
Xenomics
Abstract
Xenopus genomics, or Xenomics for short, is coming of age. Indeed, biological insight into processes such as growth factor signaling and patterning of the early embryo is now being gained by combining the value of Xenopus as a model organism for cell and developmental biology with genomic approaches. In this review I address these recent advances and explore future possibilities gained from combining this powerful experimental system with genomic approaches, as well as how our quest to understand basic biological principles will be greatly facilitated though the marriage of Xenopus and genomics.
Xenopus genomics is very much in its infancy. Although large-scale sequencing efforts were slow to be initiated in this system, in the past 3–4 yr there has been an explosion of genomic information accumulating in Xenopus laevis and its diploid relative, Xenopus tropicalis (Fig.1). Since the beginning of 2003, >320,000 sequences have been deposited in public repositories for X. laevis and >1,100,000 for X. tropicalis, mostly in the form of expressed sequence tags (ESTs). With this expansive amount of new sequence information, X. tropicalis recently jumped into third place on the list of organisms with the most EST's, behind human and mouse. During the same period of time, the Joint Genome Institute (JGI) has been sequencing the X. tropicalis genome, using a shotgun approach, and it has recently completed 8× coverage. The JGI is currently in the process of assembling the X. tropicalis genome, and it is expected that the JGI will announce its results by the end of 2005.
Now that such an extensive amount of genomic information is becoming available in Xenopus, how will this be useful in our scientific pursuits? This question is best answered by further asking, “What is the ultimate value of obtaining sequence information?” If the ultimate aim is not simply to catalog genes but to understand their function, then it would be very advantageous to find the ideal organisms to study gene function. It is here where the marriage of Xenopus and genomics will reap its full benefits, as Xenopus is perhaps the best vertebrate model organism for functional genomics.
Functional screens
Xenopus is arguably the best available vertebrate model for systematic large-scale in vivo gene function analysis. In this section I review the different types of functional screens that can be performed in Xenopus.
Gain-of-function screens
Oocyte expression screens
It has been recognized since the early 1970s that the Xenopus oocyte can be used as an in vivo test tube to study the function of biological macromolecules (Gurdon 1975). First, Xenopus oocytes can be cultured in vitro for many days, and second, microinjection of exogenous macromolecules, such as DNA or mRNA, will behave appropriately in the oocytes, resulting in their transcription and/or translation, respectively. However, before Xenopus oocytes could be used in large-scale expression screens, it was important that purified mRNAs could be generated in large quantities for any gene of interest. This next critical step was made in the mid-1980s, when a method was found for producing large quantities of synthetic mRNA in vitro by using bacteriophage promoters, and it was shown that these synthetic mRNAs would be efficiently translated when injected into Xenopus oocytes (Krieg and Melton 1984; Melton et al. 1984).
Xenopus oocytes were first exploited in large-scale functional screens by groups interested in identifying receptors for neuropeptides and neurotransmitters (Masu et al. 1987; Julius et al. 1988). The first group to successfully use oocytes to clone a novel neuropeptide receptor was Masu and colleagues (1987). This group was interested in identifying the receptor for the tachykinin neuropeptide, substance K. They knew from previous work that Xenopus oocytes injected with total mRNA isolated from bovine brain and stomach expressed functional mammalian substance K receptors on their membranes, as assayed by electrophysiological measurements. Now that a sensitive assay for receptor function and a source of mRNA encoding the receptor were available, all that was required was to develop a large-scale functional screen to clone the receptor. To do this, they constructed a cDNA library from bovine stomach in a vector containing a bacteriophage promoter (SP6), so that in vitro transcribed RNA could be made from this library. Then, they injected in vitro transcribed RNA from this library in pools and assayed for substance K activity, based on electrophysiological measurements. Once an active pool was identified, subpools were tested for activity until an individual clone encoding the receptor for tachykinin neuropeptide, substance K, was identified (Masu et al. 1987). A similar strategy was used a year later by Julius and colleagues (1988) to clone the 5HT1c serotonin receptor in Xenopus oocytes. Since these two pioneering screens, many other neuropeptide receptors and ligand-gated ion channels have been identified by using large-scale functional screens in Xenopus oocytes. A screen in oocytes has also been developed aimed at identifying secreted molecules that contain mesoderm and/or neural-inducing activities (Lustig and Kirschner 1995). In this assay, pools of in vitro transcribed RNA made from a cDNA library from tissue with inducing activity are injected into oocytes. Then an explant of competent tissue is placed on top of the oocytes. If the oocyte secretes factors that change the fate of the explant, then that pool is sib-selected until a single active clone is identified.
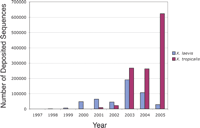
Graph of number of deposited ESTs for Xenopus laevis (blue) and Xenopus tropicalis (red) for each year since 1997. In the past 2–3 yr, there has been an explosion of sequence information in the public repositories, heralding the arrival of these two systems into the genomics era.
Gain-of-function screens in embryos
In 1991 Smith and Harland (1991) took a similar approach to the one taken to clone neuropeptide receptors but, in their case, with the aim of identifying genes that could mimic the vegetal dorsal inducer in the early frog embryo. For their assay, they irradiated the vegetal hemisphere of one-cell-stage embryos with UV, in order to remove the endogenous vegetal dorsal inducing signal, resulting in ventralized embryos. Then they prepared a cDNA library from gastrula embryos and injected in vitro transcribed RNA from this library in pools and assayed for rescue of dorsal axis in the UV-irradiated/ventralized embryos. By using this approach, they identified Xwnt8 as a potent vegetal dorsalizing factor, which could rescue a complete dorsal axis in UV-irradiated embryos (Smith and Harland 1991). A year later, Smith and Harland (1992) used the same approach to clone noggin, a novel gene that was later shown to encode a potent extracellular antagonist of Bone Morphogenetic Protein 4 (BMP4) (Zimmerman et al. 1996). Since the pioneering work of Smith and Harland, many important molecules have been identified in Xenopus, using large-scale gain-of-function screens (also known as expression cloning screens); see Table 1 for examples. Interestingly, most of these are signaling molecules, antagonists of signaling molecules, or transcription factors downstream of signaling molecules.
Genes identified in large-scale gain-of-function screens in Xenopus
The large-scale gain-of-function screens used to identify these genes were done with redundant, non-normalized libraries, with pool sizes ranging from 96 clones per pool to several thousand clones per pool (Smith and Harland 1991; Grammer et al. 2000). Although this approach has given rise to the identification of a large number of important genes during early development, the approach has been inherently inefficient for several reasons. First, most of the clones that are screened do not contain the full-length coding sequence of the protein, as any given library contains mostly truncated clones. Second, given that the libraries used for these screens were non-normalized, clones encoding the same protein product were screened multiple times, especially those genes transcribed in the embryo at high levels, while genes present at more modest levels in the embryo were screened only rarely. However, by using sequence information and bioinformatics tools, one can select a non-redundant full-length clone set, thus allowing the functional screens to be performed much more efficiently (Fig.2; Gilchrist et al. 2004; Chen et al. 2005; Voigt et al. 2005; http://www.gurdon.cam.ac.uk/informatics/Xenopus.html). By using such a streamlined full-length clone set, it has been possible to decrease the clone size per pool during the functional screens to eight clones per pool (Voigt et al. 2005). This dramatically increases the sensitivity and efficiency of the functional screens that are performed (Chen et al. 2005; Voigt et al. 2005). To date, a wealth of genes have been uncovered by using large-scale gain-of-function screens in Xenopus. Now that a non-redundant full-length clone set has been developed as a physical resource to the community, Xenopus is very likely to remain one of the ideal systems for large-scale functional genomic efforts.
Expression screens in the test tube
Xenopus eggs and embryos have also been exploited with much success for investigating many basic cell biological and biochemical principles, due to the ready availability of very large numbers of eggs and embryos from these frogs. In particular, it is possible to generate extracts from eggs and embryos, which recapitulate many cell biological processes, such as nuclear disassembly and reassembly, nuclear import and export, DNA replication, chromosome assembly and disassembly, mitotic spindle assembly and function, protein synthesis and degradation, cell cycle control, apoptosis, and microtubule and microfilament assembly and disassembly (Murray and Kirschner 1989a,b; Murray et al. 1989; Glotzer et al. 1991; Newmeyer and Wilson 1991; Pfaller et al. 1991; Smythe and Newport 1991; Dasso et al. 1992; Allan 1993; Holloway et al. 1993; Newmeyer et al. 1994; Hengartner 1995; King et al. 1995, 1996; Yu et al. 1996; Evans et al. 1997; Kornbluth 1997; Thommes and Blow 1997; Lohka 1998; Pain et al. 1998; de la Barre et al. 1999; Desai et al. 1999; Shirasu et al. 1999; von Ahsen and Newmeyer 2000; Mandato et al. 2001; Arias and Walter 2004). Since these cell-free extracts can be manipulated in many different ways, it has been possible to assess the role of specific proteins in distinct processes. For example, one can immuno-deplete the extracts of particular proteins or protein complexes and address the effect in different cellular processes. It is also possible to use Xenopus extracts in combination with expression-based screens to identify proteins that are substrates for particular biochemical pathways (King et al. 1997; Lustig et al. 1997). In this case a cDNA library is split into pools, as is done for the functional screens described above, but instead of injecting in vitro transcribed RNAs from pooled clones into oocytes or embryos, the pooled clones are transcribed and translated together in vitro, in the presence of radioactively labelled amino acids. In this way, the behavior of the labelled translated proteins can be monitored after being added to the egg extracts. Such screens have been used to identify proteins that are phosphorylated in a cell cycle–dependent manner (Stukenberg et al. 1997). They have also been used to identify proteins that are specifically degraded during mitosis (McGarry and Kirschner 1998). This general strategy has been modified in a variety of ways, as, for example, in order to identify new substrates for caspase 3 (Kothakota et al. 1997) or to identify novel uracil-DNA glycosylases (Haushalter et al. 1999). In the future, these screens will benefit from the availability of non-redundant full-length clone sets, as has been used for functional screens in embryos (Gilchrist et al. 2004; Chen et al. 2005; Voigt et al. 2005). Combining the tractability and manipulability of cell-free extracts from eggs and embryos with large-scale functional genomic approaches, such as those described above, will continue to provide valuable approaches for identifying new components involved in cell biological and biochemical processes in the future.
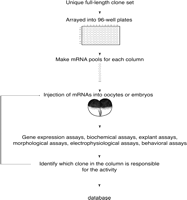
Large-scale gain-of-function screen strategy in Xenopus. A unique full-length clone set is established and arrayed into 96-well plates. Miniplasmid preps are made and pools of plasmids made for each column. Each pooled set of plasmids is transcribed into mRNA in vitro. Pooled mRNA is injected into oocytes or embryos, and then a variety of functional screens are performed, depending on the type of molecules that are being sought. Once an active pool is identified, the pool is broken down to individual clones and assayed again to identify the active clone.
Loss-of-function screens and genetic approaches
Although for many years gain-of-function screens have been very fruitful in Xenopus for identifying genes involved in cell and developmental biology processes, loss-of-function screens or genetic approaches have not been traditionally used in this system. However, such approaches have recently begun to be employed as well. One of the primary reasons that such approaches have not traditionally been adopted is that X. laevis, the more commonly used species, is allotetraploid. This means that it essentially has two nonidentical copies for each gene, which complicates loss-of-function experiments due to potential genetic redundancy. In addition, X. laevis has a long generation time of around a year, further complicating its use as a straightforward genetic system. However, in recent years X. tropicalis, a diploid relative of X. laevis, has begun to be used more and more due to its simpler genome and shorter generation time, making it a more ideal system for both genomic studies and loss-of-function experiments (Amaya et al. 1998).
For >10 yr, loss-of-function experiments in Xenopus have traditionally been based on overexpression of dominant-negative variants of genes (Amaya et al. 1991). The disadvantage of this method is that it requires previous knowledge of protein domains and function for designing appropriate dominant-negative variants of genes. In addition, dominant-negative constructs are seldom specific to only one gene product. Several years ago Janet Heasman and colleagues (2000, 2002) reported that antisense morpholino oligonucleotides (aMOs) complementary to the start of translation of the mRNA could specifically and efficiently inhibit translation in X. laevis embryos. It was also shown that aMOs can efficiently and specifically inhibit translation of mRNAs in X. tropicalis into the tadpole stages (Nutt et al. 2001). In addition, aMOs complementary to splice junction inhibit splicing of premRNA in vivo, providing another means of inhibiting gene function in the embryo (Kenwrick et al. 2004; Sivak et al. 2005). Targeting aMOs to splice junctions has the additional benefit that its effect can be monitored by RT-PCR (Kenwrick et al. 2004; Sivak et al. 2005). The disadvantage in targeting aMOs to splice junctions is that it requires knowledge of the genomic structure of the gene in question. However, given the extent of genome information now available in X. tropicalis (http://genome.jgi-psf.org/; http://www.ensembl.org/Xenopus_tropicalis/index.html), it is now possible to identify splice junctions to most genes. Given that only sequence information around the start of translation and/or around splice junctions is needed to design aMOs, it has become possible to consider performing mid- to large-scale loss-of-function screens in X. tropicalis (Kenwrick et al. 2004). Such an approach is likely to be very fruitful in the future for addressing the function of genes during early development on a large scale. Even a small-scale aMO screen uncovered a novel gene, pinhead, essential for head formation (Kenwrick et al. 2004).
Another approach likely to be valuable for identifying mutation in genes of interest is combining chemical mutagenesis screens with direct sequencing (Hurlstone et al. 2003; Stemple 2004). To this end, Lyle Zimmerman and Derek Stemple (pers. comm.) are currently using this approach to identify mutations in hundreds of genes in X. tropicalis. But what about forward genetic screens in X. tropicalis? Several groups are currently performing chemical, irradiation, and insertional mutagenesis screens, but such approaches remain in their infancy due to the lack of easily scored mutations for optimizing mutagen dose levels. However, recently two groups published the first set of naturally occurring mutations in X. tropicalis, which should help facilitate such optimization experiments (Grammer et al. 2005; Noramly et al. 2005).
Large-scale whole-mount in situ hybridization screens
One of the primary means of specifying cell fate during development is through differential gene expression. With this in mind, Christof Niehrs and colleagues began a systematic large-scale whole-mount in situ hybridization screen using Xenopus embryos in the early 1990s with the aim of identifying genes with localized expression patterns during development (Gawantka et al. 1995, 1998; Pollet et al. 2005). They started this screen several years before any large-scale sequencing projects in Xenopus had been initiated, so their approach was simply to take random clones from several embryonic staged cDNA libraries and to perform whole-mount in situ hybridizations systematically on thousands of clones on embryos at the gastrula, neurula, and tailbud stage. In the initial publication of the screen, they performed whole-mount in situ hybridization on 1765 randomly picked clones (Gawantka et al. 1998). From these, they identified 273 genes that were differentially expressed. More recently, the Niehrs group published the second phase of their large-scale gene expression screen, in this case presenting the results after performing whole-mount in situ hybridizations on 8369 cDNA clones. From these, they identified 773 genes with restricted expression (Pollet et al. 2005). Given that there was an overlap of 39 genes identified in both screens, up to now, this large-scale gene expression screen has resulted in the identification of 957 unique genes in Xenopus with restricted expression patterns. No doubt there will be more genes found in phase 3, but as it stands, the cataloging of the restricted expression patterns of nearly a thousand genes during early Xenopus development is an ambitious and exceedingly important project. A database (AxelDB) containing this information is available at http://indigene.ibaic.u-psud.fr/article.php3?id_article=48 (Pollet et al. 2000).
Several important insights have been gained from this gargantuan project. One is that a significant number of genes have restricted expression patterns. Overall, one-fourth to one-fifth of genes tested had restricted expression patterns during early development. Surprisingly, many of these genes encode proteins thought to be involved in basic cellular housekeeping functions. About one-third of the genes with restricted expression patterns are cell type and/or organ specific. These genes are immediately useful as markers, but many of these are also likely to have important roles in specifying cell fate. Although identifying cell type–specific genes was one of the aims of the large-scale gene expression screen, the more interesting insights have been gained from genes with more complex expression patterns. One is the concept of tissue relatedness, gained from correlating the cell types and/or tissue types that share common expression of genes; and the other is the concept of synexpression groups, which comprise genes with common complex expression patterns (Gawantka et al. 1998; Niehrs and Pollet 1999; Pollet et al. 2005).
Tissue relatedness
By using hierarchical clustering, the tissue types that shared common expression of genes were correlated, thus resulting in a tissue-relatedness tree (Gawantka et al. 1998; Pollet et al. 2005). The purpose of this analysis was to arrive at a relatively unbiased measure of tissue relatedness. Two types of tissue relatedness were found by using this analysis. The first is tissues that share a common lineage relationship, termed monofatic relatedness (Pollet et al. 2005). Examples of these include such tissues as the diencephalon and telencephalon, and lateral plate mesoderm and ventral blood islands. The monofatic relationships are not surprising, and indeed, most of the tissues are monofatic. However, there was another type of relatedness that came out of this analysis, termed parafatic relationships, which was less expected (Pollet et al. 2005). These are tissues that share the expression of many genes, although they do not share a common lineage relationship. In these cases it is believed that these tissues share common physiological processes. Examples of parafatic relationships include the cement gland and notochord, both being highly secretory tissues. In other cases, the underlying common physiological process underlying other parafatic relationships, such as the hypophysis (pineal gland) and several mesodermal derivatives, is less obvious. Another example of a parafatic relationship is the endocrine pancreas and the nervous system, which share the expression of many genes in common although they are not related by lineage (Edlund 2001).
Synexpression groups
Perhaps the most interesting feature to come out of the large-scale gene expression screen is the identification of groups of genes that share identical or very similar complex expression patterns, termed synexpression groups (Gawantka et al. 1998; Niehrs and Pollet 1999; Pollet et al. 2005). These are groups of genes that encode proteins likely to be involved in common functional pathways but are used at multiple embryonic stages and/or tissues, and are thus under common transcriptional control. Synexpression groups include the BMP4 group, the delta group, the chromatin group, the endoplasmic reticulum group, the karyopherin group, and the FGF8 group (Gawantka et al. 1998; Wischnewski et al. 2000; Furthauer et al. 2002; Pera et al. 2002; Tsang et al. 2002; Pollet et al. 2005). One of the most valuable aspects of the concept of synexpression groups is that one is able to predict the likely function of a gene product based on its expression, which can then form the basis of further functional experiments. In many cases this has been borne to be true. For example, the Xvent2 protein, which is a homeodomain-containing protein in the BMP4 synexpression group, is indeed a target and downstream mediator of BMP signaling (Onichtchouk et al. 1996, 1998). BAMBI, another gene in the BMP4 group, have likewise been shown to be involved in the regulation of BMP signaling (Onichtchouk et al. 1999). Similarly, the Delta synexpression group contains several enhancers of split (ESR)–like genes, known to be targets of Delta-Notch signaling (Jen et al. 1999; Li et al. 2003). However the more interesting results has arisen from experiments in other genes within this group, which had not previously been implicated in Delta-Notch signaling. One such gene, Nrarp (also know as NAP), is a novel ankyrin-containing protein, which is both a target and modulator of Delta-Notch signaling (Lamar et al. 2001; Lahaye et al. 2002). The concept of synexpression has been adopted by other systems, and one synexpression group that has arisen from a large-scale in situ screen in zebrafish is the FGF synexpression group (Furthauer et al. 2002; Pera et al. 2002; Tsang et al. 2002). This group includes FGF8 and FGF3, as well as several genes known to be involved in the regulation of FGF signaling, such as the sprouty2 and sprouty4 (Furthauer et al. 2001, 2002). However it also includes other genes, previously not implicated in FGF signaling, such as sef, which stands for similar expression to fgf genes (Furthauer et al. 2002; Tsang et al. 2002). Sef interacts with the intracellular domain of the FGF receptor and functions as a feedback inhibitor of FGF dependent ras/MAPK signaling (Furthauer et al. 2002; Tsang et al. 2002). Another gene with an expression pattern very similar to FGF8 is FLRT3, which stands for fibronectin-leucine-rich transmembrane protein 3. This gene also has been shown to be a transmembrane modulator of FGF-mediated MAPK signaling (Bottcher et al. 2004). In summary, members of synexpression groups have often been shown subsequently to be involved in common pathways, thus confirming the value of these groupings in providing good predictive value for functional analysis.
The future of large-scale whole-mount in situ hybridization screens
The gene expression screen carried out in the Niehrs group has been fantastically useful in uncovering new marker genes as well as novel protein products in established molecular pathways. What is the future of such screens? Given that a relatively small percentage of total genes (∼4000 of the predicted 30,000 genes in the vertebrate genome) have been included in this screen, there is certainly scope for the incorporation of further genes into the whole-mount in situ hybridization screen. Indeed, most of the genes selected for the published screens originated from earlystage libraries. Simply expanding the selection of clones to later-staged libraries is likely to result in the identification of many more genes with differential gene expression patterns. In addition, a large amount of genomic information is now available, in the form of ESTs in both X. laevis and X. tropicalis, as well as genome sequence information in the case of X. tropicalis. Therefore, in the next phase of screens, it would seem prudent to use a unique set of genes, rather than randomly selected clones for the screen. The Ueno group in Japan is taking essentially this approach in their large-scale in situ hybridization screen, which they make available at http://xenopus.nibb.ac.jp/. To date, they have made available the expression patterns of 801 genes in their database. A large-scale in situ screen using the unique set of full-length clones identified in X. tropicalis is similarly underway (E. Amaya and N. Papalopulu, unpubl.). The ultimate aim is to determine the expression patterns of all genes in X. laevis and X. tropicalis, but this highly ambitious goal will take many years to complete. Another essential aim for the future will be to coordinate the results of the large-scale gene expression screens in Xenopus with those underway in other chordate models systems, such as the mouse, chick, ascidians, zebrafish, and medaka (Bettenhausen and Gossler 1995; Neidhardt et al. 2000; Crosier et al. 2001; Kudoh et al. 2001; Makabe et al. 2001; Nishikata et al. 2001; Satou et al. 2001; Thut et al. 2001; Gammill and Bronner-Fraser 2002; Bell et al. 2004; Quiring et al. 2004; Thisse et al. 2004). Ideally one should be able to query the expression pattern of a given gene in a model organism, find the ortholog in another model system, and compare the expression patterns between systems. Such sort of analysis will be invaluable in correlating expression and functional data between one organism and others.
Gene expression profiling and microarrays
While assaying the expression pattern of one gene at a time, as has been done in the large-scale whole-mount in situ screen described above, is enormously useful due to the detailed information that can be gained from this sort of analysis, it is exceedingly time consuming to do this on the tens of thousands of genes present in the genome. However one way that thousands of genes can be assayed simultaneously is through the use of microarrays (Brown and Botstein 1999). Given that Xenopus embryos can be generated in the thousands and it is relatively easy to manipulate them, it can be argued that this system is ideally suited for microarray experiments. The Brivanlou laboratory was the first to recognize this potential, and in 1999 Curtis Altmann and Ali H. Brivanlou announced the first Xenopus cDNA microarray. It contained 864 gastrula cDNAs, including 768 randomly selected clones from an expression library and the rest made up of known markers (Altmann et al. 2001). This early prototype microarray heralded the first steps toward the development of genomic resources in Xenopus, 2 yr before any large concerted sequencing effort in Xenopus had come off the ground. This prototype array was used to identify new genes with regionalized expression pattern, as well as novel activin inducible genes (Altmann et al. 2001).
The Brivanlou then followed this prototype array with a second-generation cDNA microarray, containing, in addition to the clones in the prototype array, a further 4000 randomly picked gastrula clones (Munoz-Sanjuan et al. 2002). The second array again predated the availability of concerted sequencing efforts, but nevertheless, they were able to use this array to monitor the transcription changes that occur in ectodermal explants that had been neuralized by the inhibitory Smad, Smad7. By using this 5000 cDNA microarray, they identified 142 genes whose transcriptional profile changed in the neuralized explants. Surprisingly, many of the genes identified in their screen were predicted to encode proteins involved in post-transcriptional rather than transcriptional control, suggesting an important role for post-transcriptional control during the initial stages of neural development (Munoz-Sanjuan et al. 2002). They followed up the screen by performing whole-mount in situ hybridization on the candidate clones, and finally, on those that were full-length, they tested them in functional assays of neuralization, either on their own or in combination with other neuralizing factors (Munoz-Sanjuan et al. 2002). In this way, they were able to carry the experiment from profiling, to confirmation by RT-PCR, to determination of expression patterns of the candidate genes by whole-mount in situ hybridization, and finally to function analysis, confirming the value of this system to quickly go from profiling experiments using microarray to studies of gene function in vivo.
Since this early work, other groups have begun to generate more Xenopus cDNA and oligonucleotide microarrays, in some cases using sequence information to try to limit the amount of redundancy in the arrays (Tran et al. 2002; Chung et al. 2004; Konig et al. 2004; Chalmers et al. 2005; Shin et al. 2005). However, the most comprehensive use of microarray analysis in Xenopus has been done by Christof Niehrs and colleagues (Baldessari et al. 2005). In their study they performed gene profiling experiments with 37 different samples and followed this with cluster analysis to determine coregulated gene clusters. The goal was to determine whether they could identify additional synexpression groups to the ones they had identified previously in their large-scale whole-mount in situ hybridization screens (Gawantka et al. 1998; Pollet et al. 2005), described previously. To be as comprehensive as possible, they took samples from different stages of development; they dissected embryos into different domains at four stages; they also generated ventralized and dorsalized embryos; and finally they took samples from adult organs. In their analysis, they identified 15 coregulated gene clusters, ranging in size from 1328 clones to 23 clones per cluster (Baldessari et al. 2005). These gene clusters were the following: a protein biosynthesis group, a chromatin group, an RNA processing group, a respiratory chain and Krebs cycle group, a cell cycle group, an endoplasmic reticulum group, a vesicle transport group, a synaptic vesicle group, a microtubule group, an intermediate filament group, an epithelial protein group, a collagen group, a cement gland/hatching gland group, a muscle group, and a muscle and heart group. In addition, they filtered the data in order to extract new region and tissue specific genes. Undoubtedly these data will add to the wealth of information already present in Xenopus and will provide essential information for further functional experiments on these and other genes in the future.
In addition to the comprehensive analysis by the Niehrs group described above, many other gene profiling studies using microarrays have been published in the past year. These include the use of microarrays to identify gene targets of retinoid signaling (Arima et al. 2005), targets of FGF signaling (Chung et al. 2004), targets of BMP signaling (Peiffer et al. 2005), and genes activated when BMP signaling is inhibited, thus identifying neural specific genes (Shin et al. 2005). Thus in a short period of time, it has been possible to elucidate the gene targets of some of the major signaling pathways in early development in the frog, using gene profiling experiments on microarrays. In addition, microarray experiments will allow the identification of direct targets of transcription factors, as has been done already for VegT (Taverner et al. 2005). To this end, Xenopus is an excellent system for identifying direct targets of transcription factors, since it is relatively easy to combine the use of hormone-inducible transcription factors with the protein translation inhibitors, such as cycloheximide (Smith et al. 1991; Kolm and Sive 1995; Taverner et al. 2005). Therefore it will not be long before such experiments are performed on a large number of transcription factors using microarrays in Xenopus.
Eventually it will be possible to begin to build complex gene regulatory networks in Xenopus, as is being done for Drosophila and the sea urchin (Davidson et al. 2002; Levine and Davidson 2005). Indeed, such gene networks have already been initiated in Xenopus (Loose and Patient 2004; Koide et al. 2005). Although such efforts are only just beginning, there is little doubt that with the recent acquisition of extensive genomic information, together with the facility to perform elegant large-scale functional screens, large-scale whole-mount in situ hybridization screens, microarray experiments, and transgenic experiments (Kroll and Amaya 1996; Bronchain et al. 1999; Sparrow et al. 2000; Karaulanov et al. 2004), a complete understanding of the genomic program of development will soon be available in Xenopus.
Footnotes
-
Present address: The Healing Foundation Centre, Faculty of Life Science, University of Manchester, Manchester, M13 9PT, UK.
-
E-mail enrique.amaya{at}manchester.ac.uk; fax 44 161 275 1505.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3801805. Freely available online through the Genome Research Immediate Open Access option.
- Cold Spring Harbor Laboratory Press








