
Abstract
Attempts to identify regulatory sequences in the human genome have involved experimental and computational methods such as cross-species sequence comparisons and the detection of transcription factor binding-site motifs in coexpressed genes. Although these strategies provide information on which genomic regions are likely to be involved in gene regulation, they do not give information on their functions. We have developed a functional selection for promoter regions in the human genome that uses a retroviral plasmid library-based system. This approach enriches for and detects promoter function of isolated DNA fragments in an in vitro cell culture assay. By using this method, we have discovered likely promoters of known and predicted genes, as well as many other putative promoter regions based on the presence of features such as CpG islands. Comparison of sequences of 858 plasmid clones selected by this assay with the human genome draft sequence indicates that a significantly higher percentage of sequences align to the 500-bp segment upstream of the transcription start sites of known genes than would be expected from random genomic sequences. We also observed enrichment for putative promoter regions of genes predicted in at least two annotation databases and for clones overlapping with CpG islands. Functional validation of randomly selected clones enriched by this method showed that a large fraction of these putative promoters can drive the expression of a reporter gene in transient transfection experiments. This method promises to be a useful genome-wide function-based approach that can complement existing methods to look for promoters.
With the sequencing of the human genome nearly complete, intense efforts are being made to annotate the genome at sites such as the National Center for Biotechnology Information (NCBI), Ensembl, and University of California Santa Cruz (UCSC). Most of the annotation is of protein-coding regions of genes, which comprise less than 4% of the human genome. The large-scale discovery and study of regulatory sequences in the human genome remain a considerable challenge. Regulatory sequences make up a small fraction of the genome that is noncoding. Cis-regulatory elements such as promoters, enhancers, insulators, silencers, matrix attachment regions, and locus control regions play crucial roles in regulating the levels, sites, and timing of gene expression (Pennacchio and Rubin 2001). Promoters are key regulatory sequences that are located near the 5′ ends of genes and are necessary for the initiation of transcription. Promoter sequences from vertebrates have not yet been identified on a large scale, and they are poorly annotated in public databases. There are currently 1871 nonredundant human promoters in the Eukaryotic Promoter Database 73, an annotated collection of RNA polymerase II promoters, for which the transcription start site has been determined experimentally (Praz et al. 2002). This suggests that experimental analyses have identified fewer than 10% of the potential promoter regions, assuming that there are at least 30,000 promoters in the human genome, one for each gene.
Finding and dissecting critical promoter regions has been done for one or a few genes by performing deletion analyses on suspected promoters in plasmid constructs and transfecting them into cultured cells (Myers et al. 1986; Borges and Dingledine 2001). This experimental method is not easily scalable to provide whole-genome discovery of promoter elements. On a genome-wide scale, pattern-based and genomic context-based computational approaches can suggest possible transcription factor-binding regions, but the rate of false-positive predictions is very high (Fickett and Hatzigeorgiou 1997; Scherf et al. 2000; Ohler and Niemann 2001).
Sequence analysis of coregulated genes is another way of mining for putative promoters and other regulatory elements, as coexpression of genes often occurs by the use of common cis-acting sequences (Roth et al. 1998). However, the correlation between clusters of genes with similar expression profiles and sequence motifs is not perfect in either direction. Cross-species sequence comparison is yet another strategy for identifying regulatory regions, based on the assumption that they are conserved (Hardison 2000; Pennacchio and Rubin 2001). Once conserved noncoding sequences are identified, it is still a challenge to select the ones that have regulatory function, and separate them from other conserved elements, such as unidentified exons and RNA genes. Therefore, complementary high-throughput experimental strategies for identifying novel regulatory sequences and adding functional information to putative ones would be useful.
This paper describes a retroviral plasmid library-based approach to identify promoter regions in the human genome. This method selects for and detects the promoter function of isolated DNA fragments from a complex mixture in a cell culture assay. We analyzed the sequences of plasmid clones selected in this assay by using the draft sequence of the human genome. We identified promoters of known genes and predicted genes, as well as many other likely promoter regions based on the presence of features such as CpG islands. The combination of this retroviral plasmid library-based assay with computational analysis of sequences of putative promoter-containing clones promises to be a useful function-based approach that can complement existing methods to look for promoter regions and other regulatory elements.
RESULTS
We developed and tested a cell-based functional assay to identify promoter regions in the human genome. This assay uses a library of genomic DNA fragments cloned into a retroviral vector, which is advantageous because a single retroviral integration event occurs per target cell in an efficient manner. We constructed a genomic DNA library in a Moloney murine leukemia virus (MMLV) plasmid pSKF-6 (Fig. 1A) by cloning human genomic DNA fragments of 300–600 bp upstream of a promoterless green fluorescent protein (GFP) reporter gene. pSKF-6 is a self-inactivating (SIN) vector with a defective 3′ long terminal repeat (LTR) that replaces the functional 5′ LTR upon reverse transcription of the virus, resulting in the transcriptional inactivation of the provirus in the infected cells. This eliminates the possibility of transcriptional activation of GFP by viral LTRs.
Selection scheme for promoter regions. First, 300–600-bp genomic DNA fragments are subcloned into the BstXI sites in (A) retroviral plasmid pSKF-6. (B) The genomic DNA library is transfected into packaging cells that supply viral proteins encoded by gag, pol, and env, to convert DNA to the RNA virus. Target cells are infected with the viral library and screened by FACS for clones that drive the expression of the GFP reporter gene. Genomic DNA is prepared from the GFP-positive cells, and inserts are recovered by PCR amplification for sequencing and functional validation assays.
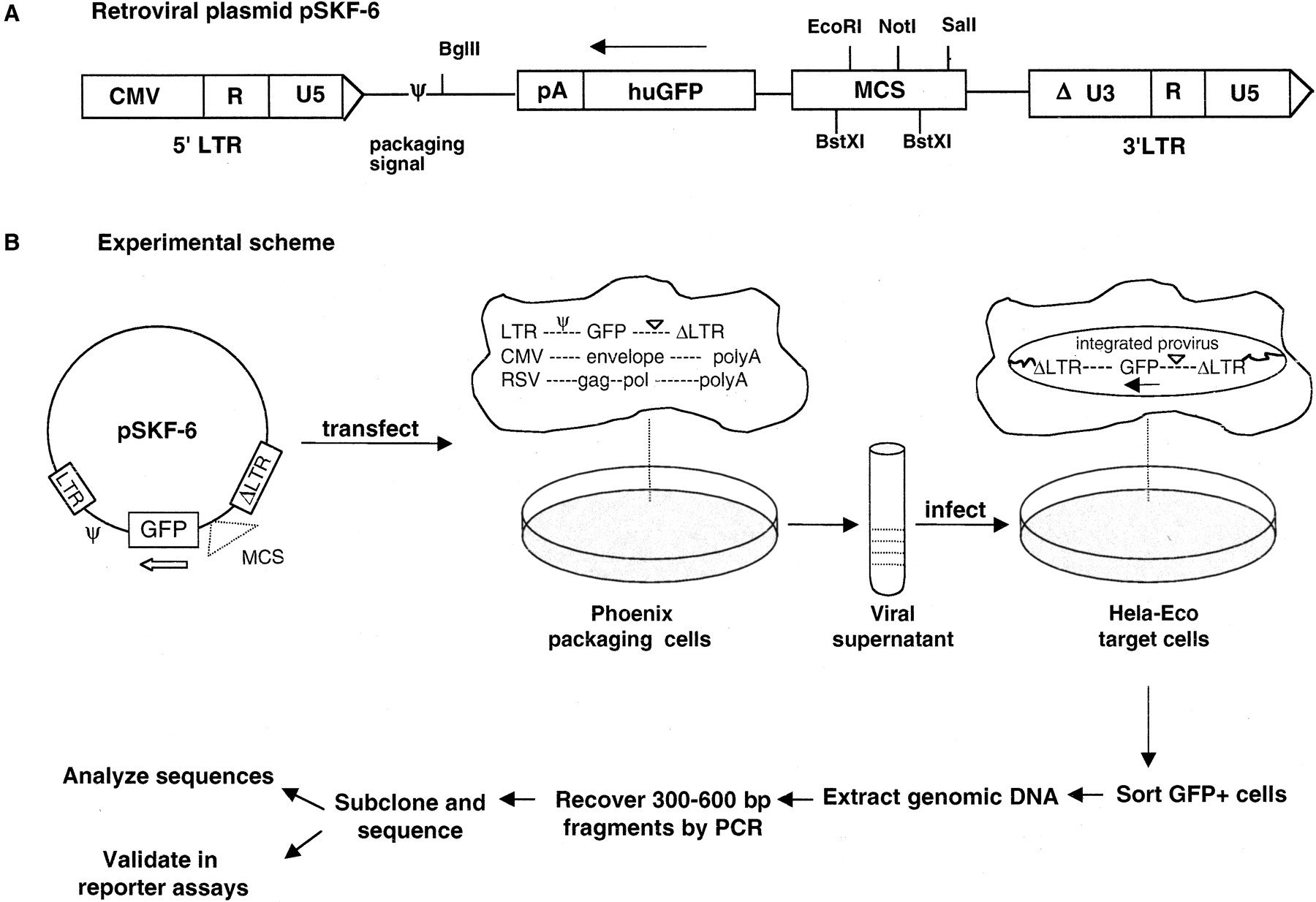
We transfected this human genomic library into the Phoenix-Eco packaging cell line to convert DNA to the RNA virus (Fig. 1B). HeLa-Eco target cell lines were infected with this viral library and screened by using fluorescence-activated cell sorting (FACS) for clones that can drive the expression of GFP. We isolated genomic DNA from sorted cells that were GFP-positive and recovered inserts containing putative promoters by PCR-amplification with primers that recognize the vector. We subcloned the pool of amplified PCR products, sequenced the insert from each clone, and performed detailed sequence analysis for 858 putative promoter-containing clones. We retested 130 clones individually for promoter activity in transient transfection reporter assays.
Selection for Putative Promoter Region Clones by FACS
We sorted the library into two fractions that we refer to as “GFP+ high” and “GFP+ low” fluorescence populations (Fig. 2B), based on the amount of GFP fluorescence in the cell pools. The GFP+ high fraction represents approximately the top 40% of fluorescent cells, and the GFP+ low fraction represents approximately the remaining 60% of fluorescent cells. To examine whether a second round of FACS improves the yield of putative promoter clones, we expanded the top 10% of GFP+ high cells from one experiment by culturing for an additional 5 d and then resorted and selected for high levels of GFP fluorescence.
FACS of the genomic DNA library into low and high fractions. (A) Histogram for retroviral plasmid pSKF-6 with no promoter and the positive control pSKF-6 with the human PGK promoter (right peak). (B) Histogram for retroviral plasmid pSKF-6 with no promoter and the 300–600-bp fragment library. Histograms represent cell number as a function of GFP fluorescence. GFP expression was measured by using flow cytometry with excitation at 488 nm and emission at 534 nm read as fluorescein.
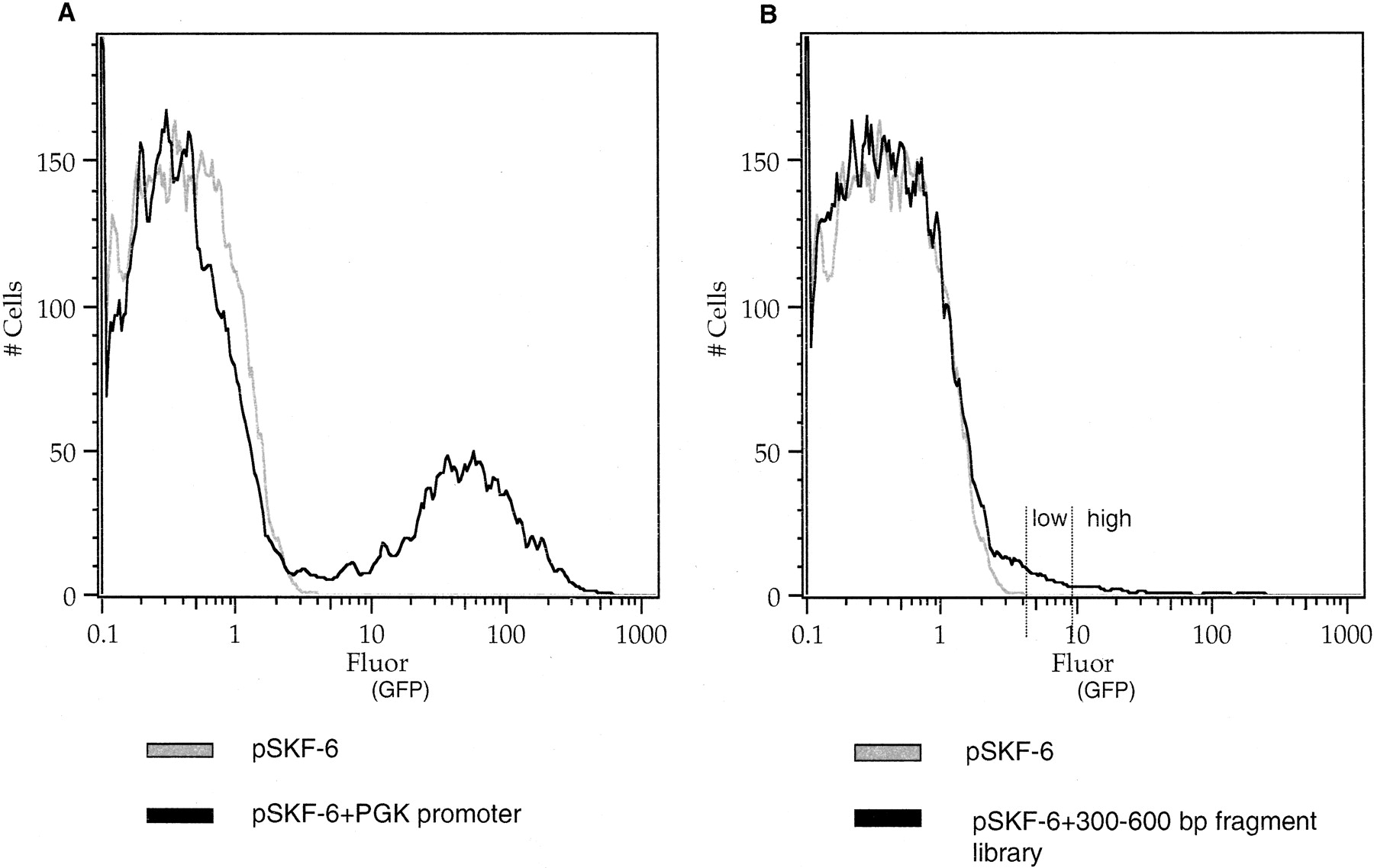
Testing our system with the vector pSKF-6 with no promoter inserts consistently gave ≤0.2% GFP+ cells (Fig. 2A). This indicates an occurrence of minimal positive position effects due to integration of the viral DNA into regions of the genome containing regulatory elements. This is in agreement with other studies that used gene- or promoter-trap retrovirus-based vectors, where the frequency of integrations that led to the expression of a reporter gene from an endogenous active promoter in the host cell was between 0.04% and 0.5% (Von Melchner et al. 1990; Jonsson et al. 1996; Medico et al. 2001). Positive controls containing the vector pSKF-6 with the human PGK promoter (Fig. 2A), or the mouse β-globin promoter (data not shown), yielded high numbers of GFP+ cells. A negative control vector with the his3-ded construct that has no promoter activity resulted in ≤ 0.1% GFP+ cells, showing that there are negligible position effects that cause the GFP reporter gene to be expressed (data not shown). The yeast his3-ded1 region is a 550-bp spacer DNA that is part of a stop cassette of vector pBS302 (Lakso et al. 1992).
Sequence Analysis of 858 GFP+ Clones
We sequenced approximately 1000 GFP+ clones and analyzed the high-quality sequencing reads by alignment to the August 2001 assembly of the human genome draft sequence with the BLAT program (Kent 2002). A total of 858 sequences had good alignment (i.e., were more than 90% identical to the genomic sequence with less than 10 base pairs of insertions or deletions) with the human genome sequence. For each alignment, we searched the annotation of the human genome to determine whether there was overlap with known genes, predicted genes, CpG islands, and mRNA sequences. For known and predicted genes, we split the alignment positions into the 2 kb of sequence located just upstream of the transcription start site of a gene, exon 1, intron 1, and the rest of the gene after the start of exon 2. In a few cases, sequence analysis with BLASTN (Altschul et al. 1990) against the nr database (http://www.ncbi.nlm.nih.gov/BLAST) revealed additional information about a sequence that we integrated with the data from the human genome BLAT and database searches. Below, we present a summary of the data obtained from computational analyses that provided evidence as to whether a clone sequence contains a promoter. Details on all analyzed sequences can be found in Supplementary Tables 1 and 2 (available online at www.genome.org).
Sequence Analysis of 858 GFP- Positive Clones
Category | GFP+ population | # of GFP+ clones/total | % of GFP+ clones | # of GFP+ clones that also hit a CpG island | # of GFP+ clones that also hit exon 1 or intron 1 of a Refgene |
|---|---|---|---|---|---|
| A | |||||
| Within 500 bp upstream of a Refgene transcription start site | GFP+ low | 15/418 | 4% | 9/15 | — |
| GFP+ high | 57/440 | 13% | 49/57 | — | |
| 500bp-2kb upstream of a Refgene transcription start site | GFP+ low | 8/418 | 2% | 0/8 | — |
| GFP+ high | 7/440 | 2% | 3/7 | — | |
| B | |||||
| Within 2 kb upstream of transcription start site of predicted genes in >2 annotation tables | GFP+ low | 37/418 | 9% | 13/37 | 19/37 |
| GFP+ high | 35/440 | 8% | 23/35 | 17/35 | |
| C | |||||
| CpG islands only | GFP+ low | 34/418 | 8% | 34/34 | 9/34 |
|
GFP+ high
|
51/440
|
12%
|
51/51
|
15/51
|
[i] Redgene refers to known genes derived from RefSeq mRNA alignments
Predicted gene annotation tables are from Genscan, Ensembl, Acembly, and Softberry
Examples of GFP+ Clones That Align With the Region 500bp Upstream of the Transcription Start Sites of Refgenes
Clone ID | Refgene ID | Gene name | Position | CpG island | Validation | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
GFP+ low sequences
| ||||||||||
| SKA13-H04 | NM_017584 | aldehyde reductase-like 6 | (-245, +88) | + | nt | |||||
| SKA05-G11 | NM_007058 | calpain 11 | (-628, -327) | + | nt | |||||
| SKA14-C06 | NM_016011 | CGI-63 protein | (-418, +35) | - | nt | |||||
| SKG03-E09 | NM_005227 | ephrin-A4 | (-335, -42) | + | LUC+++ | |||||
| SKT01-D1 | NM_000153 | galactosylceramidase | (-171, +123) | - | nt | |||||
| SKT02-B4 | NM_005517 | high mobility group protein 17 | (-5, +256) | - | LUC++ | |||||
| SKT04-A11 | NM_017721 | hypothetical protein FLJ20241 | (-552, -36) | - | LUC++ | |||||
| SKT06-B3 | NM_024643 | hypothetical protein FLJ23093 | (-264, +100) | + | LUC++ | |||||
| SKT02-C4 | NM_021732 | hypothetical protein PP5395 | (-361, +100) | + | nt | |||||
| SKA05-E05 | NM_017566 | hypothetical protein DKFZp434G0522 | (-792, -447) | - | nt | |||||
| SKT01-B3 | NM_024055 | hypothetical protein MGC5499 | (-274, +223) | + | βGAL+ | |||||
| SK04-H9 | NM_022151 | MAP-1 protein | (-448, -80) | + | nt | |||||
| SKT06-A8 | NM_002513 | non-metastatic cells 3 protein | (-829, -343) | + | nt | |||||
| SKT01-F12 | NM_012392 | peflin | (-508, -214) | + | nt | |||||
| SKA08-D03 | NM_005777 | RNA binding motif protein 6 | (-361, -90) | - | nt | |||||
|
SKG04-E05
|
NM_003420
|
zinc finger protein 35
|
(-8, +412)
|
+
|
LUC-
| |||||
|
GFP+ high sequences
| ||||||||||
| SKT08-B3 | NM-001628 | aldo-keto reductase 1 | (-268, +154) | - | LUC- | |||||
| SKG01-H10 | NM_016039 | CGI-99 protein | (-98, +284) | + | βGAL++ | |||||
| RM1-E06 | NM_021254 | chromosome 21 open reading frame 59 | (-176, +96) | + | LUC++ | |||||
| SKG01-E02 | NM_004373 | cytochrome c oxidase Vla 1 | (-315, -65) | + | nt | |||||
| SKA02-B12 | NM_005226 | endothelial diff. sphingolipid GPCR3 | (-247, +70) | - | LUC++ | |||||
| SKG01-F06 | NM_003827 | ethylmaleimide sens factor attach protein | (-595, -296) | + | nt | |||||
| SKA10-D10 | NM_007267 | expressed in activated T/LAK cells | (-429, -115) | + | nt | |||||
| SKA03-C09 | NM_005766 | Ferm, Rhogef, Pleckstrin DM protein | (-396, +94) | - | LUC++ | |||||
| SKG01-H12 | NM_020150 | GTP-binding protein SAR 1 | (-16, +190) | - | LUC++ | |||||
| RM2-A03 | NM_003516 | H2A histone family, member O | (-650, -311) | - | nt | |||||
| SKA04-G10 | NM_015699 | hypothetical protein (DJ159A19.3) | (-496, -198) | - | nt | |||||
| SKT05-C12 | NM_015383 | hypothetical protein DJ328E19C1.1 | (-124, +142) | - | LUC+ | |||||
| SKT03-B7 | NM_031904 | hypothetical protein FKSG44 | (-209, +269) | + | βGAL+++ | |||||
| SKA02-E08 | NM_017977 | hypothetical protein FLJ10040 | (-733, -442) | - | nt | |||||
| SKG02-C07 | NM_032678 | hypothetical protein FLJ13142 | (-473, -217) | + | LUC- | |||||
| SKT03-C7 | NM_024771 | hypothetical protein FLJ13848 | (-405, -59) | + | nt | |||||
| SKG01-A07 | NM_017721 | hypothetical protein FLJ20241 | (-552, -36) | - | nt | |||||
| SKT03-D8 | NM_024643 | hypothetical protein FLJ23093 | (-264, +100) | + | LUC+++ | |||||
| RM2-H12 | NM_024084 | hypothetical protein MGC3196 | (-125, +293) | + | nt | |||||
| RM4-H07 | NM_024516 | hypothetical protein MGC4606 | (-37, +384) | + | nt | |||||
| SKT08-E4 | NM_021732 | hypothetical protein PP5395 | (-361, +100) | + | nt | |||||
| SKA01-E03 | NM_014717 | KIAA0390 gene product | (-58, +169) | - | nt | |||||
| SKT08-G3 | NM_003201 | mitochondria transcription factor 6-like | (-319, -79) | + | LUC+ | |||||
| SKT07-F6 | NM_002633 | phosphoglucomutase 1 | (-330, +94) | + | βGAL++ | |||||
| SKT07-E4 | NM_007182 | Ras association domain family 1 | (-344, -16) | + | βGAL+++ | |||||
| SKG01-A09 | NM_000999 | ribosomal protein L38 | (-85, +261) | + | LUC+ | |||||
| SKA09-B10 | NM_033022 | ribosomal protein S24 | (-254, +109) | + | LUC+++ | |||||
| SKT08-D4 | NM_002966 | S100 calcium binding protein A10 | (-321, -22) | + | LUC++ | |||||
| SKT03-H6 | NM_013444 | ubiquilin 2 | (-235, +104) | + | LUC++ | |||||
|
SKT05-H3
|
NM_052821
|
WD repeat domain 5 variant 2
|
(-191, +344)
|
-
|
LUC++
| |||||
[i] Position of clone is shown relative to transcription start site, which is at position 0. +, overlap and -, no overlap with a CpG island. Functional validation in transient transfection β-galactosidase (βGAL) and luciferase (LUC) assays: fold activity, +, 3-10; ++, 10-50; +++, >50; -, negative; nt, not tested
Discovery of Putative Promoter Regions of Known Genes
Strong evidence that a clone selected by our method contains a promoter is alignment of the sequence to the known or inferred promoter region of a RefSeq gene (Pruitt and Maglott 2001). Promoters for genes transcribed by RNA polymerase II are located just upstream from transcription start sites and generally extend one hundred to a few hundred base pairs upstream. Because mRNA sequences for many known genes are incomplete at their 5′ ends, the true transcription start site of a gene may be upstream of the transcription start site listed in the annotation database. Therefore, we believe it is prudent to examine a 2-kb window upstream of the transcription start site rather than one that is a few hundred base pairs in length.
Of 858 sequences that we defined from clones selected by this method, 6% of GFP+ low clones and 15% of GFP+ high sequences aligned to the 2-kb segment upstream of the transcription start site of a known gene (category A in Table 1). Interestingly, 70% (61/87) of the sequences in category A overlap with CpG islands. Eighty-three percent (72/87) of the GFP+ sequences in category A align with the region just 500 bp upstream of the transcription start site of a known gene. Although some sequences, such as the putative promoter region of the ephrin-A4 gene, fall completely upstream of the transcription start site, others, such as the putative promoter region of the ribosomal protein L38 gene, contain the promoter and part of the 5′UTR of the gene (Table 2). This is not unexpected, as the 300–600-bp genomic DNA fragments cloned into the library are large enough to contain a promoter and extend into the 5′ UTR of a gene. The newly identified promoter sequences belong to a variety of genes coding for enzymes (e.g., phosphoglucomutase1), calcium binding proteins (e.g., peflin), membrane binding proteins (e.g., ubiquilin2), ribosomal proteins (e.g., S24), and histone family members (e.g., H2BFL; Table 2, Suppl. Table 3).
Based on the total number of genes in the Refgene table from UCSC (14,402 genes), and the size of the August 2001 assembly of the human genome (2.88 Gb), we estimate that 0.3% of the human genome sequence falls within 500 bp upstream of the transcription start site of a Refgene. In our experiments, we identified 4% (15/418) GFP+ low clones in this category. This fraction (4%) is significantly higher than the rate of 0.3% expected if there was no selection for promoters (P < 2.24 × 10-32, X2 test). This result suggests that our method significantly enriched for DNA fragments that aligned to a 500-bp region just upstream of a Refgene transcription start site. Similarly, we identified 13% (57/440) GFP+ high clones that aligned to a 500-bp region upstream of the transcription start site of a Refgene. This fraction (13%) is not only significantly higher than 0.3%, but is also significantly higher than the observed ratio for the GFP+ low clones (P < 1 × 10-6, Fisher's Exact Test), indicating that the enrichment is better when cells are selected in the higher fluorescence range. This is not surprising considering that GFP+ high cells are more distinctly separated from the background fluorescence than GFP+ low cells.
In one of our experiments, a single round of sorting of the top 10% of GFP+ high cells indicated that 8% of GFP+ clones contain putative promoter sequences that align within 500 bp upstream of the transcription start site of a known gene. Sorting the GFP+ high cells a second time after expanding them in culture resulted in 21% of GFP+ clones aligning to 500 bp upstream of the transcription start site of a known gene, with some of the putative promoter sequences being present more than once (data in Suppl. Table 4). These results indicate that a second round of selection provides better enrichment for promoters and minimizes false positives selected by FACS.
Identification of Likely Promoters Based on Gene Predictions and CpG Islands
Although identifying the promoter regions of known genes in an enrichment experiment is the best indicator that our assay system works, it is important to classify other sequences that are selected as being GFP-positive. Of 858 sequences, 9% of GFP+ low clones and 8% of GFP+ high clones aligned to the 2-kb segment upstream of the transcription start site of a predicted gene in at least two of four data sets of predicted genes from Genscan, Ensembl, Softberry (Fgenesh++), and Acembly (category B in Table 1). Half of the sequences from this category overlapped with a CpG island.
In humans, CpG islands are found at the 5′ ends of at least half of the genes and often contain the promoter and one or more exons (Cross and Bird 1995). Therefore, we examined whether sequences selected by our promoter trapping method are enriched for the presence of CpG islands. In addition to the identified putative promoter sequences aligning upstream of Refgenes or at least two predicted genes, 8% of GFP+ low clones and 12% of GFP+ high clones overlapped with a CpG island (category C in Table 1). This resulted in a total of 13% (56/418) of GFP+ low clones and 29% (126/440) of GFP+ high clones (from categories A, B, and C in Table 1) having overlap with a CpG island in the human genome. Based on the total number of base pairs in CpG islands (∼22 Mb) and the size of the August 2001 assembly of the human genome (2.88 Gb), we estimate that 0.8% of the annotated human genome draft sequence contains CpG islands. Therefore, sequences identified in our assay are distinctly enriched for CpG island-containing stretches (P < 2.38 × 10-180, X2 test). This not only confirms that CpG islands are often associated with promoters that are transcriptionally active (for review, see Antequera and Bird 1999), but also provides clues for the presence of genes and regulatory sequences in uncharacterized genomic DNA.
Detection of Other Sequences That Are Likely Promoters
Approximately 70% of the GFP+ sequences recovered from the sorted cells do not fall into any of the categories described above. Whereas many might be present as a result of experimental noise, others have interesting features that point to the possibility that they are promoters or behave as promoters in the context of our assay system. Six percent (48/858) of the sequences that are not in the categories A, B, or C in Table 1 aligned with the first exon or intron of a Refgene. In addition, 50% (36/72) of the sequences in category B and 28% (24/85) of the sequences in category C also fell within the first exon or intron of a known gene (Table 1).
The first exon and first intron are likely locations for an alternate promoter for driving the expression of an alternative transcript of a gene. Indeed, alternative promoters in exon 1 or intron 1 have been described for several genes, such as NADH-cytb5 reductase and MDM2, a gene amplified in cancer (for review, see Ayoubi and Van de Ven 1996). Several GFP+ clones that aligned with the first exon or intron in our study have more than one transcript variant described in LocusLink (NCBI) or in the Acembly gene annotation. For example, the GFP+ clone SKT06-C6 contains parts of exon 1 and intron 1 of the longest isoform of the tumor necrosis factor receptor superfamily 6B (TNFRSF6B) gene that is known to have many alternative transcripts (Bai et al. 2000).
Our analysis indicates that an additional 9% (77/858) of the sequences align within the 2-kb upstream region of a gene predicted in only one annotation database. An additional 8% (71/858) of sequences overlap with retroviral-like transposable elements that are identified by the RepeatMasker program (A. Smit and P. Green, unpubl.). About 8% of the human genome is composed of retroviral-like LTRs that contain internal transcriptional regulatory sequences for propagation by retrotransposition (International Human Genome Sequencing Consortium 2001). Although retrotransposons are often considered inert parts of the genome, recent studies have shown that some of them have retained the capacity to initiate transcription and probably have effects on gene expression (Whitelaw and Martin 2001). Forty-nine percent (418/858) of the sequences that we isolated align to the coding or noncoding part of a gene 3′ to the start of exon 2, or to a part of the human genome that is not annotated as a gene.
Additional Evidence for the Identification of Putative Promoters
We checked whether our sequences are conserved between the human and mouse genomes, as promoters are highly likely to be conserved across species due to their important function. Based on an alignment between the human and mouse genomes (International Mouse Genome Sequencing Consortium 2002), we found that 48% of the nucleotides in GFP+ low sequences and 57% of the nucleotides in GFP+ high sequences are in regions that are conserved between human and mouse (details in Suppl. Table 2). These values are considerably higher than 40%, the fraction of the human genome that can be aligned to the mouse genome at the nucleotide level. Interestingly, 47% of GFP+ sequences reside completely in conserved regions, and an additional 25% of GFP+ sequences reside partially in conserved regions.
We also searched the 858 sequences for a potential TATA-box (TA[T/A][T/A][T/A] [T/A]). Although the TATA-box is generally around -30 bp from the transcription start site, we scanned the entire sequence, because the precise transcription start sites are unknown. Notably, 50% (427/858) of sequences have this TATA-box (Suppl. Table 2). This is significantly higher than what we would expect based on the nucleotide distribution of our sequences (P < 0.05, X2 test).
Validation of 130 Clones in Transient Transfection Reporter Assays
To provide additional functional evidence for the selected GFP+ clones, we tested 116 clone inserts in dual luciferase assays of 293 or HeLa-Eco cells and an additional 14 clones in β-galactosidase reporter assay systems. This set of 130 clones was comprised of 29 randomly picked GFP+ clones from those that align to the 2-kb region upstream of the transcription start site of known genes (category A), and an additional 101 randomly chosen GFP+ clones (from categories B, C, and other GFP+ sequences).
For putative promoter regions within 2 kb upstream of a Refgene transcription start site (category A), 86% (25/29) were able to drive the expression of the reporter gene in this validation assay (Fig. 3, Suppl. Table 5). Fifty-six percent (10/18) of the clones that align to within 2 kb upstream of predicted genes in two or more annotation tables (category B) were found to be positive for luciferase activity. Seventy-three percent (19/26) of the clones that are classified as putative promoters based on overlap with a CpG island (category C) were luciferase or β-galactosidase positive. The remaining 57 randomly selected clones that we tested were not in categories A–C. These include a variety of clones, such as those that align with exon 1 or intron 1 of a known or predicted gene, and those that contain retrotransposable repeat elements (Suppl. Table 5). Of these clones, 61% (35/57) were able to drive expression of the reporter gene in this validation assay.
Distribution of GFP-positive clones that are functionally validated in transient transfection reporter assays. Clones are divided into three groups based on the fractional increase in activity of the reporter gene. Category A: within 2 kb upstream of a Refgene transcription start site, 86% luc+/β-gal+. Category B: within 2 kb upstream of predicted genes in two or more annotation tables, 56% luc+/β-gal+. Category C: overlap with a CpG island, 73% luc+/β-gal+. Other: clones not in Categories A–C, 61% luc+/β-gal+.
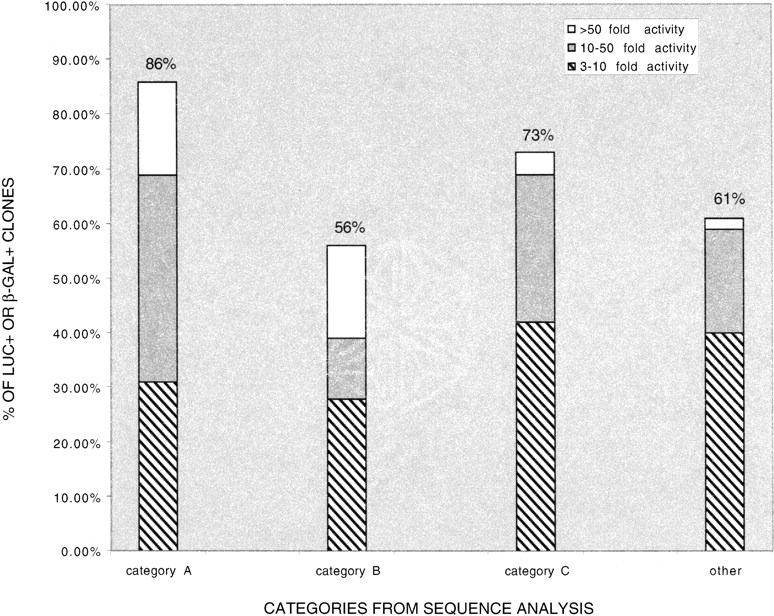
We divided positive clones into three groups based on the fractional increase in activity of the reporter gene (Fig. 3; Suppl. Table 5). We observed likely promoters of varying strengths from all sequence analysis categories. It should be noted that more than half of those sequences with retrotransposable repeat elements that we tested showed promoter activity in this assay. We found that 32% (41/130) of the clones that we retested in the validation assay were not able to drive the expression of the reporter gene. This could be because they are not true promoters, and are part of the experimental noise that might be expected in a multistep enrichment process such as our method. Alternatively, it is possible that they are indeed promoters but are inactive in transient transfection assays in 293 cells, as they were originally discovered to have promoter activity in HeLa-Eco cells from our retroviral screen. The observation that 68% (89/130) of the clones can be functionally validated on a clone-by-clone basis in transient transfection reporter gene assays provides verification that our retroviral screen indeed selects for genomic DNA fragments that have promoter activity.
DISCUSSION
We present here a functional selection for potential promoter sequences in the human genome that uses a retroviral plasmid library in which the inserts are genomic restriction fragments that are subcloned upstream of a promoterless reporter gene. By using flow cytometry, this method selects for cells that have green fluorescence as a result of integration of a provirus containing a putative promoter that drives the expression of the GFP gene. The method is versatile, because a stock of a retroviral library can be used to isolate novel sequences with potential promoter activity in different cell lines as well as primary cells, and at distinct stages of development. In addition, the approach is likely to be generally useful for isolating other transcriptional regulatory elements, such as enhancers.
Strategies that employ retroviral promoter traps have been used in mammalian cell lines and transgenic mice to isolate a few promoters, to identify developmental genes, and to study differentially regulated genes (Von Melchner et al. 1990; Friedrich and Soriano 1991; Wan and Nordeen 2002). For example, a lacZ-containing promoter-trap retrovirus was introduced into mouse myeloid progenitor cells to identify genes involved in early hematopoiesis (Jonsson et al. 1996). These strategies differ from our method in that they involve the introduction of a retroviral vector containing a promoterless reporter gene into a host cell, followed by the analysis of those integrations that allow the expression of the reporter from an active endogenous promoter in the host cell. Retroviral libraries of randomly synthesized 18-mers have been used in an attempt to identify cis-elements that modulate the activity of a minimal promoter (Edelman et al. 2000).
Although retroviruses can integrate their DNA into a large number of genomic sites, it is unclear whether they have preferred integration targets in the host genome. Some studies show that transcriptionally active DNA is not a preferred target for retroviral integration, whereas other studies reveal that retroviruses such as HIV preferentially integrate in active genes and regional hotspots (Weidhaas et al. 2000; Schroder et al. 2002). Integration of our retroviral vector into transcriptionally active regions or other chromosomal sequences such as transcriptional silencers could have led to position effects that might have influenced the expression of GFP. This may have produced a small fraction of false positive, as well as some false negative fluorescent clones. By using appropriate positive and negative retroviral vector controls, we have shown that position effects do not seem to influence GFP reporter expression significantly.
We identified promoters of known genes, predicted genes, and other sequences that are very likely to be promoters. We also obtained sequences that may not have promoter activity in the genome, but either behave as promoters in the context of our system or are background artifacts that might be generated during FACSsorting of large numbers of cells. It is likely that our method is biased towards selecting for stronger promoters, and that the presence of a suitable enhancer in our retroviral vector would improve the chances of selecting for weaker promoters.
On analyzing sequences from GFP+ high clones, we see approximately 50-fold enrichment for sequences that align to the 500-bp region upstream of the transcription start site of known genes, compared with what we would expect from random genomic sequences. This observation by itself provides convincing evidence that our approach enriches for promoters. For most known genes (Refgenes), links from the human genome browser at UCSC (Kent et al. 2002) depict promoters as being the 1-kb regions upstream of annotated transcription start sites. Our method narrows down these regions to defined 300–600-bp segments. Analysis of human/mouse alignments as well as BLAST analysis reveals sequences that are conserved in other species, such as mouse, rat, rabbit, and dog. In several instances, cross-species comparison identifies a 50–100-bp conserved segment that is likely to be the core sequence of the promoter.
A large number of analyzed sequences contain repeats such as LINES, SINES, and LTRs that comprise more than 40% of the human genome (Smit 1999). Although repeats are often viewed as selfish DNA, they contain internal RNA polymerase II and III promoters that may contribute to gene expression (Smit 1996). In addition to retrovirus-like elements, there is growing evidence that SINESand LINEScan contribute to the expression of some genes (Smit et al. 1999). B2 SINE elements found in the mouse and human genomes can provide mobile RNA polymerase II promoters (Ferrigno et al. 2001), whereas LINE/LI elements may have an antisense promoter that can drive the expression of adjacent cellular genes (Speek 2001). The data set of sequences generated from our functional screen will be useful for studying the types of repeat elements that might influence the transcription of genes nearby.
Because the method significantly enriches for promoter regions, it could be used on a more extensive scale to create databases of functional promoters in various species. It could also be useful for characterizing the DNA structure of promoters, by providing novel sequences in which to search for common motifs. Analysis of sequence data from our promoter-trapping scheme aids in the identification or confirmation of promoters, short first exons, and new genes in the genome. It also provides clues on alternative promoters that might mediate differential or tissue-specific gene expression. We are currently modifying the retroviral vector system to test whether this approach can be extended to identify other cis-regulatory sequences, such as enhancers and insulators, on a genome-wide scale. The generation of databases of regulatory sequences will be a useful resource for understanding the complexities of gene regulation.
METHODS
Construction of Suitable Retroviral Vectors
We constructed retroviral vector pSKF-6 (Fig. 1A) derived from vector pSIN (Moloney murine leukemia virus-based) obtained from Dr. Gary Nolan, Stanford University. The U3 region in the 5′ LTR, which contains transcriptional regulatory signals and the TATA box, is replaced by the CMV promoter to produce a higher titer of virus. The 3′ LTR has a deletion of enhancer sequences and part of the TATA box in the U3 region. We inserted the gene encoding humanized green fluorescent protein (huGFP) in a 3′ to 5′ orientation relative to the viral LTRs. This ensures that the expression of huGFP is driven only by DNA that is subcloned into the multiple cloning site (MCS) and not by possible residual promoter activity from the defective viral LTR. The huGFP is a 722-bp enhanced codon-substituted humanized form of GFP that is 99% identical to EGFP (Clontech). Bovine growth hormone unidirectional polyadenylation signals downstream of GFP direct proper 3′ end processing of the mRNA.
Construction of Control Vectors and a Genomic DNA Library
We subcloned a strong constitutive phosphoglycerate kinase (PGK) promoter, a moderately strong mouse β-globin promoter, and a yeast his3-ded1 region that has no promoter activity, into the MCSof pSKF-6. For library construction, we isolated 300–600-bp fragments of RsaI-digested total human genomic DNA (CEPH/Utah 1331-02 from Coriell Institute) from a 1% agarose gel. We inserted the gel-extracted DNA into the BstXI sites in pSKF-6 by using BstXI adapters (Invitrogen), and transformed the ligation mix into Max Efficiency DH10B competent cells (Invitrogen). We prepared plasmid DNA with Qiafilter midi kits (QIAGEN).
Production of High-Titer Retrovirus and Infection of Target Cell Lines
We transfected test constructs and library DNA into Phoenix-Eco 293T packaging cells (Dr. Gary Nolan, Stanford University), seeding 3 × 105 cells/well in 1.6 mL complete DMEM (Invitrogen Life Technologies) in 6-well plates. Following overnight incubation at 37°C, we transfected the packaging cells with 1 μg retroviral plasmid DNA/well by using Effectene reagent (QIAGEN). The cells were maintained at 32°C for 24–48 h posttransfection, during which time retrovirus was produced at titers of 105–106 particles/mL of viral supernatant. Viral supernatants were harvested and filtered by using a 0.45-μm syringe filter. NIH 3T3 mouse fibroblasts and HeLa-Eco human epithelial cells that express the ecotropic receptor were used for infection with the retroviral supernatant. First, 1.5 × 105 NIH 3T3 or HeLa-Eco cells/well were seeded in 1.6 mL culture media in 6-well plates. Following overnight incubation, cells in each well were overlaid with 1.5 mL of diluted viral supernatant and 1.5μL polybrene (5 mg/mL). The plates were swirled gently and then incubated overnight at 32°C for 24 h. At 24 h postinfection, we changed the media and incubated the cells at 37°C. At 48–72 h postinfection, the cells were ready for sorting. The ratio of viral particles to number of cells was optimized to ensure a high percentage of single integration events. This ratio generally resulted in infection of approximately 30% of target cells.
Selection and Sequencing of Putative Promoter Regions
We sorted GFP+ target cells in the Vanford flow cytometer (Stanford FACS facility). Dead cells were excluded using propidium iodide staining. We used FlowJo software (Treestar) for FACS data analysis. For experiments where a second round of FACS was performed, half the sorted cells from the first round were put back into the appropriate culture media for 96 h and then sorted a second time. Genomic DNA was extracted from sorted cells by using the Qiamp DNA Blood Mini Kit (QIAGEN).
Putative promoter-containing clones were recovered by genomic PCR in which we used Sin9F and R primers designed to the region flanking the BstXI sites in vector pSKF-6. Sin9F: 5′-acgcaagcttCAGTCTAGAGTCGGGCAGAT-3′ and Sin9R: 5′-catactcgagCCTATAGGTGGGGTCTTTCA-3′. We subcloned the PCR products into the pBlue-TOPO vector (Invitrogen) and sequenced the inserts by using the T7 primer (5′-TAATACGACTCACTATAGGG-3′). Sequencing was done at the Stanford Human Genome Center on ABI 377 sequencers with Big Dye Terminator chemistry (ABI).
Sequence Analyses
We aligned each sequence to the August 2001 assembly of the human genome (hg8; http://genome.ucsc.edu/downloads.html) by using the BLAT program (Kent 2002). For each sequence with high-quality alignment, we queried the annotation database to determine whether there are known genes, predicted genes, CpG islands, and mRNAs around the genomic region to which the sequence aligns (http://genome.ucsc.edu/goldenPath/06aug2001/database/). For each downloaded file, we created a relational database table, and populated it with information extracted from the file. A brief description of the annotation databases and the number of records in each table are shown in Supplemental Table 6.
For analyzing the fraction of sequences that are conserved between human and mouse, we downloaded the human/mouse alignment from http://genome.ucsc.edu/goldenpath/28jun2002/vsMm2/axtbest/. The “axtbest” alignments are filtered so that only the best alignment of any given region is kept. We checked whether the region of the human genome to which our sequences mapped was aligned to the mouse genome. To calculate the fraction within the aligned region, we divided the number of nucleotides that are in conserved regions by the total number of nucleotides that can be aligned to the human genome.
We searched sequences for the presence of a TATA-box (TA[T/A][T/A][T/A][T/A]) element. We calculated the expected number of clones in which the TATA-box element would appear by chance, based on the nucleotide frequencies of the clone sequences. The X2 test was used to determine whether the observed frequencies of the TATA-box element were significantly different (P < 0.05) from the expected.
Validation of Putative Promoter-Containing Clones
We tested a subset of sequenced clones individually for promoter activity in transient transfection reporter assays. Most clones were tested by using the Dual-Luciferase Reporter Assay system (Promega). Fragments containing the putative promoter regions were amplified by PCR from the plasmids in which they were sequenced by using Sin9 primers. These primers had HindIII and XhoI restriction sites incorporated (underlined in the sequences described above) for directional subcloning of the PCR products into the MCSof pGL3-Enhancer (Promega), a promoterless firefly luciferase reporter vector. We subcloned the mouse β-globin promoter and the yeast his3-ded1 region into the MCSof pGL3-E to serve as positive and negative controls.
Seventy thousand 293 or HeLa-Eco cells were seeded in 350 μL Complete DMEM in each of the wells of a 24-well plate. Following overnight incubation at 37°C, we used the Effectene reagent to transfect cells with 400 ng/well of firefly luciferase plasmid DNA containing the fragments to be tested. To normalize for transfection efficiency, the cells were cotransfected with 40 ng/well of Renilla luciferase control plasmid pRL-TK (Promega). Forty-eight h later the cells were lysed, and the lysates were assayed for firefly and Renilla luciferase activity in a Wallac 1420 plate luminometer (PerkinElmer). Plasmid pGL3-E+his3-ded was used as a reference standard where the average ratio of firefly luciferase counts to Renilla luciferase counts was set to 1. A putative promoter-containing clone was considered to be positive for luciferase if its activity threshold was greater than three times that of pGL3-E+his3-ded.
We tested a small number of clones by using the Galacto-Light Plus system (Applied Biosystems), a chemiluminescent reporter gene assay for the detection of β-galactosidase. As described earlier, we subcloned PCR fragments into pBlue-TOPO, a promoterless β-galactosidase reporter vector. We used a portion of the plasmid DNAs that were used as sequencing templates, for transfections into 293 cells following the protocol described above. We lysed the cells 48 h later and assayed the lysates for β-galactosidase activity. A putative promoter-containing clone was considered to be positive for β-galactosidase if its activity threshold was greater than that of pBlue-TOPO. If a clone tested negative for β-galactosidase activity, we subcloned it into pGL3-E and tested for luciferase activity, as the luciferase assay has about threefold greater sensitivity. We performed all reporter gene assays in duplicate or triplicate.
Notes
[4] Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.529803.
[5] Present address: Pharmacogenomics, Bristol-Myers Squibb Pharmaceutical Research Institute, Princeton NJ 08543, USA.
[6] Corresponding author. E-MAIL [email protected]; FAX (650) 725-9689.
We thank Gary Nolan and his laboratory for providing us with the SIN retroviral vector, packaging cells, and advice on using the retroviral system; the Stanford FACS facility, the Sequencing Team of the Stanford Human Genome Center, Jun Li, and Gregory Ford for helpful advice, and Amit Indap for bioinformatics assistance. S.K.F. was supported by a Stanford University School of Medicine Dean′s Postdoctoral Fellowship award for a portion of this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- ↵Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410.
- ↵Antequera, F. and Bird, A. 1999. CpG islands as genomic footprints of promoters that are associated with replication origins. Curr. Biol. 9: 661-667.
- ↵Ayoubi, T. and Van de Ven, W. 1996. Regulation of gene expression by alternative promoters. FASEB J. 10: 453-460.
- ↵Bai, C., Connolly, B., Metzker, M.L., Hilliard, C.A., Liu, X., Sandig, V., Soderman, A., Galloway, S.M., Liu, Q., Austin, C.P., et al. 2000. Overexpression of M68/DcR3 in human gastrointestinal tract tumors independent of gene amplification and its location in a four-gene cluster. Proc. Natl. Acad. Sci. 97: 1230-1235.
- ↵Borges, K. and Dingledine, R. 2001. Functional organization of the GluR1 glutamate receptor promoter. J. Biol. Chem. 276: 25929-25938.
- ↵Cross, C. and Bird, A.P. 1995. CpG islands and genes. Curr. Opin. Genet. Dev. 5: 309-314.
- ↵Edelman, G.M., Meech, R., Owens, G.C., and Jones, F.S. 2000. Synthetic promoter elements obtained by nucleotide sequence variation and selection for activity. Proc. Natl. Acad. Sci. 97: 3038-3043.
- ↵Ferrigno, O., Virolle, T., Djabari, Z., Ortonne, J., White, R.J., and Aberdam, D. 2001. Transposable B2 SINE elements can provide mobile RNA polymerase II promoters. Nat. Genet. 28: 77-81.
- ↵Fickett, J.W. and Hatzigeorgiou, A.G. 1997. Eukaryotic promoter recognition. Genome Res. 7: 861-878.
- ↵Friedrich, G. and Soriano, P. 1991. Promoter traps in embryonic stem cells: A genetic screen to identify and mutate developmental genes in mice. Genes & Dev. 5: 1513-1523.
- ↵Hardison, R.C. 2000. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 16: 369-372.
- ↵International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921.
- ↵International Mouse Genome Sequencing Consortium. 2002. Initial sequencing and analysis of the mouse genome. Nature 420: 520-562.
- ↵Jonsson, J., Wu, Q., Nilsson, K., and Phillips, R.A. 1996. Use of a promoter-trap retrovirus to identify and isolate genes involved in differentiation of a myeloid progenitor cell line in vitro. Blood 87: 1771-1779.
- ↵Kent, W.J. 2002. BLAT—The BLAST-like alignment tool. Genome Res. 12: 656-664.
- ↵Kent, W.J., Sugnet, C.W., Furey, T.S., Roskin, K.M., Pringle, T.H., Zahler, A.M., and Haussler, D. 2002. Human Genome Browser at UCSC. Genome Res. 12: 996-1006.
- ↵Lakso, M., Sauer, B., Mosigner, B., Lee, E.J., Manning, R.W., Yu, S.H., Mulder, K.L., and Westphal, H. 1992. Targeted oncogene activation by site-specific recombination in transgenic mice. Proc. Natl. Acad. Sci. 89: 6232-6236.
- ↵Medico, E., Gambarotta, G., Gentile, A., Comoglio, P.M., and Soriano, P. 2001. A gene trap vector system for identifying transcriptionally responsive genes. Nat. Biotech. 19: 579-582.
- ↵Myers, R.M., Tilly, K., and Maniatis, T. 1986. Fine structure genetic analysis of a β-globin promoter. Science 232: 613-618.
- ↵Ohler, U. and Niemann, H. 2001. Identification and analysis of eukaryotic promoters: Recent computational approaches. Trends Genet. 17: 56-60.
- ↵Pennacchio, L.A. and Rubin, E.M. 2001. Genomic strategies to identify mammalian regulatory sequences. Nat. Genet. Rev. 2: 100-109.
- ↵Praz, V., Perier, R., Bonnard, C., and Bucher, P. 2002. The Eukaryotic Promoter Database, EPD: New entry types and links to gene expression data. Nucleic Acids Res. 30: 322-324.
- ↵Pruitt, K.D. and Maglott, D.R. 2001. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 29: 137-140.
- ↵Roth, F.P., Hughes, J.D., Estep, P.W., and Church, G.M. 1998. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat. Biotech. 16: 939-945.
- ↵Scherf, M., Klingenhoff, A., and Werner, T. 2000. Highly specific localization of promoter regions in large genomic sequences by PromoterInspector: A novel context analysis approach. J. Mol. Biol. 297: 599-606.
- ↵Schroder, A., Shinn, P., Chen, H., Berry, C., Ecker, J., and Bushman, F. 2002. HIV-1 integration in human genome favors active genes and local hotspots. Cell 110: 521-529.
- ↵Smit, A.F.A. 1996. The origin of interspersed repeats in the human genome. Curr. Opin. Genet. Dev. 6: 743-748.
- ↵Smit, A.F.A. 1999. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr. Opin. Genet. Dev. 9: 657-663.
- ↵Speek, M. 2001. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Mol. Cell. Biol. 21: 1973-1985.
- ↵Von Melchner, H., Reddy, S., and Ruley, H.E. 1990. Isolation of cellular promoters by using a retrovirus promoter trap. Proc. Natl. Acad. Sci. 87: 3733-3737.
- ↵Wan, Y. and Nordeen, S.K. 2002. Identification of genes differentially regulated by glucocorticoids and progestins using a Cre/loxP-mediated retroviral-promoter-trapping strategy. J. Mol. Endocrinol. 28: 177-192.
- ↵Weidhaas, J.B., Angelichio, E.L., Fenner, S., and Coffin, J.M. 2000. Relationship between retroviral DNA integration and gene expression. J. Virol. 74: 8382-8389.
- ↵Whitelaw, E. and Martin, D.I.K. 2001. Retrotransposons as epigenetic mediators of phenotypic variation in mammals. Nat. Genet. 27: 361-365.
WEB SITE REFERENCES
- ↵http://www.ncbi.nlm.nih.gov/BLAST/; NCBI BLAST.
- ↵http://genome.ucsc.edu/downloads.html/; UCSC Genome Bioinformatics.
- ↵http://genome.ucsc.edu/goldenPath/06aug2001/database/; Golden Path build of draft human genome sequence.