
Pooled genomic indexing of rhesus macaque
- Aleksandar Milosavljevic1,4,
- Ronald A. Harris1,
- Erica J. Sodergren1,
- Andrew R. Jackson1,
- Ken J. Kalafus1,
- Anne Hodgson1,
- Andrew Cree1,
- Weilie Dai1,
- Miklos Csuros2,
- Baoli Zhu3,
- Pieter J. de Jong3,
- George M. Weinstock1, and
- Richard A. Gibbs1
- 1 Human Genome Sequencing Center, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA
- 2 Department of Computer Science and Operations Research, Université de Montréal, Montréal, Québec H3C 3J7, Canada
- 3 Children's Hospital Oakland Research Institute, Oakland, California 94609, USA
Abstract
Pooled genomic indexing (PGI) is a method for mapping collections of bacterial artificial chromosome (BAC) clones between species by using a combination of clone pooling and DNA sequencing. PGI has been used to map a total of 3858 BAC clones covering ∼24% of the rhesus macaque (Macaca mulatta) genome onto 4178 homologous loci in the human genome. A number of intrachromosomal rearrangements were detected by mapping multiple segments within the individual rhesus BACs onto multiple disjoined loci in the human genome. Transversal pooling designs involving shuffled BAC arrays were employed for robust mapping even with modest DNA sequence read coverage. A further innovation, short-tag pooled genomic indexing (ST-PGI), was also introduced to further improve the economy of mapping by sequencing multiple, short, mapable tags within a single sequencing reaction.
Bacterial artificial chromosomes (BACs) have emerged as the large-insert cloning system of choice for mammalian genome projects. The restriction fingerprint mapping method (Coulson et al. 1986; Olson et al. 1986) provided a sequence-ready physical map of human BACs (Marra et al. 1997; Soderlund et al. 2000) used for the initial assembly of the human genome (Lander et al. 2001). Genomic sequences of humans and a large number of other organisms have enabled novel strategies for the design of hybridization probes for systematic identification of BACs from one species that map onto specific genomic regions of a related species (Kim et al. 2001; Thomas et al. 2003). Moreover, sequences obtained from individual BACs from a particular species can now be used for mapping the BACs onto homologous loci within genomes of related species and for detecting genomic rearrangements spanned by individual BACs.
Two basic BAC sequencing methods are now well established: sequencing of small-insert shotgun libraries prepared from individual BACs (Lander et al. 2001; Waterston et al. 2002; Gibbs et al. 2004) and sequencing of the ends of BAC clone inserts (Zhao 2000; Zhao et al. 2000, 2001; Fujiyama et al. 2002; Larkin et al. 2003; Poulsen and Johnsen 2004). Sequencing of BAC ends does not require relatively costly preparation of shotgun libraries from individual BACs. BAC-end sequencing economically provides limited but useful information for a number of applications. The sequence-tagged connector (STC) method for genome assembly (Mahairas et al. 1999; Siegel et al. 1999, 2000) employs sequences from the ends of BAC clone inserts for the scaffolding of sequence contigs. The BAC-end sequence (BES) mapping method computationally anchors end-sequences onto reference sequences of related species. Because of a generally small number of large chromosomal rearrangements between mammalian genomes, the order of BACs from any particular mammal can now be tentatively inferred by mapping the BACs onto their orthologous positions in the human genome (Fujiyama et al. 2002; Larkin et al. 2003). Mapped BACs can also be selected for targeted comparative sequencing of regions of highest biomedical interest.
In addition to interspecies applications, BACs are useful for the study of variations across strains of model organisms, direct study of haplotype structure within populations, and disease-causing germline rearrangements characteristic of genomic disorders (Lupski 1998). A newly proposed end-sequence profiling (ESP) method (Raphael et al. 2003; Volik et al. 2003) extends applications to the somatic level by employing anchored BAC-end sequences to examine the anatomy of chromosomal aberrations in cancer cells. The unprecedented level of detail of aberrations revealed by ESP opens new opportunities for understanding the progression of cancer and the mechanisms of the development of drug resistance due to a cancer cell's adaptive response to therapy.
The key limitation of all BAC-end sequence mapping methods such as STC, BES, and ESP is their exclusive reliance on the BAC-end sequences. Specifically, BES has limited efficiency even across relatively closely related species such as mammals. About 10% of bovine BAC-end reads can be anchored onto their homologous positions in the mouse genome and only ∼30% onto human (Larkin et al. 2003). Another problem is that BACs such as those created from cancer genomes may contain multiple internal segments from multiple disjoined genomic loci that cannot be sampled by ESP (Volik et al. 2003).
An obvious, albeit much more costly, alternative to the sequencing of BAC ends is direct shotgun sequencing of BACs. Depth of direct shotgun sequencing can in principle be adjusted, allowing detection of chromosomal rearrangements across species or chromosomal aberrations in cancer cells at arbitrary levels of resolution. The key problem with direct shotgun sequencing is the prohibitive cost and effort involved in shotgun library preparation. Even a modest 3× random BAC clone coverage of a mammalian genome would require shotgun library preparation from ∼60,000 BACs at a cost of several million US dollars in library preparations alone, without counting the sequencing cost.
The pooled genomic indexing (PGI) method described here obviates the need for library preparation for individual BACs while enabling the same fine resolution of mapping achievable previously only via direct shotgun sequencing of individual BACs. Short-tag pooled genomic indexing (ST-PGI) further improves the economy of mapping by sequencing multiple, short, mapable tags within a single sequencing reaction.
Shotgun sequencing of pooled BACs has been previously proposed in the context of the clone-array pooled shotgun sequencing (CAPSS) method (Cai et al. 2001). Probabilistic models for CAPSS and the related clone-array pooled shotgun mapping (CAPS-MAP) method have also recently been developed and validated in genome-scale simulation experiments (Csuros et al. 2003). In contrast to CAPSS and CAPS-MAP, which rely solely on the assembly of shotgun reads for deconvolution, PGI employs a reference sequence to reduce the amount of necessary sequencing per BAC. Consequently, if a reference sequence of a related organism is available, the deconvolution of reads and mapping of BACs can be accomplished via PGI with much less effort and at a lower cost.
Mathematical aspects of pooling design for PGI have been previously developed (Csuros and Milosavljevic 2002, 2004), and potential applications of PGI in the context of comparative genomic sequencing have been outlined (Milosavljevic et al. 2003). Here we present the results of the first full experimental validation and characterization of the performance of PGI and ST-PGI. In a key experiment, a total of 3858 randomly selected BAC clones from the CHORI-250 library, representing ∼24% of the rhesus macaque (Macaca mulatta) genome, were mapped onto 4178 homologous loci in the human genome.
Results
Principle of method
The PGI method (Fig. 1) reduces the overhead cost of library preparation by shotgun sequencing BAC pools, each pool typically consisting of 24, 48, or 96 BACs. BACs are conceptually arranged in rectangular array patterns, 24 × 24, 48 × 48, or 96 × 96, respectively, and are pooled by row and by column. Short-insert clone libraries prepared from BAC pools by the shotgun method are sequenced to a defined depth. Reads obtained by shotgun sequencing are computationally anchored onto homologous segments within reference sequences. Shotgun reads from different pools that map close to each other, forming a cluster of mappings in the reference sequence, are deconvoluted to the BAC at the intersection of the pools. The BAC is simultaneously mapped onto the reference sequence at the location of the cluster. A cluster of as few as two reads from two different pools may suffice for mapping.
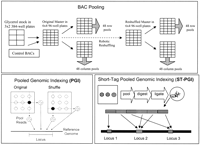
Pooled genomic indexing (PGI). BACs are arrayed in a conceptual rectangular array (top). The original glycerol stock is used to inoculate the Original Master array, which is then grown and used to inoculate two sets of plates, one used to grow cultures for pooling by row and another for pooling cultures by column. For sufficient yields of cells, BACs were grown in duplicate plates, as described in the Methods section. Striped arrows indicate inoculation and growth of cultures in the 96-well format. A shuffled array is also inoculated by using a specially designed single-tip rearraying robot. Shotgun libraries are constructed for each row- and column-pool. Reads from the pools are deconvoluted to the original BACs using the reference genomic sequence (bottom left). If reads from at least two pools map close to each other within the reference sequence, the reads are deconvoluted to the BAC at the intersection. The BAC is also mapped onto the segment (index) between the mapped locations. ST-PGI (bottom right) sequences multiple, short, mapable sequence tags of length between 50 and 200 bp within a single reaction.
The same set of BACs is first arrayed in an original and then in a shuffled array pattern (Fig. 1). The shuffling is performed according to the transversal design, which guarantees that any two pools across the two arrays have at most one BAC in common (Csuros and Milosavljevic 2004). Transversal design has two key benefits: First, it allows unique indexing even if a BAC is sampled in any two out of four possible pools; second, up to three identical or highly overlapping BACs can be unambiguously deconvoluted even if they reside on the same array. Transversal designs also compare favorably in the context of PGI with other well-known combinatorial designs (Csuros and Milosavljevic 2004).
The ST-PGI method (Fig. 1) further improves the economy of PGI by sequencing multiple mapable tags in the 50-200-bp range within a single sequencing reaction. The tags are obtained by digesting DNA prepared from pooled BACs with a cocktail of three enzymes, Bfa I, Mse I, and Csp6 I. The enzymes recognize different 4-bp sites and produce compatible 5′-TA overhangs. Concatemers are formed by ligating the tags and are then sequenced using standard methods. The tags within a sequenced concatemer are computationally parsed using the reference sequence to recognize tag boundaries and are individually anchored onto the reference sequence. The details of the general concatemer sequencing and parsing method, which was originally developed in the context of PGI but has applications well beyond it, are described in the Supplemental material.
Shotgun sequencing and mapping of pooled BACs in controlled experiments
Given the importance of shotgun sequencing of pooled BACs, we sought to characterize this step in controlled experiments. One aim of the experiment was to determine whether locations of constituent BACs can be detected by anchoring shotgun reads from the pools. The second aim was to determine the efficiency of sampling of individual BACs within pools under ideal conditions by full reads and short tags.
A total of 96 BACs were selected from the rat BAC library (CHORI-230). The selected BACs were sequenced in the course of the rat genome sequencing project (Gibbs et al. 2004), and their locations in the assembly were known. While the rat genome was assembled only to a draft level and thus provided an imperfect target for read anchoring, working with rat BACs in the context of the ongoing rat genome sequencing project provided an opportunity to perform experiments efficiently in ideal conditions and on quality-controlled reagents.
Pools containing subsets of the 96 BACs were obtained by the normalized DNA pooling method as described in the Methods section. Four 24-BAC pools contained four disjoint subsets of 24 BACs, two 48-BAC pools contained two disjoint subsets of 48 BACs, and one 96-BAC pool contained all 96 BACs. DNA samples were prepared for each of the pools independently. Four small-insert shotgun libraries were independently prepared from each DNA sample. Counting each small-insert shotgun library preparation as a single experiment, there were a total of sixteen 24-pool experiments, eight 48-pool experiments, and four 96-pool experiments.
A total of 5805 shotgun reads from four 24-BAC pools were uniquely anchored, as well as 6316 reads from two 48-BAC pools and 5734 reads from the 96-BAC pool. As expected, the reads anchored in clusters. Shotgun reads across all sixteen 24-pool experiments formed 344 clusters (out of expected 384), 48-pool reads across all eight experiments formed 387 clusters (out of expected 384), and 96-pool reads across all four experiments formed 344 clusters (out of expected 384). The clusters localized to known genomic locations of the BACs and to homologous regions. Distribution of reads across clusters indicated slightly larger sampling bias in 96-BAC pools than in 48- and 24-BAC pools due to oversampling of a small number of BACs in the pooled DNA (Fig. 2).
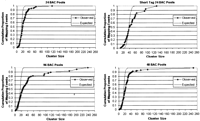
Mapping of reads and tags from pooled rat BACs. Observed and theoretically expected cumulative distributions of mapping events as a function of cluster size are shown for 24-, 48-, and 96-BAC pools and for the ST-PGI method on 24-BAC pools. Points on the observed curve correspond to actual observed clusters. The expected curve is calculated based on the Poisson approximation (lambda = n * p) of the binomial distribution based on observed number of mapping events (n) and clusters (k) and average number of mapping events per cluster (p = n/k). Deviation between observed and theoretically expected distributions indicates bias introduced by oversampling of certain BACs within the pools.
Two DNA samples from 24-BAC pools were also used to validate the performance of the short-tag concatemer sequencing method. Concatemers were formed, sequenced, and parsed into tags, and the tags were mapped onto genomic sequence as described in the Supplemental material. A total of 1834 tags formed 48 clusters (out of expected 48) that collocated with clusters obtained by mapping full-length reads from the same pools. Slightly more tags ended up in highly abundant clusters compared with full-length reads from 24-BAC pools (Fig. 2). A total of 3.2 (SD 0.6) unique mapping events were observed per quality concatemer read versus 0.72 (SD 0.03) such events for quality full-length read, an improvement in mapping efficiency of 4.4 (Supplemental material).
In summary, results of controlled experiments indicated that locations of constituent BACs can be detected by anchoring shotgun reads from the pools, that a 48-BAC pool design provides good uniformity of sampling of individual BACs, and that efficiency of mapping can be increased when using short tags. These results established parameters for the design of PGI experiments for mapping rhesus BACs, as described next.
Mapping rhesus BACs onto the human genome
A total of 4512 randomly selected rhesus BACs from the CHORI 250 library were arrayed across two 48 × 48 arrays, denoted Array 1 and Array 2. A total of 48 wells in Array 1 and 24 wells in Array 2 were left empty and served as negative controls. A set of 24 control BACs of known chromosomal localization was present in both arrays. Each of the two original arrays was shuffled using transversal design. BACs were pooled by row and by column, each pool consisting of 48 BACs and each BAC occurring in four different pools. The two-array transversal design reduces the number of shotgun library preparations to twice the square root of the number of arrayed BACs. Specifically, 192 library preparations, 96 for the original and 96 for the shuffled version of an array, were attempted for each of the 48 × 48 arrays, a total of 384 library preparations for a total of 4608 elements on Arrays 1 and 2. Two library preparations for Array 1 failed.
A total of 100,733 reads were obtained, 37,992 from Array 1 and 62,741 from Array 2. Quality reads were defined as having a length of at least 100 bases of Phred 20 nonvector sequence. When the total number of reads is divided by the total number of pools and then by the number of BACs per pool, one obtains about four reads per BAC per pool in Array 1 and about seven reads per BAC per pool in Array 2. On average, 5.5 quality reads were obtained per BAC per pool, 4.3 (78%) could be mapped by the BLAT program, 3.2 (58%) uniquely mapped (second-best BLAT score was <98% of the top score), and 2.9 (53%) deconvoluted (Fig. 3). Pool-based read statistics can be obtained by multiplying the read numbers by 48, the number of BACs per pool. BAC-based read statistics can be obtained by multiplying the read numbers by four since each BAC occurs in four pools. A total of 53,029 reads, were deconvoluted to individual BACs across both arrays (Fig. 3).
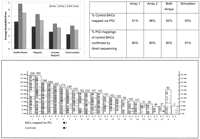
Mapping rhesus BACs onto human genome. On average, 5.5 quality reads were obtained per BAC per pool, 4.3 (78%) could be mapped, 3.2 (58%) uniquely mapped, and 2.9 (53%) deconvoluted (top left). Because each BAC was pooled four times, in two row pools and two column pools using the transversal design, the read numbers need to be multiplied by four to obtain number of reads per BAC. A total of 3858 BACs mapped onto 4178 loci (bottom; an interactive version of this figure is available at www.genboree.org). Number of mappings appears above each chromosome. Performance of PGI was evaluated by using 99 randomly selected BACs and by simulation experiments (top right), as described in the Methods section. Each of the 99 BACs was directly sequenced and mapped. A PGI mapping of a BAC was counted as confirmed if the BAC was mapped to the same position by directly obtained sequence.
A total of 3858 rhesus macaque BACs across the two arrays were mapped onto 4178 homologous positions within the human genome sequence. A total of 1927 BACs from Array 1 mapped onto 2062 loci and a total of 1955 BACs from Array 2 mapped onto 2116 loci. The number of BAC mappings per human chromosome was proportional to chromosome length, except for X and Y which both had fewer mappings, as expected since the BAC library was made from a male (Fig. 3).
To test the accuracy of mapping, a total of 103 BACs from Array 1 were randomly selected as controls and directly sequenced. As discussed in the Methods section, four of them could not be mapped onto the human genome. The failure to map may be due to the loss of ancestral genomic DNA in the human lineage after branching of macaque or due to the incompleteness of the current version of the human genome assembly. In either case, ∼4% of rhesus BACs may not be mapable onto human genome due to reasons unrelated to PGI.
A total of 99 BACs that could be mapped were used as controls. A subset of 24 of those BACs was also included in Array 2. The control BACs allowed us to estimate bounds on the accuracy of BAC mapping via PGI (Fig. 3). Because of potential errors in the confirmation process such as incomplete shotgun sequencing of the BACs, sample tracking, and genomic duplications, the confirmation rate provided only a lower bound on the accuracy of PGI. PGI mapping of simulated rhesus BACs made from mutated human sequence was performed as described in the Methods section to factor out the potential errors inherent in the confirmation process. The controls and simulation indicate that between 91% and 93% of BACs that are mapable by direct shotgun sequencing can also be mapped by PGI at an estimated accuracy of between 80% and 91%.
Despite the fact that Array 2 was sequenced at seven reads per BAC per pool and Array 1 only at four reads per BAC per pool, the confirmation rate for both was the same (80%) and completeness in Array 2 improved only by 5% from 91% to 96%. This indicates that genomes of Old World monkeys can be efficiently mapped onto human even at relatively low coverage of 16 reads per BAC.
In order to compare consistency of mapping obtained by ST-PGI and PGI methods, a total of five row pools and five column pools from Array 2 were also subjected to the three-enzyme ST-PGI protocol (K. Kalafus et al., in prep.). Out of the total of 25 BACs at the intersection of row pools and column pools, a total of 20 (80%) were mapped to identical loci by PGI and ST-PGI. Out of the five remaining BACs, one was mapped to different loci by PGI and ST-PGI and four were mapped by one method but not the other. Tags from the 10 pools (containing 455 distinct BACs) mapped into 411 clusters. Most (88.3%) clusters could be confirmed by an independently obtained full-read PGI index mapping onto the same locus.
The number of mapping events per concatemer varied across libraries prepared from the 10 pools. Concatemers from four of the libraries exhibited between four and 5.5 (4.65 average) more unique mapping events per read compared with the number of unique mapping events per full-length quality read (0.54). The remaining six pools exhibited improvement factor in the one to three range (2.43 average). There were no pools with an improvement factor in the three to four range, indicating bimodal distribution, possibly due to instability of the protocol or due to lack of control of amounts of input DNA in this experiment. Development of a protocol that would consistently achieve improvement factors in the four to 5.5 range is underway. When comparing performance of BES and ST-PGI below, we conservatively assume the current improvement factor of 3.3 over full-length reads.
Detecting chromosomal rearrangements between rhesus and human
Mapping of a rhesus BAC onto multiple disjoined human chromosomal loci indicates a potential chromosomal rearrangement. A total of 63 BACs in the two arrays mapped to two or more disjoined human intrachromosomal loci by three-pool or four-pool indices using an earlier less optimized version of the deconvolution algorithm as described in the Methods section. Of the 63 BACs, a total of 56 mapped to two loci, six to three, and one of them to four loci. In order to confirm that multiple mappings indeed point to rearrangements, 14 BACs from Array 1, each mapping to two disjoined intrachromosomal human loci by a three-pool or four-pool index, were directly sequenced and assembled into contigs, and the contigs were mapped onto the human sequence. Multiple intrachromosomal mappings of four of the BACs—CH250-268P6, CH250-268B13, CH250-268E11, and CH250-272A22—were confirmed by the mapping of directly sequenced contigs (Fig. 4).

Chromosomal rearrangements detected by PGI. Four rearrangements detected by PGI were confirmed by direct sequencing of BACs. The direct sequencing confirmed that each of the BACs mapped to two chromosomal locations in human. Mappings of one of the four BACs, denoted CH250-272A22, indicate either an interchromosomal rearrangement or BAC chimerism; CH250-268B13 and CH250-268E11 span tandemly duplicated regions on chromosome 16; CH250-268P6 (enlarged on the right) spans a 254-kbp interchromosomal duplication in human. By using mouse and rat as outgroups for chimpanzee and human, one can hypothesize that the duplication occurred in the human lineage after the branching of chimpanzee.
BAC CH250-268P6 mapped to two loci on chromosome 4 with a gap of 254 kb between the loci. A gap of similar size is present in orthologous level 1 alignment chains in the mouse, rat, and chimpanzee genomes (Fig. 4). Direct mapping of the rhesus BAC onto the chimpanzee genome assembly did not reveal this gap. This evidence indicates an insertion in the human lineage after the branching of chimpanzee. The putative insert contains the MGC26143 gene. MGC26143 and the KARP-1-binding protein (KAB) are members of the Ensembl Protein Family ENSF00000001155 and are highly similar. The only mappings of MGC26143 and KAB mRNA to chimpanzee place them on chimpanzee chromosome 1, which is orthologous to human chromosome 1. This evidence indicates an interchromosomal duplication event in the human lineage after the branching of chimpanzee.
Direct mappings of contigs from BACs CH250-268B13 and CH250-268E11 revealed that many of the contigs had nearly equivalent mappings to two loci on chromosome 16. These loci on chromosome 16 are known to contain a number of large segmental duplications (Loftus et al. 1999; Eichler et al. 2001).
BAC CH250-272A22 mapped to two loci at a large distance on either side of the known fusion region of chromosome 2. Human chromosome 2 was formed by the telomeric fusion of two ancestral chromosomes, corresponding to chimpanzee chromosomes 12 and 13, and occurred in the human lineage after the branching of great apes (Yunis and Prakash 1982). The rhesus orthologs to chimpanzee chromosomes 12 and 13 are still separate chromosomes (Haig 1999). The rhesus BAC mapped to chimpanzee chromosomes 12 and 13, and the mappings were consistent with their mappings to the fused human chromosome 2. Sequence comparison did not reveal duplicated regions in human or chimpanzee that would explain multiple mappings of the rhesus BAC. Consequently, this BAC mapping pattern can be explained most parsimoniously by hypothesizing (1) a rearrangement between chromosomes in the macaque lineage, (2) a rearrangement between chromosomes in the hominoid lineage after branching of macaque and before the branching of chimpanzee, or (3) chimerism of BAC CH250-272A22.
In summary, out of the 14 BACs with multiple intrachromosomal mappings from Array 1, four were confirmed by the mapping of directly sequenced contigs and at least three of them point to evolutionary rearrangements. One of the four correctly mapped BACs may be chimeric. We should also note that a number of BACs mapped to multiple loci between chromosomes. Attempts to confirm such mappings by sequencing seven BACs with the interchromosomal mappings failed.
As described in the Methods section, a new, more optimized, and accurate deconvolution method has been used to produce the final mappings of rhesus BACs reported in this article. The new method still maps ∼8% of rhesus BACs onto multiple loci. A total of 6% of BACs would be predicted to map to multiple loci due to known intrachromosomal segmental duplications in the human genome (calculation is described in the Methods section). Accounting for imperfect sensitivity of PGI, these results still indicate that a major fraction of rearrangements may be explained by intrachromosomal segmental duplications.
Resolution of chromosomal rearrangement detection
A BAC that spans a breakpoint induced by a chromosomal rearrangement such as a large insertion, inversion, or fission will typically contain two segments that map more than a BAC-size apart on the reference genomic sequence. Much more complex patterns of rearrangements involving more than two segments per BAC were observed in cancer cell lines (Volik et al. 2003). At a sufficient level of sampling by short tags, ST-PGI allows mapping of each of the constituent segments of a BAC to their original chromosomal locations, thus revealing the detailed structure of rearrangements within individual BACs. As the depth of shotgun sequencing of the pools increases, segments of ever shorter lengths within a BAC can be mapped onto their specific homologous regions.
We sought to characterize the resolution of mapping as a function of depth of shotgun sequencing of the pools. A simulated experiment involving a single 24 × 24 array was performed, as described in the Methods section. Simulated BACs were mapped onto the genome of the same species, as would be the case when mapping rearrangements in cancer genomes, and the results were compared with theoretical calculations (Csuros and Milosavljevic 2004). Chimeric BACs containing segments of various lengths from different parts of the genome were simulated in order to establish probability of mapping sequence segment of particular length. Results of the simulation experiments and theoretical calculations are in Figure 5. Table 1 summarizes sample values.

Detecting rearrangements via ST-PGI. In order to evaluate resolution of mapping via ST-PGI, simulated experiments involving a single 24 × 24 array were performed and compared with theoretical calculations (for details, see Methods section). Sampling was performed at 40 tags per 200 kbp BAC in the first experiment (left) and at 80 tags per 200 kbp BAC in the second experiment (right). The percentage of mapped segments of particular lengths was observed. The theoretical calculation is overly optimistic because it assumes that every concatemer can be correctly parsed and every tag mapped.
Average distances between CASTS-s and mapping probability of BAC segments
Comparison with direct shotgun sequencing and BAC-end sequencing methods
The performance of the various methods for comparative BAC clone mapping such as PGI and direct shotgun sequencing or BAC-end sequencing cannot be reduced to a single metric without gross oversimplification. Comparison is further complicated by somewhat different outputs of the methods. Relative performance by methods may also depend on numerous other parameters such as evolutionary distance, repetitive structure of the reference genome, state of completion of genome assembly, and quality of BAC libraries. Acknowledging problems inherent in attempting direct comparisons, in the following we provide a rough comparison highlighting distinct advantages of PGI over other methods in specific contexts.
For the purpose of comparison with BES, we assume that the cost of 50 sequencing reactions equals the cost of a single shotgun library preparation, a total of 100 cost units for direct shotgun sequencing of a BAC at the depth of 50 reads per BAC. Assuming a 48 × 48 array format, PGI reduces the cost of library preparation by a factor of 12 with 48 BACs per pool, each BAC being pooled four times. ST-PGI reduces the cost of shotgun sequencing by a factor of 3.3 when mapping a rhesus BAC onto human, thus allowing the mapping of three BACs and 12.8 CASTS via ST-PGI for the cost of mapping one BAC and two CASTSs via BES. Details of this calculation are provided in the Methods section.
Table 2 summarizes output of BAC-end sequencing, PGI and ST-PGI at fixed cost. Key assumptions made for the purpose of this comparison and the formulas used are described in detail in the Methods section. The following two outputs were considered: number of mapped BACs and number of CASTSs. Table 2 gives amounts of each type of output produced by BES, PGI, and ST-PGI for the same cost.
Output of BES, PGI, and ST-PGI for the same cost
Discussion
The mapping of BACs covering 24% of the rhesus genome onto human demonstrates the practicality of PGI. Comparative mapping of BACs provides a physical map of higher resolution than previously achievable in macaque (Murphy et al. 2001). The key to the efficiency of PGI is multiplexing with the shotgun library preparation step being performed on pools of BACs rather than on individual BACs. ST-PGI introduces an additional level of multiplexing by sequencing multiple mapable tags within a single sequencing reaction.
In contrast to BAC-end sequencing, PGI samples the whole BAC, thus allowing more robust mapping. For example, if the recognition site of a restriction enzyme used for preparing a BAC library is present within a repetitive element, <1% of BACs may be mapable by both ends (Larkin et al. 2003). In contrast, PGI maps mostly based on internal sequences and is thus not as significantly affected by repeats. Generally, BACs can be mapped via PGI even onto problematic regions at an increased cost of deeper sequencing of BAC pools. By virtue of mapping segments within a BAC, PGI can map efficiently onto genomes that are not finished. Since many of newly sequenced organisms such as that of rat are not slated for finishing (Gibbs et al. 2004), this property of PGI is likely to be increasingly significant.
In addition to BAC mappings, PGI also economically produces closely spaced CASTSs (Larkin et al. 2003). By producing multiple CASTSs per sequencing reaction, PCR-able distances between CASTSs can be achieved much more economically for a genome by ST-PGI than by other methods. Targeted sequencing of PCR products across putative breakpoints allows targeted study of rearrangements at the base-pair level without incurring the cost of shotgun sequencing of individual BACs.
PGI and ST-PGI are suitable for comparative genomic sequencing projects such as comparative sequencing and mapping of Old World monkeys using the human genome as a reference. Even at low read coverage, ST-PGI can provide a valuable “comparative scaffold” that can be either used to guide global assembly of whole-genome shotgun reads or for targeted sequencing of genomic regions of highest importance.
In addition to comparative sequencing across species, PGI can also be employed for comparative mapping and targeted sequencing of BACs within species for the purpose of direct detection of genetic variability. Sequencing of mapped BACs across human populations would directly reveal haplotype structure, and the sequencing of various strains of model organisms such as mouse, rat, or populations of rhesus would reveal genetic variation relevant for the design and interpretation of experiments using those organisms.
We have demonstrated that a number of chromosomal rearrangements between rhesus and human can be detected via PGI even at relatively shallow levels of shotgun sequencing. This is particularly relevant in view of the fact that the frequency of chromosomal duplications and other rearrangements in the human lineage (Eichler 2001) and across mammalian species (Kent et al. 2003) appears to be higher than anticipated. In addition to the mapping of evolutionary rearrangements, the method can also be applied to the study of chromosomal aberrations in cancer (Volik et al. 2003) and of genomic disorders (Lupski 1998). Whereas the depth of sampling by ESP is limited to the two end sequences, the depth of sampling by PGI can be adjusted to the desired level of mapping resolution. At high resolution, PGI provides fine mapping of chromosomal aberrations at only a fraction of the cost required for direct shotgun sequencing of individual BACs. At low resolution, ST-PGI provides an economical alternative to ESP. In contrast to ESP, which requires mapping of at least two BACs across a chromosomal breakpoint for accurate detection, PGI can accurately map a breakpoint based on reads from a single BAC. This is particularly relevant for applications to BAC libraries prepared from heterogeneous tumor samples where the chances of capturing the same rearranged chromosomal segment in two sequenced BAC clone inserts may be low.
Finally, implementation of PGI requires only incremental changes in the production at a typical genome center such as Baylor's Human Genome Sequencing Center. The currently existing production lines, which include BAC handling, small-insert library preparation, and sequencing, need only be augmented by the pooling process and informatics.
Methods
BAC libraries
Rat BACs used in control experiments came from the collection of BACs from the arrayed CHORI-230 library of Brown Norway rat (Rattus norvegicus) that were sequenced at the Human Genome Sequencing Center at Baylor College of Medicine during the Rat Genome Sequencing Project (Gibbs et al. 2004). Rhesus BACs came from 384-well plates from the EcoRI segment of the CHORI-250 library of rhesus macaque (Macaca mulatta). Both CHORI-230 and CHORI-250 were produced at the Children's Hospital of Oakland Research Institute (CHORI) and are available through BACPAC Resources at CHORI (bacpacorders{at}chori.org).
BAC pooling and shotgun sequencing
Two pooling methods were employed: one for the controlled mixing of rat BAC clones and the second for producing the rhesus macaque arrays of BACs. Rat BACs were extracted by phenol chloroform with centrifugation, and BAC DNA was purified by an alkaline SDS-based procedure. The DNA concentration of each preparation was determined by using a fluorometer. Pools of 50 μg DNA were constructed containing 24, 48, or 96 clones. Each clone in a pool contributed 1/24, 1/48, or 1/96 of the DNA. Since the original set of 100 clones was used for all the pools, clones in the 96-clone pool were also present in a 48-clone and a 24-clone pool. Four 24-clone, two 48-clone, and one 96-clone pools were prepared. Finally, each 50 μg pool was divided into five 10-μg aliquots. Random shotgun libraries were prepared (Gibbs et al. 2004) on four replicates of each constructed pool and sequenced. The remaining 10-μg aliquot was used in the preparation of short tag libraries.
The rhesus macaque 48 × 48 clone arrays were constructed with BAC clones grown separately. Cultures were pooled without normalization, and a DNA extraction of the BAC plasmids was carried out on each pool. Each array contained clones from six 384-well library plates from the CHORI-250 library (http://bacpac.chori.org/libraries.php). Four 96-well growth boxes containing 1 mL Luria broth, 0.15 mM KNO3, and Chloramphenicol in each well were inoculated from each library plate. This master set of cultures arrayed in twenty-four 96-well boxes was grown overnight for 16-18 h. Each 96-well box had a predetermined sequenced control clone in the A1 well and had blank media in the H12 well. Duplicate 96-well boxes were inoculated from the master set for cultures to be pooled as rows of the 48 × 48 array. Thus, each clone contributed 2 mL of culture to each pool of 48 clones. A second set of duplicate boxes was inoculated and grown for the preparation of the column pools. BAC DNA was extracted and purified from the pools using the same procedure as applied to individual rat BACs. After analyzing the complexity of a HindIII restriction enzyme digest of the purified DNA, a random shotgun library was created and sequenced using standard methods (Gibbs et al. 2004).
The shuffled array consisting of twenty-four 96-well boxes was inoculated (Fig. 1, top) from the master array by using a Seiko robotic arm equipped with a single pin. Software operating the robot was written in Visual Basic. The robot shuffled according to a supplied spreadsheet, which is generated from a database. For optimal deconvolution, shuffling follows the transversal design where the intersection of any two pools across two arrays contains at most one BAC. Detailed mathematical analysis of pooling designs for PGI is provided elsewhere (Csuros and Milosavljevic 2004). The process for preparing DNA from the shuffled array is the same as that used for the original array. Unprocessed sequence reads are available from the National Center for Biotechnology Information (NCBI) Trace Archive (ftp://ftp.ncbi.nih.gov/pub/TraceDB/macaca_mulatta/).
Anchoring of reads and tags
Pool reads were masked for vector content and contaminants. To reduce false mappings due to repeats, RepeatMasker (A. Smit, R. Hubley, and P. Green, unpubl.; http://ftp.genome.washington.edu/RM/RepeatMasker.html) was used to mask both simple and primate specific repeats in pool reads. The masked reads were then mapped to the human genome UCSC build hg16 (NCBI build 34) by using BLAT gfServer version 23. The following BLAT (Kent 2002) parameters were used: tileSize = 11, minMatch = 2, maxGap = 2, minScore = 20, minIdentity = 0, maxIntron = 50. Results returned from BLAT were filtered to determine the best hit.
Concatemers containing short-tags were parsed using human genome sequence by the Tagamizer program (Supplemental material). Tagamizer detects boundaries between tags by identifying absence of relatively rigid alignments against the human sequence that bridge the junctions between tags. Tagamizer is a script written around the BLAT (Kent 2002) program and is available for download at http://www.brl.bcm.tmc.edu/castl/tagamizerDownload.html.
Deconvolution of reads and mapping of BACs
A preliminary index of order k (k = 2,3,4) was formed whenever reads from k different pools mapped via BLAT within a 200-kbp window and the intersection of the k pools contained a BAC. The 200-kbp window was chosen based on the average clone insert size of 163 kbp. The initial mapping resulted in a relatively high false-positive rate for indices, particularly three- and two-pool indices. A number of filtering steps were applied in the final most optimized version of the method to remove low-confidence indices. First, indices were made to “compete” for reads by allowing a read to contribute only to its highest order index. In the case of ties due to multiple highest order indices, the read was retained in both indices. In the other direction, indices were made to “compete” for BACs by allowing a BAC to be mapped only by its highest order index. In the case of ties due to multiple highest order indices, all highest order indices were used to map the BAC. Consequently, indices mapping a BAC to multiple loci are all of the same order. Additional filtering was performed to remove all two-pool indices in which both pools only contained a single read. Prior to filtering, the control BACs in Array 1 across all index orders demonstrated an accuracy of 14% and a completeness of 100%. After filtering, the control BACs had an accuracy of at least 80% and a completeness of 91%.
The 14 BACs mapping to multiple disjoined human loci, as described in the Results section, that were selected for direct sequencing were mapped using an earlier less optimized version of the deconvolution method, which did not include “competition” of indices for BACs and reads or the filtering of two-pool indices in which both pools only contained a single read. The 10 BACs without confirmed multiple mappings had a single mapping after filtering; however, the four BACs with confirmed multiple mappings also had a single mapping.
Selection of control BACs
A total of 111 BACs along the main diagonal and two neighboring diagonals of the Array 1 original array were selected for sequencing, and the sequencing of 103 of them succeeded. Sequenced BACs were assembled into contigs. A total of 99 BACs containing contigs could be reliably mapped onto the human genome by BLAT using a minimum requirement of 600 matching bases and a minimum percentage identity of 40%, and those BACs were selected as controls for Array 1. The remaining four BACs did not contain contigs that met these criteria. A subset of 24 of the 99 BACs that had highly reliable mappings were chosen as controls for Array 2.
PGI mapping of simulated rhesus BACs
A simulation was performed in order to determine the accuracy and completeness of PGI without confounding factors such as low-quality sequences and incorrect control mappings. This simulation was performed by computationally creating 2304 BACs from the human genomic sequence and then creating reads from the BACs. Reads were mutated by 10% in order to simulate the divergence between human and rhesus. The simulated BACs were assigned to pools in a 48 × 48 array, and reads from the BACs were distributed among the pools. Read coverage and pool bias were simulated based on the observed coverage and bias within the BAC pools of Array 1.
Detection of rearrangements
Potential chromosomal rearrangements were predicted by identifying BACs that reliably mapped by four- or three-pool indices to two distinct loci within a human chromosome. Only the loci that were >200 kbp apart were considered.
Simulating detection of rearrangements
A total of 540 BACs were generated by using randomly selected segments from the finished human genome. The following four types of BACs were simulated: 144 non-rearranged BACs consisting of a single continuous segment 200 kbp long, 288 chimeric BACs consisting of two distant segments, each 100 kbp long, 72 chimeric BACs consisting of four distant segments, each 50 kbp long, and 36 chimeric BACs consisting of eight distant segments, each 25 kbp long. The simulated BACs were then placed within a 24 × 24 array, and the deconvolution process was employed to map the BAC segments. The probabilities of mapping 10-kbp segments were obtained by extrapolation using a theoretical model (Csuros and Milosavljevic 2004).
Estimating the probability that a BAC maps to multiple locations due to intrachromosomal duplications
We assume that a BAC will map to multiple loci if it spans an intrachromosomal segmental duplication. Following the method of Cheung et al. (2003), we assume that there are 1530 distinct intrachromosomal segmental duplications of total length 80.3 Mbp and average length 52 kbp. Assuming for simplicity that each of the 1530 segments is of average length, the probability that a randomly selected BAC-sized segment of size 163 kbp would completely span one of them is 0.06.
Comparison of PGI, ST-PGI, and BES
A number of assumptions were made for the purpose of comparing PGI, ST-PGI, and BAC-end sequencing (Table 2). First, it is assumed for convenience that the cost unit equals the cost of obtaining a single shotgun read. Second, it is assumed that each BAC-end sequence costs four units due to costlier up-stream BAC DNA preparation compared with plasmid DNA preparation, lower yield of quality reads, and larger cost of sequencing reagents for the BAC-sized clone insert. The cost of shotgun library preparation equals 50 read units and is amortized over 24/4 = 6 BACs, 12 BACs, and 24 BACs for 24 × 24 arrays, 48 × 48 arrays, and 96 × 96 arrays, respectively. Based on the results of mapping rhesus BACs onto human, we further assume that the cost of an ST-PGI tag equals 0.3 units and that 85% of BACs will be mapped using 16 PGI reads or 16 ST-PGI tags (at cost of 16 * 0.3 units). Based on the fact that about half of random rhesus reads can be uniquely mapped onto human loci, we further assume that 25% of rhesus BACs would have been mapped onto human genome using both BAC-ends.
As an example, consider calculation of the performance of BES and ST-PGI in the 48 × 48 array format on BACs of average size of 200 kbp. While the size of BACs varies by library, 200 kbp is selected because it is typical for the libraries used in this article (average size of a rhesus BAC is 163 kbp, while the average size of a rat BAC is 200 kbp). The cost of mapping a 200 kbp BAC and two CASTSs by BES equals (1/0.25)*2*4=32 units. The cost of mapping a 200-kbp BAC and 16 * 0.53 = 8.5 CASTSs (0.53 is calculated as 2.9/5.5 since 2.9 out of 5.5 reads can be mapped and deconvoluted) is (50/12 + 16 * 0.3)/0.85 = 10.5. It follows that for the cost of mapping one BAC and two CASTSs by BAC-end sequencing, one can map 32/10.5 = 3.0 BACs and 3.0 * 8.5 = 25.6 CASTSs by ST-PGI.
Following this 48 × 48 array ST-PGI example, corresponding values for PGI are obtained by entering the sequencing cost of 16 units per BAC instead of 16 * 0.3 units in the ST-PGI example. Corresponding values for 24 × 24 and 96 × 96 arrays are obtained by entering respective amortized library preparation costs of 50/6 and 50/24 units per BAC.
Program availability
Code and licenses for software used including Tagamizer, PGI deconvolution software, and related methods are available free of charge for academic use. Current access and licensing information is posted at http://www.brl.bcm.tmc.edu/castl/tagamizerDownload.html.
Acknowledgments
This work has been supported by the National Institutes of Health grants R01 HG 02583-01 from the National Human Genome Research Institute and U01 RR 18464 from the National Center for Research Resources to A.M., in part by the grants from Natural Sciences and Engineering Research Council of Canada and the Fonds québécois de la recherche sur la nature et les technologies to M.C., and in part by the National Institutes of Health grant U54 HG 02051 from the National Human Genome Research Institute to R.A.G.
Footnotes
-
[Supplemental material is available online at www.genome.org and www.genboree.org.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3162505.
-
↵4 Corresponding author. E-mail amilosav{at}bcm.tmc.edu; fax (713) 798-4373.
-
- Accepted October 13, 2004.
- Received August 17, 2004.
- Cold Spring Harbor Laboratory Press








