
Quantitative Assessment of Transcriptome Differences Between Brain Territories
- Michel de Chaldée1,
- Marie-Claude Gaillard1,
- Nicolas Bizat2,
- Jean-Marie Buhler1,
- Olivier Manzoni3,
- Joël Bockaert3,
- Philippe Hantraye2,
- Emmanuel Brouillet2, and
- Jean-Marc Elalouf1,4
- 1 Service de Biochimie et de Génétique Moléculaire, Département de Biologie Joliot-Curie, Commissariat à l'Energie Atomique (CEA) Saclay, 91191 Gif-sur-Yvette Cedex, France
- 2 Unité de Recherche Associée CEA—Centre National de la Recherche Scientifique 2210, Service Hospitalier Frédéric Joliot, Département de Recherche Médicale, Orsay, France
Abstract
Transcriptome analysis of mammalian brain structures is a potentially powerful approach in addressing the diversity of cerebral functions. Here, we used a microassay for serial analysis of gene expression (SAGE) to generate quantitative mRNA expression profiles of normal adult mouse striatum, nucleus accumbens, and somatosensory cortex. Comparison of these profiles revealed 135 transcripts heterogeneously distributed in the brain. Among them, a majority (78), although matching a registered sequence, are novel regional markers. To improve the anatomical resolution of our analysis, we performed in situ hybridization and observed unique expression patterns in discrete brain regions for a number of candidates. We assessed the distribution of the new markers in peripheral tissues using quantitative RT–PCR, Northern hybridization, and published SAGE data. In most cases, expression was higher in the brain than in peripheral tissues. Because the markers were selected according to their expression level, without reference to prior knowledge, our studies provide an unbiased, comprehensive molecular signature for various mammalian brain structures that can be used to investigate their plasticity under a variety of circumstances.
The mammalian brain is subdivided into distinct territories defined by anatomical and functional characteristics. A unique gene expression pattern in each brain region is expected to convey a large part of its functional features. Great progress in the understanding of cerebral functions will therefore be achieved by deciphering brain transcriptomes. Comparison of expression patterns between different brain structures was first undertaken on a modest number of genes by use of subtractive hybridization (Usui et al. 1994), and differential mRNA display (Nagano et al. 1998; Mizushima et al. 2000). In contrast, global approaches currently allow large-scale comparative analyses. So far, they have been used mainly to detect molecular alterations in neurodegenerative (Luthi-Carter et al. 2000; Napolitano et al. 2002) and neuropsychiatric (Mirnics et al. 2000; Sun et al. 2001) conditions. Curiously, relatively few studies have addressed the regional diversity of gene expression within the normal adult brain (Sandberg et al. 2000; Pavlidis and Noble 2001; Siu et al. 2001; Zirlinger et al. 2001; Bonaventure et al. 2002), and many regions remain to be explored.
In the present work, we used a microadaptation (Virlon et al. 1999) of the serial analysis of gene expression (SAGE) method (Velculescu et al. 1995) to determine mRNA expression profiles in adult mouse whole brain, striatum, nucleus accumbens, and somatosensory cortex. SAGE provides permanent, potentially exhaustive and quantitative data, and makes possible the discovery of new genes (Velculescu et al. 1995). Among a total of 35,732 distinct transcripts, 135 are differentially expressed from one brain region to another. We further analyzed a sample of these transcripts, and studied their distribution within the brain and in peripheral tissues using a combination of in situ hybridization, real-time quantitative RT–PCR, and Northern hybridization. The majority of the differentially expressed transcripts are novel regional markers and provide molecular tools to study the physiology of different brain structures.
RESULTS
Brain Transcriptomes
The SAGE method relies on the sequencing of 10-nucleotide tags isolated from a defined region of each polyadenylated transcript. Tag abundance provides a quantitative measure of mRNA level, and high throughput is achieved by concatenating several tags into a single DNA molecule. We generated 5 SAGE libraries, from which we sequenced 118,622 tags, 26,053 from the whole brain, 22,401 from the lateral striatum, 23,648 from the medial striatum, 21,960 from the somatosensory cortex, and 24,560 from the nucleus accumbens. We detected a total of 35,732 different tags, corresponding to an average number of 11,178 tags in each structure.
In all of the regions investigated and in the whole brain, we observed a high expression level of the mitochondrial genome, reflecting the energy demand of the neural tissue; 15.9% of the tags sequenced in the entire project are derived from mitochondrial transcripts. The 50 most prevalent tags of nuclear origin reach abundances ranging from 0.5% to 0.1% of the tags sequenced (Supplemental Table A, available online at www.genome.org). This list of tags corresponds to transcripts encoding both brain-specific and ubiquitous proteins. Overall, the identity of the most abundant transcripts agrees with previous data published for the mouse brain (Chrast et al. 2000). The regional marker showing the highest expression level, namely the striatum-specific phosphodiesterase 1B (Ca2+-calmodulin-dependent, 63 kD), is the 48th most abundant nuclear transcript in the project.
Region-Specific Transcripts
We identified transcripts specifically expressed in each brain region through a two-step procedure. First, we considered only the tags significantly more frequent in a given library than in the whole brain library (Monte-Carlo simulations; P < 0.05). Then, to further guarantee biological relevance and regional specificity, we selected the tags exhibiting a fivefold enrichment as compared with two libraries. According to these criteria, we detected 35 to 45 markers in each region investigated (Table 1). The total number of markers was 135, due to overlapping between libraries. Among the 125 markers matching registered cDNAs or ESTs, we considered that 78 are novel, as the literature provides no clue as to their abundance in the region in which we found them. Of these 78 novel markers, 48 have no demonstrated function in the brain.
Number of Markers Detected in Each Brain Region
As an illustration, the most abundant striatal markers are displayed in Table 2. Some have been described specifically as striatum enriched, for example, phosphodiesterase 1B, Ca2+-calmodulin-dependent, 63 kD (Polli and Kincaid 1994), proenkephalin (Tang et al. 1983), cyclic AMP-regulated phosphoprotein, 21 kD (Ehrlich and Greengard 1991), striatum-specific G-protein coupled receptor 88 (Mizushima et al. 2000), striatum-enriched protein tyrosine phosphatase, 61 kD (Sharma et al. 1995), Ras homolog enriched in striatum (Usui et al. 1994; Falk et al. 1999), preprotachykinin A (Goedert and Hunt 1987), dopamine and cyclic AMP-regulated phosphoprotein, 32 kD (Kurihara et al. 1988), regulator of G-protein signaling 9, isoform 2 (Rahman et al. 1999), adenylyl cyclase 5 (Mons and Cooper 1994), G-protein, γ 7 subunit (Watson et al. 1994), and dopamine receptor 2 (Bunzow et al. 1988). Expression in the striatum has also been reported for calcineurin catalytic subunit, α isoform (Polli et al. 1991), Kv channel-interacting protein 2 (An et al. 2000), focal adhesion kinase 2 (Xiong et al. 1998), synaptoporin (Ovtscharoff et al. 1993), hippocalcin (Grant et al. 1996), and Ca2+-independent receptor of α-latrotoxin (Krasnoperov et al. 1997). The ability to detect such known markers validates our selection procedure. In addition, comparison with SAGE data from mouse kidney and total embryo revealed that most of the markers are not significantly expressed in either tissue, suggesting that they might be brain restricted. A noticeable exception concerns dopamine and cyclic AMP-regulated phosphoprotein 32 kD, also present in the kidney (Meister et al. 1989). Strikingly, a number of striatum-enriched transcripts take part in information processing. They encode neuropeptides (proenkephalin, preprotachykinin A), receptors (G-protein coupled receptor 88, dopamine receptor 2, Ca2+-independent receptor of α-latrotoxin), or proteins involved in signal transduction (phosphodiesterase 1B Ca2+-calmodulin-dependent 63 kD, cyclic AMP-regulated phosphoprotein 21 kD, calcineurin, striatum-enriched protein tyrosine phosphatase 61 kD, Rap1 GTPase-activating protein, Ras homolog enriched in striatum, dopamine and cyclic AMP-regulated phosphoprotein 32 kD, regulator of G-protein signaling 9 isoform 2, adenylyl cyclase 5, G-protein γ 7 subunit). Transcripts specific to the other brain regions investigated are displayed in Supplemental Table B.
Most Abundant Striatal Markers
Expression Pattern of Novel Markers
We further analyzed the regional specificity of novel markers by in situ hybridization using radiolabeled oligonucleotide probes (Fig. 1) and real-time quantitative RT–PCR (Fig. 2). Hybridization patterns and RT–PCR results were consistent with the SAGE data for 26 of the 42 novel markers that we tested. Particularly good agreement was observed for 20 of them, shown in bold in Table 2 and Supplemental Table B. Incompleteness of the cDNA databases, resulting in tag misassignment, may account for some of the nonvalidated candidates. Expression levels of the confirmed markers ranged from high to moderate. For instance, we could validate the regional specificity of cortical marker CxM42, which we found only five times among the 21,960 tags sequenced in the somatosensory cortex library. In situ hybridization provided additional information on the distribution of the markers within the brain. In particular, it allowed intra-regional heterogeneity to be observed: CxM3 and CxM11 are expressed mostly in cortical layer VI, and CxM20 is absent from cortical layer V. Hybridization, especially to sagittal brain sections, also provided a means of exploring cerebral structures not investigated by SAGE. For instance, nucleus accumbens marker AcM7 is also a relevant marker of the cingulate cortex, and CxM20 is expressed in the hippocampus and the thalamus. Using a probe against CxM3, we observed an intense and specific staining in the hippocampus (CA1, part of CA3, and dentate gyrus) and the granular layer of the cerebellar cortex, in addition to the cortex and the thalamus.

Expression of new regional markers in the brain visualized by in situ hybridization. (A) A probe against proenkephalin messenger (left) was used as a positive control. A mutated proenkephalin probe with preserved thermodynamic characteristics (right) provided a negative control. (B–H) In situ hybridizations to coronal (left) and sagittal (right) sections are shown for StM3 (B), StM6 (C), StM23 (D), AcM7 (E), CxM3 (F), CxM11 (G), and CxM20 (H). (Ac) Nucleus accumbens; (Cb) cerebellum; (Cg) cingulate cortex; (Cx) cortex; (Hp) hippocampus; (Ot) olfactory tubercles; (St) striatum; (Th) thalamus.
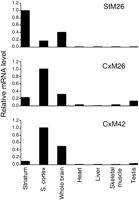
Expression of StM26, CxM26, and CxM42 within the brain and in peripheral tissues assessed using real-time quantitative RT–PCR. mRNA levels were calculated relative to the tissue sample in which expression is maximum (i.e., the striatum for StM26, and the somatosensory cortex for CxM26 and CxM42). For each tissue, the same cDNA sample was used to study the expression of the different markers. (S. cortex) Somatosensory cortex.
Most of the validated markers match either recently identified mRNAs without known function (striatal markers StM11, StM23, StM26, StM39, and markers AcM8, CxM11, CxM20, CxM27, CxM34, CxM42, CxM45), or ESTs (StM3, StM6, StM20, CxM3, CxM26). Mu-crystallin (AcM7) was characterized initially in marsupials as a major lens protein (Kim et al. 1992). It has been identified as an NADP-regulated thyroid hormone-binding protein in the kidney (Vié et al. 1997), but its role in the brain remains unknown. Chemokine CCL27 (CxM33) has two splice variants. One is chiefly expressed in the skin and codes for a secreted protein that may act as a chemoattractant for cutaneous T-cells. The other is expressed mostly in testis and brain. It lacks a signal peptide, and its function has not been determined yet (Baird et al. 1999).
We then studied the tissue distribution of the validated markers. Data were available from the literature for a number of them (Isobe et al. 1991; Yoshida et al. 1995; Lee et al. 2000; Yayoshi-Yamamoto et al. 2000; Perera et al. 2001; Sugita et al. 2002). For the remaining markers, we used Northern hybridization (Fig. 3) and real-time quantitative RT–PCR on total brain and peripheral tissues. We systematically performed Northern hybridizations for ESTs (StM3, StM6, StM20, CxM3, CxM26) to gain information on the size of the transcripts and any splice variants. Although the markers were selected for their heterogeneous distribution within the brain, we could detect a signal in total brain mRNA extracts in all cases. Except for StM3, which proved to be more abundant in the skeletal muscle, expression was stronger in the brain than in peripheral tissues.

Tissue distribution of novel markers assessed using Northern hybridization. The size of the hybridizing fragments is shown on the left.
DISCUSSION
Using a microadaptation of the SAGE method, we found an average of 40 region-specific transcripts in each of 4 territories of the mouse brain. We set the depth of our analysis to detect the most abundant transcripts, and we selected and sorted the regional markers on the basis of their expression level. Each marker fulfills two criteria. First, its frequency is higher in one brain region than in the whole brain. Different tests have been developed to perform such comparisons, but they have been shown to give equivalent results (Ruijter et al. 2002). Second, each marker is five times more frequent in one library than in any two others. This enrichment criterion is essential to discard ubiquitous transcripts. Because of their regional specificity and relative abundance, the markers that we found may be functionally significant for the regions investigated. Furthermore, their combination can be considered as a molecular signature of a given structure, both allowing its identification and providing a standard for future comparisons. However, because tag identification remains a critical step in the SAGE method, experimental validations are recommended before planning functional studies. Here, we provided confirmatory experiments for 20 novel markers, which are now ready to undergo further characterization.
Because our selection procedure does not rely on previous knowledge, it is not surprising that many markers that we detected were so far unknown as such. The proportion of new markers is highest in the nucleus accumbens and lowest in the lateral striatum, reflecting extensive study of this structure. The lateral and medial parts of the striatum share 17 markers in common, in agreement with the lack of a clear functional specialization of striatal subregions. With only six markers shared with the striatum, the nucleus accumbens displays a more distinct molecular profile. We observed a reliable difference between both parts of the striatum for the ventromedial marker AcM7, which is expressed in the nucleus accumbens and the medial striatum, but not significantly in the lateral striatum. This expression pattern is reminiscent of calbindin distribution (Gerfen et al. 1987). Known markers that we detected in the somatosensory cortex, such as oxysterol-binding protein-related protein 1 (Laitinen et al. 1999) and olfactomedin 1 (Nagano et al. 1998), are also expressed in other parts of the cortex. Likewise, in situ hybridization experiments did not reveal transcripts restricted to the somatosensory area. Future comparison to other cortical regions will be needed to highlight such area-specific markers. For this purpose, we could rely on the classical divisions of the brain, and generate libraries, for instance, from the motor and cingulate cortices. This approach is supported by recent microarray analyses of the amygdala (Zirlinger et al. 2001) and the hippocampus (Zhao et al. 2001), which provided a molecular confirmation for the anatomical boundaries between the subregions of each structure. The alternative is to divide the brain into cubes, and build an unbiased expression map from the expression profile of each cube. The relevance of this strategy was demonstrated recently (Brown et al. 2002).
Transcriptome analysis allowed us to identify a large number of mRNAs expressed predominantly in different brain regions. Some of them display a very specific expression profile, and may therefore serve as markers for a variety of applications. For instance, the expression level of StM3 is comparable with that of proenkephalin, but unlike proenkephalin, it is absent from the nucleus accumbens. Such highly discriminative markers are most interesting for cell typing or purification. Novel markers may also shed new light on the functions of the cerebral regions in which they are expressed. In particular, screening their expression levels under various pathophysiological conditions should allow the characterization of a unique set of constitutively expressed and regulated genes, and thus provide appropriate diagnostic or prognostic candidates.
METHODS
Tissue Samples
Experiments were performed on 8–12-week-old male C57BL/6 mice (Iffa Credo). After anesthesia (sodium pentobarbital, 140 μg/g of body weight), the animals were decapitated and the brain was removed. The somatosensory cortex and the striatum were scalpel dissected from 1-mm brain sections cut in a coronal matrix (Pelco International). In each hemisphere, the striatum was divided into a lateral and a medial part. A mouse brain atlas (Franklin and Paxinos 1997) was used to delimit the somatosensory area of the cortex. The nucleus accumbens was punched from 1-mm parasagittal brain sections using 1.1–1.2-mm internal diameter glass capillaries (Propper Manufacturing Co. Inc.).
SAGE Libraries
Approximately 15 mg of each structure, sampled from three to seven animals, were homogenized in a 1-mL Dounce containing 600 μL Lysis and Binding Buffer from Dynabeads mRNA DIRECT kit (Dynal Biotech) supplemented with 60 μg glycogen. For the whole brain, an amount of lysate corresponding to 60 mg of fresh tissue was used. Poly(A) mRNAs were purified directly through hybridization to oligo(dT)25 covalently bound to magnetic beads. SAGE libraries were then constructed as described by Virlon et al. (1999), using Sau3A I as anchoring enzyme. Sequencing reactions were carried out on DNA minipreps, either in our laboratory or by Genome Express, using automated DNA sequencers.
Data Analysis
Sequence files were all checked to control for base calling, then analyzed using SAGE 2000 software. Tags corresponding to linker sequences were discarded, and duplicate ditags were counted only once (Velculescu et al. 1995). Region specificity was conferred by a two-step procedure. In each library, the tags that were significantly more frequent than in the whole-brain library were selected (P < 0.05 using Monte-Carlo simulations). Then, only the tags that were at least five times more frequent than in any other two libraries were retained (when a tag was missing from a library, the default tag count was set to one). Tag identification was performed using SAGEmap resource (http://www.ncbi.nlm.nih.gov/SAGE/), and BLASTN algorithm (http://www.ncbi.nlm.nih.gov/BLAST/). Multiple tags matching the same gene were mentioned (e.g., StM5), unless they discriminate between proven splice variants (e.g., CxM43). Non-neuronal references were provided by SAGE libraries generated from mouse 6.5 days post coitum whole embryos (Zakin et al. 2000) (24,271 tags) and adult mouse kidney (27,003 tags) (Virlon et al. 1999).
In Situ Hybridization
Brain fixation was performed by intraventricular perfusion of 15 mL of a 4% paraformaldehyde solution. After removal, brains were fixed for 3 h more at 4°C, and cryoprotection was achieved after two nights in a 20% sucrose solution at 4°C. Brains were frozen in isopentane at -60°C, and 20-μm sections were cut in a cryostat and mounted onto SuperFrost Plus slides (Menzel-Gläser). After thawing, sections were postfixed for 5 min in a 4% paraformaldehyde solution at 4°C, then rinsed twice in 1× PBS for 15 min at 4°C. Dehydration was achieved through a series of ethanol baths of increasing concentration, followed by air drying. Sections were then overlaid with the hybridization mix, containing 50% Rapid-hyb buffer (Amersham Biosciences), 40% formamide, 0.5 μg/μL poly(A), 100 mM DTT, and 100 fmole/μL of 3′-end [35S]dATP antisense oligonucleotide probe. Probe size ranged from 32 to 37 nucleotides. Thermoresistant parafilm or HybriWell hybridization chambers (Grace Bio-Labs) were used to prevent evaporation. Hybridization was carried out overnight at 48°C in a humid chamber or at 53°C in an SM30 device (Grant Boekel). Slides were submitted to conventional β-autoradiography. The list of probes used for in situ and Northern hybridizations is available in Supplemental Table C.
Real-Time Quantitative RT–PCR
Total RNAs were extracted from mouse brain structures and mouse tissues using either a standard guanidium thiocyanate-based method or Trizol LS reagent (Invitrogen). Real-time quantitative RT–PCR was performed in triplicate using 0.5% of random-primed cDNAs generated from 200 ng of total RNA. Targets were amplified in a 25-μL reaction volume containing 400 nM of sense and antisense primers (see Suppl. Table C), 0.1× SYBR Green I (BMA), and Platinum quantitative PCR SuperMix-UDG (Invitrogen). PCR, carried out in an iCycler (BioRad), consisted of a uracil DNA glycosylase activation step (55°C, 5 min), followed by a denaturation step (95°C, 5 min), and 40 amplification cycles (95°C, 20 sec; 60°C, 1 min). The amplification rate of each target (range, 1.91–2.00) was evaluated from the cycle threshold (Ct) numbers obtained for serial cDNA dilutions. For a given target, differences among tissue samples were calculated using the amplification rate and the Ct difference, then expressed as relative mRNA level. RT–PCR conditions were considered appropriate only if a large difference (≥7 Ct, corresponding to ≥100-fold difference in initial DNA amounts) was obtained for reactions carried out in the absence and presence of reverse transcriptase, and if agarose gel electrophoresis of the PCR product revealed a single band of the expected size.
Northern Blotting
Five micrograms of poly(A)+ RNAs purified using oligo(dT)25 paramagnetic beads (Dynal Biotech) were denaturated in a glyoxal-dimethylsulfoxide solution, electrophoresed on a 1% agarose gel, then transferred onto a Hybond N+ nylon membrane (Amersham Biosciences). Final concentrations of the hybridization buffer were 5× SSC, 0.2% SDS, 5× Denhardt's, 5 mM EDTA, 50 mM sodium phosphate buffer (pH 7.4), 10% Dextran, 100 μg/mL poly(A), 200 μg/mL salmon testis DNA, and 4.106 cpm/mL (250 fmole/mL) of 3′-end [32P]dATP antisense oligonucleotide probe. Hybridization was carried out overnight at 50°C. The membrane was washed at 60°C down to 0.5× SSC, 0.05% SDS prior to autoradiography.
Acknowledgments
We thank Ms. E. Billon and Mrs. V. Berthonaud for DNA sequencing, Mr. L. Wiart and Dr. M.-H. Montané for their contribution in generating two libraries, and Mrs. F. Condé for histological analysis. We also wish to thank Dr. A. Sentenac for his constant enthusiasm about this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
[Supplemental material is available online at www.genome.org. SAGE data will be available at GEO (http://www.ncbi.nlm.nih.gov/geo/) under accession nos. GSM4871, GSM4873, GSM4874, GSM4875 and GSM4876, which correspond to the whole brain, lateral striatum, medial striatum, nucleus accumbens and somatosensory cortex raw SAGE data.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1173403.
-
↵3 Present address: CNRS Unité Propre de Recherche (UPR) 9023, Montpellier, France.
-
↵4 Corresponding author. E-MAIL elalouf{at}dsvidf.cea.fr; FAX 33-1-69-08-47-12.
-
- Accepted March 26, 2003.
- Received January 15, 2003.
- Cold Spring Harbor Laboratory Press








