
Identification of programmed translational -1 frameshifting sites in the genome of Saccharomyces cerevisiae
Abstract
Frameshifting is a recoding event that allows the expression of two polypeptides from the same mRNA molecule. Most recoding events described so far are used by viruses and transposons to express their replicase protein. The very few number of cellular proteins known to be expressed by a -1 ribosomal frameshifting has been identified by chance. The goal of the present work was to set up a systematic strategy, based on complementary bioinformatics, molecular biology, and functional approaches, without a priori knowledge of the mechanism involved. Two independent methods were devised. The first looks for genomic regions in which two ORFs, each carrying a protein pattern, are in a frameshifted arrangement. The second uses Hidden Markov Models and likelihood in a two-step approach. When this strategy was applied to the Saccharomyces cerevisiae genome, 189 candidate regions were found, of which 58 were further functionally investigated. Twenty-eight of them expressed a full-length mRNA covering the two ORFs, and 11 showed a -1 frameshift efficiency varying from 5% to 13% (50-fold higher than background), some of which corresponds to genes with known functions. From other ascomycetes, four frameshifted ORFs are found fully conserved. Strikingly, most of the candidates do not display a classical viral-like frameshift signal and would have escaped a search based on current models of frameshifting. These results strongly suggest that -1 frameshifting might be more widely distributed than previously thought.
Sequencing programs, along with various projects in the pharmaceutical, agricultural, aquacultural, and forestry industries, are creating an explosion of DNA sequence data. With this abundance of data, there is a growing need for more effective tools and methods to extract vital information from raw DNA sequences. Algorithms for identifying protein-coding regions and predicting complete genes are of particular importance. Since the early 1990s, a number of computer programs for eukaryotic gene identification have been developed: GENMARK (Borodovsky and McIninch 1993), FGENEH (Solovyev et al. 1994; Solovyev and Salamov 1997), GeneParser (Snyder and Stormo 1995), GeneWise (Birney et al. 1996), GenScan (Burge and Karlin 1997), and Procruts (Gelfand et al. 1996; Mironov et al. 1998). Most of these programs make use of sophisticated pattern recognition techniques, such as linear discriminant analyses, neural networks, or Hidden Markov Models (HMM) to identify coding regions. Some programs also make use of database sequence alignment methods, such as BLAST (Altschul et al. 1990), to further improve their predictions. Generally, these algorithms classify out-of-frame ORFs as either a sequencing error or a pseudogene signature (Harrison et al. 2002). Up to now only a few algorithms assign a frameshift as a possible regulatory process. However, frameshifting, together with readthrough of stop codons and ribosome hopping, is part of the reprogrammed genetic decoding (“recoding”) events that allow expression of several polypeptides from the same mRNA (Gesteland et al. 1992). Although most of the recoding events described so far have been found in small autonomous genetic elements (Baranov et al. 2002a, 2003; Bekaert and Rousset 2005), a few cellular genes are known to be expressed by this mode of control (Namy et al. 2004), most of them found by chance.
Twenty years ago, Jacks and Varmus described the first programmed -1 ribosomal frameshifting event, from which they established the canonical model of the eukaryotic -1 frameshifting site (Jacks and Varmus 1985; Jacks et al. 1988). Today, several tens of viruses and one mouse nuclear gene (Shigemoto et al. 2001; Manktelow et al. 2005) have been identified as bearing such a -1 frameshifting site. A typical eukaryotic site contains a slippery heptamer, where both A- and P-site tRNAs slip by one nucleotide upstream, followed by a stimulatory structure (stem loop, or pseudoknot) downstream (Brierley et al. 1989). The slippery heptamer is separated from the stimulatory structure by a short sequence, the so-called spacer. Based on this model, studies have been undertaken to identify -1 frameshifting sites in the nuclear genome of the yeast Saccharomyces cerevisiae (Hammell et al. 1999; Liphardt 1999). However, none of these made it possible to identify with certainty authentic expressed genes controlled by -1 frameshifting. Two reasons might be proposed to explain this situation: First, the model might not be precise enough, leading to the identification of too many false-positive candidates (Bekaert et al. 2003); conversely, the model might be too rigid, failing to identify true-positive candidates. The latter would be the case, for example, if a -1 frameshift could be directed by a more “degenerate” structure, or by mechanisms that rely on other types of signals.
Although most translational recoding events are found in viruses and transposons, a few cellular genes have been identified that use this mode of expression (Namy et al. 2004). These genes are involved in a variety of biological processes and are sometimes subject to a self-regulatory mechanism. Recoding is also widely distributed between organisms; it is thus likely that numerous novel recoded cellular genes remain to be discovered. However, the prediction of recoding sites from genomic databases is currently a difficult task. Since most recoding events generate a premature in-frame stop codon, this is generally categorized as an error by computer programs, leading to improper gene annotation. Bioinformatics strategies have been developed to identify recoded genes, based on the knowledge of the recoding mechanism (model-based approach). In this case, genomic sequences are searched for regions exhibiting an already known recoding signal. Such analyses have allowed the identification of several candidate recoded genes (Hammell et al. 1999; Baranov et al. 2002b; Namy et al. 2003). These approaches suffer major drawbacks: an imprecise model leading to a high number of false-positive candidates and too rigid a model failing to identify truly positive candidates. For this reason, we and others have undertaken to develop bioinformatics approaches that do not depend on models of recoding sites and can be performed without a priori knowledge of the mechanism involved (Harrison et al. 2002; Sato et al. 2003). These approaches seek genomic configurations compatible with recoding, such as two ORFs overlapping or separated by a unique stop codon. The high number of candidates is then filtered by secondary constraints (length, presence of protein motifs, etc.). Several candidate recoded genes have already been identified in S. cerevisiae (Harrison et al. 2002; Namy et al. 2003) and in Drosophila (Sato et al. 2003) in this way. However, except for one study (Namy et al. 2003), no biological validation has been performed to assess whether the candidate regions actually induce recoding in vivo.
The goal of the present work was to set up a comprehensive strategy, based on complementary bioinformatics and molecular approaches, and on functional in vivo analyses, to identify -1 ribosomal frameshifting sites in cellular genomes, without a priori knowledge of the mechanism involved. We devised two independent methods to look for frameshifting sites in silico. The first is based on the search for genomic regions in which two domains, each carrying a protein pattern, can be associated on the same polypeptide by a single -1 frameshifting event. The second is performed by a two-step selection with HMM. The first step identifies potential candidates likely to possess a constrained coding region after their stop codon. The second step ranks the candidates by likelihood ratio, based on available biological knowledge. These two approaches do not rely on any model of the frameshifting site and thus are well adapted for de novo detection of frameshift events. We validated these methods by analyzing the genome of S. cerevisiae. Indeed, the sequence information about S. cerevisiae is highly reliable because of multiple sequencings and careful annotation maintenance. Furthermore, the availability of several other ascomycetes genome sequences offers a unique opportunity to explore eukaryotic genome evolution by comparative analysis of several species (Dujon et al. 2004).
A total of 189 frameshifted candidate regions (fsORFs) were found. We assessed the presence of a full-length mRNA and quantified -1 frameshift efficiency for a subset of the highest ranked candidates. Among the 58 characterized regions, 28 were analyzed for their ability to induce -1 frameshifting in vivo; 11 showed a frameshift efficiency 50-fold higher than the background. Several of these candidates correspond to genes with known functions, which will allow further analysis of the physiological role of the frameshifting event. Overall, these results strongly suggest that -1 frameshift might be a more widely used strategy of controlling gene expression than previously thought.
Results
General strategy
Figure 1 shows the pipeline of our -1 identification strategy. We first download and parse the nucleic acid sequences, the intron/exon data, and their position on chromosomes. We stock them in a local database for more reliability. Our system seeks genomic configurations compatible with a -1 ribosomal frameshifting event using the following criteria: two open reading frames, one in the 0 frame (ORF0), the other in the -1 frame (ORF-1), that overlap along an intermediate shared region (Step 1).
The second step was to filter undesirable low-complexity sequences that may overload the next levels. The remaining sequences were classified according to whether the 0 and/or -1 frames are already annotated as an ORF, in order to perform the subsequent HMM step. We define four classes, “left” (ORF0 is annotated), “right” (ORF -1 is annotated), “both” (both ORFs are annotated), and “none” for all the others (Step 2). This classification is necessary, as the model with a frameshift will be compared either to a coding one (if there is yet any annotation), or to a noncoding model.
Two analyses were then carried out. Regions containing known protein motifs in both ORF0 and ORF-1 were retained (Step 3). In parallel, HMM filtering and estimation were performed to predict coding regions that may continue in the -1 frame after the stop codon of ORF0 (Step 3′). This was followed by a ranking step in which we compared the likelihood ratio of each selected candidate structure on the two following assumptions: “the sequence possesses a frameshift” and “the sequence does not possess any frameshift,” taking into account the class of the candidate defined in Step 2 (Step 3′).
We then tested the candidate regions for expression in vivo by looking for the presence of a full-length polyadenylated mRNA, using oligo(dT)-primed RT-PCR (Step 4). Finally, for the remaining candidates, -1 frameshifting efficiencies were determined in vivo, using a dual reporter system (Step 5).
Creating a data set of potential -1 frameshift regions
The goal of this step was to identify structures exhibiting a genomic organization compatible with a translational -1 frameshift mode of expression. We chose to search first for overlapping ORFs. We fixed a length of at least 99 nucleotides (nt) for both ORF0 and ORF -1 areas, and at least 150 nt for the entire structure (Fig. 2). Preliminary analysis (data not shown) had shown that decreasing this size by twofold (51 nt) increased by five times the numbers of retrieved structures. Thus, although a biologically pertinent candidate might have been obtained with less stringent length constraints, this limit was chosen to keep the number of candidates compatible with the biological validation step. All searches were performed independently on four sets of data: the S. cerevisiae genome (12 Mbp); the genome of the S. cerevisiae L-A virus (4579 bp), known to bear an authentic -1 ribosomal frameshifting site; and artificial genomes that exhibit the same hexamer frequencies as the S. cerevisiae and L-A genomes, respectively. The artificial genomes were generated using Markov chains (see Methods). These sequences were used to generate negative controls to estimate, both quantitatively and qualitatively, the background or fortuitous candidates. All possible frameshifted structures were then automatically extracted. Among all potential -1 frameshifts, some are DNA microsatellites (Hamada et al. 1984), i.e., tandem repeats of the same triplet that are read as repetitions of two different amino acids, depending on the reading frame. Such sequences were excluded by using mdust software, which removes low-complexity sequences. From this analysis 22,445 regions were found in the S. cerevisiae genome, 24,248 in the artificial genome, 10 in the S. cerevisiae L-A virus genome, and eight in the artificial L-A genome.
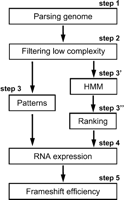
Pipeline of frameshifting candidate identification strategy.
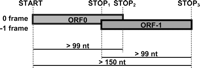
Schematic representation of the genomic configurations compatible with -1 ribosomal frameshifting.
Assessing functional frameshifting by InterproScan
All of the hit sequences were then subjected to a protein motif search. Each candidate sequence was kept only if it exhibited, in both frames, a pattern featured by the InterPro database and InterProScan (http://www.ebi.ac.uk.interpro/). Since this step was the most time-consuming of the whole analysis, it was first performed on the smallest of the two ORFs in each putative frameshifted candidate. This database includes BlastProDom, FPrintScan, HMMPIR, HMMPfam, HMMSmart, HMMTigr, ProfileScan, ScanRegExp, and SuperFamily. The default parameter settings were used for the search.
This approach was validated since the only actual frameshifting region was retrieved from the L-A virus genome. Moreover, 84 candidates were found in the S. cerevisiae genome and only 11 in the S. cerevisiae artificial genome. Among these 84 S. cerevisiae genomic regions, three categories could be defined. In the first category, 69 exhibited domains that contain stretches of repeated amino acids in each of the two frames. These are not low-complexity sequences that were already discarded at Step 2, but correspond to an area with a high density of a given amino acid, not a linear repetition of the same amino acid. Notably, no such candidates were found in the random genome. The second category is composed of regions in which the two ORFs bear similar protein patterns, or two distinct but functionally compatible motifs (e.g., a sugar transporter and a sugar binding site). We found six such regions in the S. cerevisiae genome and none in the random genome. The third category includes eight regions that bear functional regions in one ORF and amino acid repetitions in the other ORF. All 11 candidate sequences from the random genome belong to this category.
Obtaining structure candidates by HMM
One of the more efficient methods to segment sequences in coding and noncoding regions (allowing for different phases and genes on both strands) is HMM. It was introduced by Rabiner for speech recognition (Rabiner 1989). This method is now commonly used in bioinformatics, from gene detection to prediction of protein domains (Burge and Karlin 1997; Sonnhammer et al. 1998; Nielsen et al. 1999).
For each step to be performed in a HMM framework, one has to completely specify a model, i.e., a probability law on the hidden state's structure and a law for the emission of observed letters within each state. One has to note that the aim here is not simply to detect genes, but rather to select candidates for which the extension after the stop, in the -1 frame, is similar to that of coding regions. As far as we know, existing software designed for gene detection does not offer such flexibility: At present they are designed to detect nonoverlapping genes and are surely not able to detect a coding sequence with a frameshifting site. The beginning of such a gene may be missed if the length between the start codon and the frameshift is too short. Even when it is found, the program will probably decide on a false end, based on the presence of a stop codon. In addition, the part after the frameshift will hardly be detected because of the lack of a start codon. In the following paragraph, we detail the construction of the HMM and the strategy used for detection and ranking.
First, one needs to describe a model fitting with gene structure constraints. The simplest structure is summarized in Figure 3, and corresponds to the one used by common gene detectors (Burge and Karlin 1997). A gene begins with a start codon, continues by stretches of three bases corresponding to sense codons, and ends on a stop codon. Previous studies have demonstrated the distribution of codon—and thus amino acid—heterogeneities within genes (Nicolas et al. 2002). To take this type of heterogeneity into account, we allowed the model to alternate between up to three different laws for codons. All parameters of this model were first estimated on a similarity reduced set of 3158 ORFs (see Methods for details).
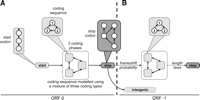
Illustration of the HMM structures used for estimation and testing. (A) Estimating a suitable model for coding regions. The gene model estimated on all nonredundant ORFs of S. cerevisiae is fitting. (B) Estimating additional parameters before the filtering step, to test the possibility of a frameshifted coding region after the first stop (dashed arrow).
Then, to adapt our model for the detection of frameshifted genes, we allowed coding regions to appear in the -1 frame after the stop. For this purpose, we inserted a transition from the state corresponding to the last base of the stop to the -1 coding frame of each coding type. We kept only those sequences for which the sum θ of the corresponding transition probabilities was >0.95, which corresponds to the clear-cut threshold shown in Figure 4.
As a positive control, we tested this step of our approach on the L-A virus. This virus is selected with a probability θ of 1.0 (this is only due to approximation errors), whereas the other candidates from the L-A virus, as well as the candidates from the S. cerevisiae random genome, reach at most a probability of 0.5.
Using this criterion, a final set of 110 candidates was retrieved. To incorporate for each selected candidate the known coding
status of the two possible coding frames, we separately treated the sequences in the four classes defined above: left, right, both, and none. In each class, we then ranked the sequences according to the likelihood ratio, which is a measure of the confidence we may
assign to the claim “X contains a frameshift” in comparison with “X does not contain a frameshift”: 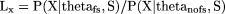
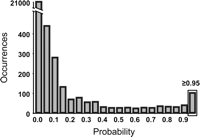
Distribution of the probability of transition from the 0 to the -1 frame for the candidate regions compatible with a -1 frameshifting event. As evidenced in this distribution, a clear peak was observed at the 0.95 limit. This threshold value was thus chosen as a cutoff to choose the candidates to be ranked.
Where theta_fs and theta_nofs stand, respectively, for the parameters of the model under the two following assumptions: “a frameshift exists” and “no frameshift exists” conditionally on the status of the ORF. More details about the models used conditionally on the subset can be found in the Methods section.
Candidates with their rank are summarized in Table 1. From these scores, we selected 23 candidates to be tested (seven from the none class, seven from the both class, five from the left class, and four from the right class). Figure 5 shows a representation of a “good” (fsORF 25) and a “bad” (fsORF 36) candidate.
Biological investigations of candidates
Common candidates
Finally, we crossed the results obtained using the protein motifs search and the HMM search. Five common candidates were identified by comparing the 84 regions obtained in the first approach with the 110 regions obtained in the second approach. As the two methods are independent, these five common candidates together with 25 candidates from the protein motifs approach and the 18 best ranked candidates from the HMM approach were selected for further biological investigation. We also selected the 10 worst candidates to serve as a control of the relevance of the ranking procedure (Table 1).
Genomic sequence of the candidates
Since an authentic frameshifting is indistinguishable from a sequencing error, we first verified the sequence of the genomic region spanning the overlap between ORF0 and ORF -1. Among the 58 candidate sequences analyzed, five did not show the presence of the expected frameshift. Since the strain used here (FY1679–18B) is different from the strain that has been used for the S. cerevisiae sequencing project (S288C), either a sequencing/annotation error or a gene polymorphism could explain this discrepancy.
Expression of candidate sequences
The next step was to test whether the candidate sequences correspond to expressed ORFs. Since most of these regions were previously considered as intergenic regions, they have not been included in systematic expression analyses. However, for those that are constituted of at least one previously annotated ORF right, left, and both classes), partial information was available and is indicated in Table 1. However, even in the cases in which the two ORFs were previously identified (both class), the presence of an mRNA corresponding to each ORF was tested independently. Thus, we checked whether an mRNA spanning the two ORFs is actually expressed. We examined the 53 remaining candidate sequences by RT-PCR, using first a reverse transcriptase step with an oligo(dT) primer that allows amplification of primarily polyadenylated mRNAs. The second PCR step was performed with an upper primer located 5′ of the first ORF (0 phase) and a lower primer located near the stop codon in the second ORF (-1 phase) to ensure that a full-length message is actually present in the cell. For a few exceptionally long regions, a random primer was used in the reverse transcriptase step and two pairs of internal primers were used for the secondary PCR instead. No signal observed in the absence of reverse transcriptase, and a unique specific amplification was obtained for 31 candidate sequences (Fig. 6; Table 1). We retrieved 16 amplifications out of the 23 HMM candidates and one out of the 10 worst candidates from HMM controls (p-value < 0.01). The finding of many more putative frameshifting sites in the highly ranked candidates than in the lowest ranked candidates is a very strong argument in favor of their biological significance.
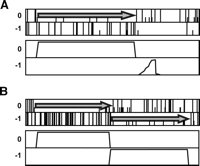
A posteriori probabilities plot of the coding states of a “Bad” (fsORF 39; A) and “Good” (fsORF 28; B) candidate from the both subset. (Top) Symbolic representation of the reading frames (plain bars, stop codon; half bars, initiation codon). (Bottom) Probability of coding in each frame. Arrows indicate coding frames.
These results demonstrate that the same molecule of mRNA covers both ORFs and that these mRNAs are polyadenylated. The region of overlap of the cDNAs corresponding to all the bicistronic mRNAs was analyzed by gel electrophoresis and subsequently sequenced (data not shown). For three candidate regions, the presence of an unexpected intron was demonstrated (Table 1). Close examination of the sequence revealed that the regions harbor a degenerate intron boundary pattern. For the remaining candidates there was no evidence of length or sequence polymorphism, suggesting that no splicing or editing event had taken place.
Quantification of -1 frameshift efficiency
It cannot be predicted whether ribosomes can actually shift from ORF0 to ORF -1 for 28 of these candidate expressed sequences, since none of them carries a canonical -1 frameshift signal, i.e., a heptamer followed by a secondary structure. To quantify -1 frameshift accurately, each fragment (about 50 nt on either side of the overlapping areas) was amplified by PCR from genomic DNA of a wild-type S. cerevisiae strain (FY1679–18B) and cloned into the pAC99 dual reporter vector (Namy et al. 2002). In this reporter system, each translating ribosome gives rise to β-galactosidase activity, whereas only those that frameshift into the overlapping region spanning ORF0 and ORF -1 would give rise to luciferase activity. Frameshifting efficiency is estimated by dividing the luciferase to β-galactosidase ratio obtained from the test construct by the corresponding ratio obtained from an inframe control construct (see Methods). Eleven fragments (Table 2) displayed a -1 frameshift efficiency ≥50-fold over the background (0.1%). Values ranged between 5% and 13% and correspond to those obtained when well documented frameshift sites are tested in the same experimental system (Bekaert and Rousset 2005).
fsORF with more than 5% of -1 frameshifting
Ascomycetes conservation
In order to determine if the organization of the 11 fragments directing frameshifting in vivo is preserved in other yeasts, we carried out alignments of the sequences against the genomic sequences of other ascomycetes. We found four structures in which only ORF0 is conserved (-∞ ≤ e-value ≤ 4.3 × 10-23), one in which ORF0 is present only in the Candida glabrata genome (fsORF 12, e-value = 5.1 × 10-13), and two in which no homolog could be found (Table 3). Interestingly, four structures (fsORF 33, 44, 51, and 52) are completely preserved (ORF0, ORF -1, and frameshifted organization). Surprisingly, fsORF 33 and 35 were reported to have a polymorphism (frameshift mutation) in S. cerevisiae and to present only one open reading frame (Brachat et al. 2003; Cliften et al. 2003; Kellis et al. 2003). Very recently, these two ORFs have been reannotated in-frame. Although strain to strain polymorphism can account for this observation, our results showing that the frameshifted structures are conserved in other ascomycetes strongly suggest that the frameshift is biologically significant.
Schematic profile of ORF0 and ORF-1 conservations in ascomycetes
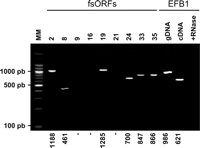
RT-PCRs. Total RNA was extracted as described in the Methods and treated with DNase I. RT-PCR was carried out in two steps. First, reverse transcription was carried out using an oligo(dT) primer, allowing only reverse transcription of poly(A) mRNAs. Then a standard PCR was performed on the mRNA after reverse transcription. The PCR products were visualized on a 1.5% agarose gel stained with ethidium bromide. A single amplification product was seen in positive lanes; the expected size is indicated (in nucleotides) for each product at the bottom of the gel. The control sample was EFB1 mRNA, which includes an intron. Specific PCR of genomic DNA and cDNA exhibits two different products. Reverse transcription after RNase shows no DNA contamination during the process.
Discussion
Here, we describe a comprehensive analysis of the S. cerevisiae genome that attempted to identify cellular recoding events occurring during translational -1 frameshifting. We developed a genomic approach, seeking genes with an extended coding potential, without prior constraint from existing ideas on the -1 frameshift mechanism.
In a first step, 22,445 genomic structures were extracted from the genome of S. cerevisiae. This value relies on two strong assumptions. First, we chose to collect only extensions of polypeptide but no premature ending, although biologically pertinent frameshifting events, such as in Escherichia coli DnaX, could lead to the synthesis of a shortened product (Tsuchihashi and Kornberg 1990). Second, we specified the minimal size of each ORF as 99 nt (33 amino acids).
Our approach identified 189 candidates in the S. cerevisiae genome. None of them had previously been found using a similar approach developed by Harrison et al. (2002). This study involved a pattern-based method, followed by a sequence comparison step against Genolevure (http://cbi.labri.fr/Genolevures/), MIPS (http://mips.gsf.de/genre/proj/yeast/), or SGD-annotated ORFs (http://www.yeastgenome.org/). Neither had any of our candidates also been found by Hammell and coworkers (1999), using a model-driven approach based on canonical frameshift signals.
Among the 189 candidate regions, 58 were analyzed further. Fifty of them showed the expected sequence, of which 31 directed transcription of an mRNA spanning the two overlapping ORFs. These 28 regions were cloned in a dual reporter vector, and 11 directed a -1 frameshifting efficiency 50-fold higher than background. To detect a possible mRNA editing mechanism, we sequenced the RT-PCR products for each of them. No RNA post-transcriptional modification was identified (Table 2). Moreover, from the amplification of the mRNA using a poly(dT) primer in the reverse transcription step, we concluded that these mRNAs are polyadenylated and not rapidly degraded.
No candidate conformed to the canonical model of the -1 frameshifting sites of Jacks et al. (1988). Three candidates exhibited a shifty heptamer in the appropriate frame but no detectable secondary structure (Table 2). Others might correspond to a -1 frameshifting event carrying a more degenerate site or even correspond to a completely different mechanism that ends with an apparent -1 frameshift, such as ribosome hopping, +2 frameshifting, or minority alternative splicing. In this latter case, the intron should be small since no differences in cDNA length were observed in the RT-PCR experiments. Some of these candidates might also turn out to be irrelevant with respect to frameshifting. In particular, some may correspond to pseudogenes or long 5′ or 3′ UTRs. Previous experiments have demonstrated that minimal frameshift signals from prokaryotic genomes can trigger ribosomes to shift to either the +1 or -1 frame in vitro (Gurvich et al. 2003). However, the same sequences in their genomic context failed to induce significant frameshifting, probably due to the sequence surrounding the frameshift site that may have evolved to suppress this phenomenon. Although this could apply to some candidates, we think this explanation is unlikely since our candidate sequences have been tested in vivo and with their surrounding sequence. Furthermore, during the last several years, we have tested several dozens of constructs for basal frameshifting efficiency and found systematically a background value between 10-4 and 10-3.
Among the candidates, three carry compatible protein patterns in the two ORFs, which suggests that they might actually be biologically significant. More precisely, Sco2 contains “electron transport” and “bipartite nuclear localization signal” motifs in ORF0 and ORF-1, respectively. It is similar to Sco1p and may have a redundant function with Sco1p in delivery of copper to cytochrome c oxidase; it interacts with Cox2p (Lode et al. 2002). Surprisingly, yeast two-hybrid assays also show it interacts with Cyclin-B (Ito et al. 2001). Both activities are consistent with the inferred motifs. Aad6 is homologous to an aryl-alcohol dehydrogenase (Delneri et al. 1999) and accordingly bears aldo/keto reductase motifs in both frames. YMR084W has glutamine amidotransferase and sugar isomerase motifs in ORF0 and ORF -1, respectively, and could be involved in amino acid and carbohydrate metabolic pathways. Perhaps the strongest argument in favor of the biological significance of a subset of the putative frameshifted ORFs identified here is their conservation in other ascomycetes.
In conclusion, the combination of two simple approaches has allowed us to identify several candidate genes potentially controlled by a -1 frameshift mechanism. Up to now frameshifting in chromosomal genes has been considered as a rare event, except in the case of +1 frameshifting found in a high proportion (>5%) in the ciliates such as Euplotes (Klobutcher and Farabaugh 2002). The promising strategy described here can possibly be extended to other organisms, both eukaryotic as well as prokaryotic (Bertrand et al. 2002), and to other recoding events. Finally, we hope that the identification of new cellular recoded genes will also tell us whether they share similar properties or play common physiological roles in the cell.
Methods
Data sources
The system uses entire chromosome sequences from the GenBank/RefSeq database (Maglott et al. 2000, http://www.ncbi.nlm.nih.gov) as inputs. S. cerevisiae chromosomes (NC_001133 to NC_001148) were downloaded on March 5, 2003, and S. cerevisiae virus L-A (NC_003754) was downloaded on December 25, 2003.
Random sequences
To define random background to be compared with real genome analyses, searches were performed independently on artificial genomes that exhibit the same hexamer frequencies as the S. cerevisiae genome or the L-A virus genome. We used GenRGenS software v1.0 (Denise et al. 2003, http://www.lri.fr/~denise/GenRGenS/) for random generation of genomic sequences, using Markov chains of order 5.
Implementation
The main system is implemented in Perl, Bioperl 1.1 (Stajich et al. 2002, http://bioperl.org/), and PostgreSQL. To detect protein signatures in the sequences, the motif database InterPro release 7.0 (Mulder et al. 2003, http://www.ebi.ac.uk/interpro/) was used along with InterProScan version 3.1 (Zdobnov and Apweiler 2001).
In terms of family coverage, the protein signature databases are similar in size but differ in content. While all the methods share a common interest in protein sequence classification, some focus on divergent domains (e.g., Pfam), some focus on functional sites (e.g., PROSITE), and others focus on families, specializing in hierarchical definitions from superfamily down to subfamily levels in order to pinpoint specific functions (e.g., PRINTS). TIGRFAMs focus on building HMMs for functionally equivalent proteins, and PIR SuperFamilies produces HMMs over the full length of a protein and have protein length restrictions to gather family members. SUPERFAMILY is based on structure using the SCOP superfamilies as a basis for building HMMs. ProDom uses PSI-BLAST to find homologous domains that are clustered in the same ProDom entry. The clustered resources are derived automatically from the UniProt databases.
Low-complexity filtering
The mdust algorithm (available from TIGR) was used to mask nucleic acid low-complexity regions, in particular from microsatellite areas, that enhance background noise and false positives.
HMM specification and estimation
Each estimation and computation on HMM was done using the software SHOW (Nicolas et al. 2002, http://www-mig.jouy.inra.fr/ssb/SHOW/). For the estimation of the coding parameters (defined as coding state; Fig. 3A), the ORF list of 5861 sequences available on the SGD Web site was used. As S. cerevisiae is known to possess a large proportion of paralogous genes, we then wiped out proteins presenting more than 70% of full-length similarity. All of these alignments were done using the FASTA program (Pearson 1990) using a BLOSUM62 matrix. Proteins were then clustered using a p-value threshold of 10-3, leading to a set of 3526 sequences. The estimation of the intergenic state (composed of one state of order two) was performed on the entire S. cerevisiae genome after masking of all of the annotated ORFs.
For the filter step (3′), the added links starting from the stop add three degrees of freedom to the model (the probabilities of shifting to the three possible coding states). In addition, three other parameters were added that correspond to the three coding state's length laws from STOP2 to STOP3. We chose to estimate these three new length parameters only on the left, right, and both subsets. It was necessary to set up such a conservative fashion, since an important proportion of the 22,445 sequences considered could possibly influence the length estimation through an atypical composition in their intergenic regions. More precisely, some intergenic regions appear to be better fitted by a mixture of two or three coding regions than by the intergenic law (Fig. 3B). Probabilities of transition from the stop to the shifted coding regions were then deduced with a classical forward–backward algorithm on the 22,445 candidate structures to achieve step 3′.
For the ranking step, the likelihood of filtered sequences was calculated under the two assumptions: “the sequence contains a frameshift” and “the sequence contains no frameshift.” Whereas the first assumption corresponds to the same model for all of the candidates, different models were designed for each of the classes left, right, none, and both for the second assumption. These correspond to the following facts:
-
none: “all the sequence is intergenic”;
-
left: “coding is followed by intergenic after STOP1”;
-
right: “coding ending on STOP3 is preceded by intergenic”;
-
both: “coding ends on STOP1, followed by intergenic and coding ending on STOP3”.
The sequences were then ranked within each class on the log odd-ratio of the two concerned assumptions, rescaled by their length.
Ascomycetes comparison
FASTA (Altschul et al. 1990) was used for the ascomycetes comparison. The FASTA search was executed (the e-value threshold was set to 1e-10) against the entire sequence of the following genomes retrieved from the GenBank/RefSeq database (Maglott et al. 2000): Candida glabrata (NC_005967–68 & NC_006026–36), Debaryomyces hansenii (NC_006043–49), Eremothecium gossypii (NC_005782–88), Kluyveromyces lactis (NC_006037–42), Schizosaccharomyces pombe (NC_003421, NC_003423 & NC_003424), and Yarrowia lipolytica (NC_006067–72).
Yeast strains and media
The S. cerevisiae strain used for this work was FY1679–18B (Mat α his3-Δ200, trp1-Δ63, ura3-52, leu2-Δ1). The strain was grown in minimal medium (0.67% yeast nitrogen base, 2% glucose) supplemented with the appropriate amino acids to allow maintenance of the different plasmids under standard growth conditions. Yeast transformations were performed by the lithium acetate method (Ito et al. 1983).
Plasmids
The pAC99 reporter plasmid has been previously described (Namy et al. 2002). Constructs were obtained by inserting a PCR fragment containing the full overlapping region into the MscI cloning site, between the lacZ and luc genes in plasmid pAC99. For -1 frameshift measurements, an in-frame control was used that allowed the production of 100% fusion protein (β-galactosidase–luciferase). The region including the inserted fragment was sequenced in the newly constructed plasmids. Each construct was then sequenced to check that no error occurred during PCR amplification.
Enzymatic activities and -1 frameshift efficiency
The yeast strains were transformed with the reporter plasmids using the lithium acetate method (Ito et al. 1983). In each case, at least five independent assays were performed under the same conditions. Cells were broken using acid-washed glass beads; luciferase and β-galactosidase activities were assayed in the same crude extract, as previously described (Stahl et al. 1995). Efficiency of -1 frameshift is defined as the ratio of luciferase activity from the test construct to the luciferase activity of the in-frame control construct. To account for variations in the levels of expression between different experiments, it was normalized by the ratio of the β-galactosidase activity from the test to the control constructs. To establish the relative activities of β-galactosidase and luciferase when expressed in equimolar amounts, the ratio of luciferase activity to β-galactosidase from an in-frame control plasmid was taken as a reference. Efficiency of -1 frameshift, expressed as a percentage, was calculated by dividing the luciferase to β-galactosidase ratio obtained from each test construct by the same ratio obtained with the in-frame control construct. In these conditions, reporter plasmids carrying no frameshifting sites give a background value of 0.1%.
Molecular biology procedures and RT-PCR
Each overlapping fragment corresponding to the candidate sequences was amplified from FY1679–18B genomic DNA by PCR, using Pfu polymerase (Promega), and cloned into the pAC99 vector and checked by sequencing.
Total RNA was extracted from 5 mL of exponential yeast culture (Schmitt et al. 1990). Each RNA sample was digested with 10 U of RNase-free DNase I (Boerhinger) for 1 h at 37°C. DNAse I was inactivated by heating for 5 min at 90°C, as recommended by the manufacturer. RNA was reverse-transcribed with oligo(dT) or random primer by Superscript II Kit (Invitrogen) for PCR amplification with Taq polymerase (Amersham). PCR fragments were visualized on a 1.5% agarose gel.
Acknowledgments
We are very grateful to Florent Bourassé, Alain Denise, Jean-Paul Forest, Christine Froidevaux, Michel Termier, and members of the G.M.T. laboratory for stimulating discussions. We are especially grateful to Anne-Lise Haenni for critically reading the manuscript and to Michael DuBow for proofreading the work prior to resubmission.
Footnotes
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.4258005.
-
↵3 Corresponding author. E-mail jean-pierre.rousset{at}igmors.u-psud.fr; fax 33 (0) 1 69 15 46 29.
-
- Accepted July 18, 2005.
- Received June 10, 2005.
- Cold Spring Harbor Laboratory Press








