. Morrison,L.M. Pertz,E. Haugen,H. Hayden,D. Albertson,D. Pinkel,(2005) Fine-scale structural variation of the human genome. Nat. Genet. 37:727–732.
- ↵A.E. Urban,J.O. Korbel,R. Selzer,T. Richmond,A. Hacker,G.V. Popescu,J.F. Cubells,R. Green,B.S. Emanuel,M.B. Gerstein,(2006) High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays. Proc. Natl. Acad. Sci. 103:4534–4539.
- ↵Y. Wang,M. Moorhead,G. Karlin-Neumann,M. Falkowski,C. Chen,F. Siddiqui,R.W. Davis,T.D. Willis,M. Faham,(2005) Allele quantification using molecular inversion probes (MIP) Nucleic Acids Res. 33:e183.
- ↵X. Zhao,C. Li,J.G. Paez,K. Chin,P.A. Janne,T.H. Chen,L. Girard,J. Minna,D. Christiani,C. Leo,(2004) An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 64:3060–3071.
Notes
[1] [Supplemental material is available online at www.genome.org. The array data from this study have been submitted to GEO under accession nos. GSE5013 and GSE5173.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.5629106