
Abstract
Manual curation has long been held to be the “gold standard” for functional annotation of DNA sequence. Our experience with the annotation of more than 20,000 full-length cDNA sequences revealed problems with this approach, including inaccurate and inconsistent assignment of gene names, as well as many good assignments that were difficult to reproduce using only computational methods. For the FANTOM2 annotation of more than 60,000 cDNA clones, we developed a number of methods and tools to circumvent some of these problems, including an automated annotation pipeline that provides high-quality preliminary annotation for each sequence by introducing an “uninformative filter” that eliminates uninformative annotations, controlled vocabularies to accurately reflect both the functional assignments and the evidence supporting them, and a highly refined, Web-based manual annotation tool that allows users to view a wide array of sequence analyses and to assign gene names and putative functions using a consistent nomenclature. The ultimate utility of our approach is reflected in the low rate of reassignment of automated assignments by manual curation. Based on these results, we propose a new standard for large-scale annotation, in which the initial automated annotations are manually investigated and then computational methods are iteratively modified and improved based on the results of manual curation.
The RIKEN Mouse Gene Encyclopedia Project aims to identify and sequence every transcript encoded by the mouse genome. The usefulness of this resource, and the analysis of the set of transcripts, are clearly dependent upon providing the most informative possible functional annotation for each sequence and making these data readily accessible to the scientific community. As eukaryotic genomes contain genes and gene families with diverse functions beyond the expertise of any one individual, it has become common to bring together a group of experts for an “annotation jamboree” (Adams et al. 2000). The FANTOM1 (Functional Annotation of Mouse) meeting held to functionally annotate the first 21,076 RIKEN mouse cDNA clones rapidly came to grips with the logistical problems of large-scale annotation, and devised computational interfaces to expedite human curation. As we began to prepare for the much larger task of annotating the 60,770-clone FANTOM2 cDNA set, which was to include reannotation of the original FANTOM1 clones to provide the best and most current annotation, we realized that a preliminary automated annotation using a well defined protocol and a controlled vocabulary would greatly expedite the task. To that end, we developed an automated cDNA annotation pipeline, which determined putative initial name, symbol, and synonyms of cDNA clones, and provided relevant evidence and annotation status, with the goal of creating and optimizing a protocol that could be used for automated reannotation of clones in the future. Our objective was the development of an automated pipeline that could serve as an alternative to manual curation, and we sought to supply computational assignments that closely matched those arising from expert human annotation. However, this is not a simple task, because best-hit sequences found by the similarity search program are often not the most appropriate sources of annotation.
Our second realization was that if human curation is necessary, an annotation jamboree is not a practical approach to annotate a very large set of cDNAs. With a well designed Web-based annotation interface, expert curation can be undertaken at a more leisurely and considered pace, and can be reviewed and revisited over time by individual experts. We therefore carried out the first large-scale on-line annotation jamboree, FANTOM2 MATRICS (Mouse Annotation Teleconference for RIKEN cDNA Sequences). During the MATRICS process, the automated annotation allowed individual expert curators to either choose sets of transcripts reflecting their interest and expertise or be assigned a randomly chosen set of clones. They were then asked to accept an automated annotation or choose their own alternative based on information provided in a carefully designed interface. Further, they assigned other types of annotation to each cDNA in the on-line jamboree, including CDS (coding sequence) regions in the mRNA, gene ontology terms (The Gene Ontology Consortium 2001) for its protein product, status of the cDNA (e.g., full-length, chimera, immature), and notes by experts. Table 1 shows details of annotation determined in FANTOM2 MATRICS. The interface must be designed to make it possible to finish annotation of 60,770 cDNA clones in two months with about 100 curators participating from various countries.
Annotation Fields in the FANTOM2 Database, Their Corresponding Title in the CAS, and a Description of the Information They Carry
Qualifier | Displayed title | Description |
|---|---|---|
| gene_name | Curated gene name | FANTOM gene name, which shows the function of protein products or the status of mRNA and is described in a sentence |
| gene_symbol | Curated gene symbol | gene symbol, if available, which shows the function in the several letters |
| synonym | Synonym | synonyms, if available |
| match_status1 | Match Status 1 | flags indicating whether a sequence represents a complete or partial gene, or a problem with the clone exists |
| match_status2 | Match Status 2 | flags for possible splice variants, and antisense transcripts |
| match_status3 | Match Status 3 | flags for possible frame shifts, unspliced introns, and chimeric sequences |
| cds_start | CDS start | CDS start position within the clone, in bp |
| cds_stop | CDS stop | CDS stop position within the clone, in bp |
| cds_status1 | CDS status 1 | flags for clones which may represent 5′UTR, 3′UTR, non-coding RNAs, or other artifacts |
| cds_status2 | CDS status 2 | flags for possible reverse complemented, 5′-truncated, or 3′-truncated sequences |
| cds_status3 | CDS status 3 | flags for possible frame shifts, immature transcripts or unexpected stop codon |
| cds_note | CDS Note | curator's note about the CDS |
| utr | UTR | curator's comment about UTR |
| expression_note | Expression Note | curator's comment about expression profile |
| antisense_hit | Antisense Hit | FANTOM gene names derived from a matched entry on the reverse complement strand |
|
note |
Note |
curator's general comments |
In this paper, we describe the rationale and design of the automated annotation pipeline and the MATRICS Web interface (CAS; cDNA Annotation System). We review the value of human curation and the extent to which the ultimate goal of fully computational annotation can be achieved.
RESULTS AND DISCUSSION
An Overview of the FANTOM2 Annotation Pipeline
The objective in designing an automated annotation pipeline is to provide each cDNA sequence with the most informative possible gene name with the greatest possible indication of function based on all of the available data; for example, nucleotide, protein, and homology data. One of the most obvious applications for assigning such a name is in cDNA microarrays, where one might generate lists of names of coregulated transcripts, and at a glance gain some idea of what function is encoded by each member of a cluster.
The basis for the annotation pipeline we developed is the use of a large number of precomputed analyses of the sequences, including prediction of potential coding sequences by ProCrest (CDS features), DNA and protein database searches, searches of a variety of motif databases, and UniGene and TIGR Gene Index clustering analysis. The pipeline filters these results to assign each cDNA sequence to one of 19 distinct categories using a sequential decision tree, which sorts the sequences based on classes with decreasing functional information content. Assignment to a particular category is reflected in the putative gene name assignment, which uses a predefined controlled vocabulary that indicates both the level of confidence of the assignment and the likelihood that the cDNA clone is complete. Figure 1 shows a decision tree of the automated annotation pipeline used in the FANTOM2 MATRICS precomputation.
The FANTOM2 annotation pipeline. White numbers in black boxes represent category numbers assigned to cDNA clones. Numbers attached to arrows indicate how many sequences passed through each stage when running the system using the 60,770 FANTOM2 sequences.
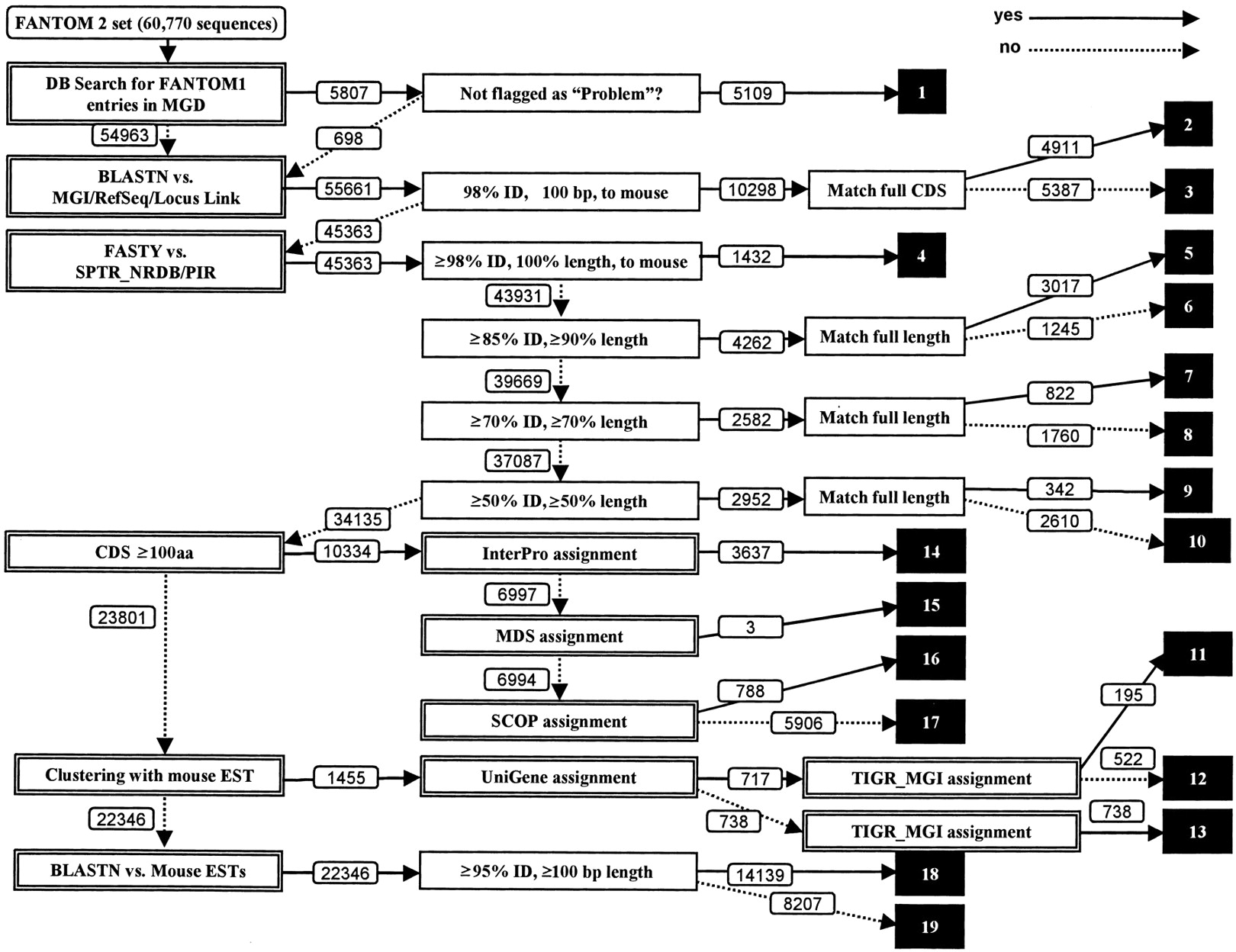
In the analysis of the FANTOM1 sequences, the Mouse Genome Informatics (MGI) group from The Jackson Laboratory analyzed sequences representing known mouse genes and linked these to the existing data in the MGI resources. They maintain and update information about the FANTOM1 sequences as part of MGI's ongoing mission of curating all gene-specific information about the mouse. Thus our annotation process should be consistent with the MGI assignment.
To begin the annotation process, all sequences are examined to determine whether an MGI gene name has been assigned without a “problem sequence” flag in the MGI database. If the query satisfies the condition, the sequence is placed in “category 1,” and the MGI annotation is assigned to the clone.
Step 2 in the annotation process involves direct similarity searches against the major DNA and protein databases using either BLASTN (DNA) or FASTY (protein). If there are significant DNA hits (≥98% identity, ≥100 bp length), the query sequence is assigned to “category 2” or “category 3” based on whether the sequence contains a predicted CDS region in the matched area. If there are no DNA hits, but significant protein hits, the query is placed into one of “category 4” through “category 10” based on the search similarity scores and fraction of the subject protein length matched: category 4, ≥98% identity, 100% length, mouse; category 5, ≥85% identity, 100% length; category 6, ≥85% identity, ≥90% length; category 7, ≥70% identity, 100% length; category 8, ≥70% identity, ≥70% length; category 9, ≥50% identity, 100% length; category 10, ≥50% identity, ≥50% length matches.
If informative DNA and protein hits are not found, the length of the predicted coding sequence (CDS) is examined. Here, we chose a minimum of 100 amino acids. This choice is empirically based upon the observation that CDS prediction below this level is unreliable, but careful human curation of predicted CDS in the range from 50–99 amino acids has led to identification of a number of additional short protein-coding transcripts; obviously the pipeline could be modified to include these. Sequences with CDS regions greater than 100 amino acids in length are passed to step 3 for motif assignment; otherwise, sequences are passed to step 4 for indirect, cluster-based assignments.
In step 3, predicted CDS regions are analyzed for InterPro, MDS, or SCOP motifs and the assignments are prioritized in this order; if motifs are found, the proteins are put into “category 14,” “category 15,” or “category 16,” respectively. If no motifs are found, the cDNA is assigned to “category 17” and the name “hypothetical protein” is assigned to the sequence.
In step 4, query DNA sequences are examined to determine whether they could be assigned putative functions based on UniGene or TIGR Gene Index clustering analysis performed using the FANTOM2 set and public sequences. This step is useful when the query sequence is truncated and only the 3′ portion, or less frequently the 5′ portion, of the full-length transcripts is represented. If the query sequence is assigned an informative name based on either clustering analysis, this annotation is used as the gene name for the sequence. Based on the clustering approach that assigns an informative name, sequences are assigned to “category 11,” “category 12,” or “category 13.”
The remainders of the query sequences are passed onto step 5 in the pipeline, where they are searched against public EST databases. Sequences that had significant hits (≥95% identity, ≥100 bp length) to previously sequenced ESTs are assigned as “unknown ESTs” and placed into “category 18.” The remaining sequences, which have failed classification in any of the previous steps, are placed into “category 19” and assigned the name “unclassifiable.”
Avoiding Uninformative Assignments
Our goal at each stage in the pipeline is to assign the most informative gene name possible. Unfortunately, the “best hit” based on sequence similarity searches does not always give the best functional annotation, because the process may identify genes with uninformative names such as “hypothetical protein.” Human curation often assigns a particular sequence a function from a hit which is nearly as significant as the best hit, but which has a more biologically relevant annotation. With this realization, we set out to obviate the need for human judgment by creating an annotation filter that would select the most useful possible name for each sequence.
By inspecting DNA and protein databases, we found that uninformative descriptions can generally be categorized into two groups. The first group consists of those that directly indicate that the function is unknown. In the SWISS-PROT + TrEMBL database, “HYPOTHETICAL 30 KDA PROTEIN” is an example of this category. The second group consists of names that are assigned from large-scale sequencing projects, such as “RIKEN 0610005K03 gene”. Many of these names consist of keywords (e.g., “RIKEN,” “gene,” or “hypothetical”) and variable words (e.g., “0610005K03”). Through a careful analysis and cataloging of such assignments, we constructed an “uninformative rule” filter based on a set of approximately 50 regular expressions; the list of expressions is shown in Supplementary Information 1, available online at www.genome.org. One advantage of this approach is that the list can evolve over time as human curators identify additional information-poor terms.
In our pipeline, uninformative names assigned in Steps 1 through 4 including MGI name assignment, DNA- and protein homology-based assignments, motif identification, and names based on clustering were ignored, and any available secondary assignments were assigned if they were deemed significant.
Output of Annotation Pipeline
Prior to human curation, we subjected all 60,770 FANTOM2 cDNA sequences to automated annotation using this pipeline. Figure 1 shows the number of sequences assigned a name in each category at each stage; the data are summarized in Table 2, along with a summary for the 33,409 representative sequences chosen after the sequences were clustered to collapse redundant sequences (The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I and II Team 2002).
The Number of Sequences Assigned to Each of Nineteen Categories Based on the Results of Our Automated Annotation Pipeline for the 60,770 FANTOM2 Sequences cDNA and the 33,409 Representative Sequences Determined to Be Unique Through Our Cluster Analysis
Category | FANTOM2 60,770 all sequences | FANTOM2 33,409 rep. sequences |
|---|---|---|
| 1. MGI assigned | 5,109 | 2,044 |
| 2. DNA hit (complete) | 4,911 | 2,354 |
| 3. DNA hit (partial) | 5,387 | 2,063 |
| 4. Protein hit (≥98% ID, 100% length, mouse) | 1,431 | 650 |
| 5. Protein hit (≥85% ID, ≥90% length, complete) | 3,017 | 1,351 |
| 6. Protein hit (≥85% ID, ≥90% length, partial) | 1,245 | 519 |
| 7. Protein hit (≥70% ID, ≥70% length, complete) | 822 | 409 |
| 8. Protein hit (≥70% ID, ≥70% length, partial) | 1,760 | 719 |
| 9. Protein hit (≥50% ID, ≥50% length, complete) | 342 | 153 |
| 10. Protein hit (≥50% ID, ≥50% length, partial) | 2,610 | 1,166 |
| 11. TIGR/UniGene clusters | 195 | 38 |
| 12. UniGene clusters | 522 | 147 |
| 13. TIGR clusters | 738 | 297 |
| 14. InterPro domain/motifs | 3,637 | 1,858 |
| 15. MDS domain/motifs | 3 | 2 |
| 16. SCOP domain/motifs | 788 | 351 |
| 17. hypothetical protein | 5,906 | 3,113 |
| 18. unknown EST | 14,139 | 8,689 |
|
19. unclassifiable |
8,207 |
7,486 |
Development of the FANTOM2 MATRICS Interface
During FANTOM1, we developed an interface that was used extensively and progressively refined during a two-week annotation jamboree (Quackenbush 2000). For FANTOM2, we decided that the most effective method for manual curation was to assemble a large number of specialist curators and allow them to review and refine the annotation remotely in a process referred to as MATRICS (Mouse Annotation Teleconference for RIKEN cDNA Sequences). To facilitate this process, we developed a Web-based annotation system CAS, the cDNA Annotation System.
In developing the annotation system, there were essential requirements. The first was providing mechanisms to review automated annotation and to reannotate them with our controlled vocabulary. Although we had great confidence in the automated annotation, we realized that there was still a need for additional human review and curation to evaluate and confirm these assignments. In order to carry out this task, the system should provide all information used in the automated annotation pipeline. Furthermore, the system must have an interface to replace a gene name with another derived from other evidence when automated annotation was not suitable. A point to note was that the corrected gene name had to satisfy our rules of gene names.
The second key requirement was providing an integrated view of the annotation and its evidence that could be used to modify the annotation as appropriate. At a minimum, this view had to include tools that allowed the CDS in the mRNA to be viewed and edited, the status of cDNA, assigned gene ontology terms, and experts' comments. In addition to the information available from the pipeline, genome mapping coordinates are useful, as they indicate whether the transcript is appropriately spliced and provide the evidence for alternative splicing analysis. Other experimental and analysis results also provide evidence for manual annotation, including cluster analysis provided by NCBI and TIGR. In addition, sequence quality and contig assembly information is essential for assessing the quality of any annotation, a fact that became clear during FANTOM1, where sequence quality allowed curators to determine whether the discrepancies between cDNA sequences and matches in target databases were likely to be sequencing errors or genuine polymorphisms, mutations, or closely related isoforms.
Another essential element of any annotation system is the ability to search the database to retrieve both additional annotation as well as sequences of a particular class for further analysis. The interface allowed experts to select clones that related to classes of their specialties and interests in the MATRICS.
One additional requirement was an intuitive and effective presentation of the various types of evidence that were available for each cDNA. To make a reliable judgment, curators need a wide range of information in an appropriate structured order to allow them to make an informed decision at each stage in the process. Structuring the presentation appropriately is an underappreciated but fundamentally important issue in creating an effective annotation system. Individuals annotated as many as 1000 sequences, which is a very large task even if each sequence requires only one minute of viewing the evidence to arrive at a conclusion. In addition, we wanted to create a tool that could be used by members of the broader scientific community who wish to access the evidence underlying our assignments.
Summary Images
The FANTOM2 interface was designed to meet these challenges by allowing curators access to the results of all of the analyses that have been performed on the FANTOM2 data set, including DNA homology search hits, protein hits, repetitive regions, predicted CDS regions, identified motifs, EST hits, and genome mapping coordinates. The first view provided by the system is a graphic “summary image,” in which all of these analyses are compactly integrated (Fig.2). A black line at the top of the images represents query cDNA sequences, and the results of the various searches are shown as parallel colored lines with a representation of the relevant coordinates within the query sequence. By pointing at a particular annotation line in the summary image, additional information is supplied to the user, including the relevant database ID/accession number, sequence descriptions, matched positions, and percent match identities. Clicking on any particular bar opens a window containing detailed information about a particular assignment including information such as sequence alignments, public data base entries and annotations, links to a variety of additional annotation viewers, and links to a “Reflect” interface (see below) that is used for replacing the current annotation.
The CAS (cDNA Annotation System) presented as a graphic summary of the evidence supporting the annotation of the clone. Shown here are two panels with the evidence assigned to the forward and reverse strands, respectively. Black lines at the top of each panel represent query (cDNA) sequences. The color code is as follows: DNA hits, red; protein hits, blue; repetitive hits, dark gray; predicted CDS, green; motifs, purple; EST hits, yellow; and genome mapping segments (for identifying splicing), ocher. Light gray bars in the DNA or protein hits category indicate gapped regions in subject sequences.

In the main page for each sequence, additional information is provided, including data on gene expression from microarray assays, information from protein-protein interaction studies, predictions of cellular localization, noncoding RNA analysis, antisense analysis, clustering data, and genome mapping data.
Curation Mechanism
The annotated gene names are assigned using a controlled vocabulary that reflects the type of annotation, the evidence supporting the assignment, the relative level of confidence, and other information. Each annotation item consists of a primary name, its “qualifier,” “annotation text,” “data source,” and “evidence.” “Qualifiers” representing the various annotation items are summarized in Table 1. Curators can input any annotation with appropriate qualifiers using a curation window. “Annotation text” is a description of the annotation, “data source” is the associated source of the evidence used in support of the annotation, and “evidence” lists the precise evidence used to arrive at the annotation.
To facilitate the entry of annotation using our controlled vocabulary, we implemented a “Reflect” mechanism in CAS that allowed curators to transfer the evidence and its descriptive qualifiers, along with an informative name, to the appropriate fields in the annotation forms, such as gene name, gene symbol, CDS start/stop, and CDS status. Figure 3 shows an example of the “Reflect” interface.
The “Reflect” interface allows curators to transfer gene names, gene symbols, and synonyms parsed from the results of annotation searches, as well as a reference to the target database and the evidence associated with the assignment. The “Add” buttons allowed curators to transfer annotation to the clone. The “override” buttons opened an annotation editing form that allowed curators to modify the annotation.

Quality and Contig Viewer
CAS uses a utility called ITOP (Inspecting Transcript Object in Phred/Phrap), which provides three views of the data. ESECONSED, like CONSED (Gordon et al. 1998), provides views of the sequence with quality scores. MOSAIC presents a view of the sequence contigs using both Scalable Vector Graph (SVG) and XML-representations, with each component sequence shown within the context of the assembly. Graph View presents a graphic view of quality value, allowing users to rapidly assess whether regions of apparent polymorphism represent low-quality regions. Although these tools are components of the CAS, they can also be used independently. The ITOP system itself is available at http://fantom2.gsc.riken.go.jp/ITOP/.
Search Interface
Table 3 shows a list of searches that are currently enabled. In the MATRICS annotation phase, this system allowed curators to select particular clones in their area of expertise based on the preliminary annotation from our pipeline, and this laid the foundation for subsequent analysis of gene discovery in the FANTOM cDNA collection, including many of the manuscripts in this issue.
Available Searches in the CAS with Descriptions
Name | Description |
|---|---|
| ID search | Search for clones using various identifiers: clone ID, sequence ID, rearray ID, and DDBJ accession number. |
| Keyword search with curated annotations | Search for entries with specified keywords in the curated annotations. |
| Keyword search with automated annotations | Search for entries with specified keywords in the curated annotations. |
| Category search with automated annotations | Search for entries in specified categories assigned during automated annotation. |
| InterPro/Pfam domain search. | Search for entries by InterPro/Pfam domains. Users can specify an Interpro/Pfam ID or select from a list of all domains. |
| GO ID search | Search for entries by computationally assigned GO terms. Users can specify a GO ID or select from a list of all GO terms. |
| Repeat search | Search for entries by repeats content. Users can specify repeat ID or select from a list of all repeats. |
| Library search | Search for entries by source library. Users can specify a library ID or select from a list of libraries. |
| IPSORT prediction search | Search for entries by IPSORT results. |
| DNA length search | Search for entries by nucleotide sequence lengths. Users can specify both lower and upper bounds. |
|
Amino acid/predicted CDS length search |
Search for entries by coding amino acid sequence or predicted CDS lengths. Users can specify both lower and upper bound of their length; for predicted CDS length search, they can select a CDS prediction method. |
Comparing Automated and Human-Curated Annotation
The human annotators who participated in the MATRICS annotation of the FANTOM2 clone set represent a broad crosssection of background and experience. As noted in the introduction, an ideal automated annotation pipeline would require no human curation, and could be updated regularly as new information and tools become available. To assess the performance of our pipeline, we compared annotation assigned by our automated pipeline with the results of human curation for 33,409 FANTOM2 sequences selected as being representative of unique transcripts in the collection (described in Methods). From this comparison, we found that 25,089 annotations (75.1%) were unchanged by the human curators, and that 26,257 (78.6%) could be considered to be matched in that the annotation did not alter the proposed biological context of a clone, but only made minor semantic changes.
To understand the differences between automated and human-curated annotations, we analyzed the changes in the remaining 7152 sequences. One relatively common change reflects the preference of human curators to give any “name” to a sequence rather than using terms such as “unknown EST” or “hypothetical protein”. For example, in the case of clone ID “0610016J10”, the human curated annotation was “inferred: RIKEN cDNA 0610016J10 gene/HYPOTHETICAL PROTEIN CGI-27 [Human] [Homo sapiens],” whereas automated annotation was “unknown EST”. Examination of the evidence suggests that the clone sequence is homologous to the 3′ UTR of a hypothetical human protein coding sequence based on the UniGene/TIGR EST clustering analysis. In this example, one of many, the curator may have felt that additional information based on the fact that there is a potential human ortholog, and that this transcript is likely to be the 3′ UTR of a protein-coding transcript, may have been important. Such information could be represented by subdividing the categories annotated as “unknown EST” and “hypothetical proteins” to provide an indication of where there is a related sequence in another species (i.e., similar to human EST). However, it is clear that this annotation does not add any insight into actual gene function, although it might help to prioritize attention given to expressed genes on a microarray output in a particular disease model.
Human curators were generally adept at correcting absurdities. An example is the annotation for clone ID “0610012K07,” “KDP OPERON TRANSCRIPTIONAL REGULATOR PROTEIN KDPE homolog [Escherichia coli],” which was selected because it passed our automated informative annotation. The human curators changed this to “unknown EST”.
Many other changes made by human curation were subtle, and the information content is not changed by the choice. For example, the two assigned annotations of clone ID “0610030E04” were “sulfotransferase family 1A, phenolpreferring, member 1” (human-curated annotation) and “aryl sulfotransferase (EC 2.8.2.1) p1 homolog [Mus musculus]” (automated annotation). The latter is, in our view, better.
True judgment calls were comparatively unusual. One example is the alternative names ascribed to clone ID “4732474G14” which were “ESTROGEN REGULATED LIV-1 PROTEIN” (human annotation) and “sema domain, transmembrane domain (TM), and cytoplasmic domain, (semaphorin) 6A” (automated annotation). Both names are appropriate, but the human choice provides a direct link to a gene that already has a name in humans, and is therefore a better choice.
One example where human curation was frequently unreliable was in dealing with the open-reading frames encoded by the mouse B2 repeat. In many cases, curators chose annotations such as “serine proteinase 33,” or “5′ nucleotidase, 1A,” which are transcripts that contain the B2 repeat in their 3′ UTR and give a 100% match to the query but only for a small percentage of their length. Following the FANTOM2 annotation meeting, the automated annotation pipeline was modified to avoid such repeats, and human curation was redone to reannotate gene names derived from repetitive elements.
As alluded to previously, the most common bias observed in the human annotation was to replace assignments with little information content.
Most often, human curation changed annotation associated with clones whose sequence had relatively little associated information. There were 2912 members of the 33,409 representative sequence set that were annotated as “hypothetical protein,” “unknown EST,” or “unclassifiable” in both human-curated and automated annotation, but the choice was different in each case. There are a number of reasons that could be addressed with minor modifications to the automated pipeline. The major one is the choice of the CDS region, which affects whether a name is assigned in one of these uninformative classes. The automated annotation pipeline used CDS regions predicted by ProCrest, whereas curators could choose from among a number of alternative CDS predictions. In other instances, curators correctly chose an alternative assignment based on correction of a sequencing error that caused a frame shift; in other cases, annotation changes were based on alignment with another hypothetical protein. Some curators systematically changed “unknown EST” to “unclassifiable,” when the number of matched ESTs was small and the whole query sequences were not covered by ESTs. Neither represents a particularly informative choice, and both were included in the pipeline to distinguish between sequences that were observed as transcribed in other studies and therefore likely to be something other than “junk” or unprocessed nuclear RNA. In a separate comprehensive cDNA microarray analysis of a subset of the FANTOM2 cDNAs (Bono et al. 2002; Bono et al. 2003), we have seen relatively little difference in the number of “unknown ESTs” and “unclassifiable” sequences that are expressed in at least one major mouse tissue.
The Importance of Filtering Uninformative Annotation
Overall, the automated pipeline performed quite well. We believe that this was due, in part, to the use of the “uninformative rule” filtering. To assess its impact, we reran our automated annotation pipeline without it. Using only the “best hit” shows that 16,450 annotations (49.2%) were completely matched and, using the same criteria as before, 17,270 (51.7%) annotations can be considered to be matched to an equivalent term. This finding indicates that 25%–30% of automated annotations were accepted by human curators because the “uninformative rule” changed the initial choice of “best hit” to a name with greater information content that was relevant to the curators.
Combination of Automated Annotation and Human Curation
In a large-scale annotation, the quality of manual annotation is mixed, with both high and low, and computational annotation is settled but of intermediate quality. At the present time, it is clear that we need both an automated annotation pipeline to provide preliminary annotation and human curation to review and validate the initial assignments. Following the human review, differences between the preliminary and final annotation should be carefully investigated and wherever patterns can be discerned, integrated into the next-generation automated annotation pipeline. These iterative efforts are essential to the goal of making computational annotation a “gold standard.”
As the current best solution for large-scale annotation is incorporating manual and automated annotation, some additional systems might be helpful: (1) a system to combine the pipeline and curation system dynamically, in which the information about rejected automated annotation is automatically gathered and can become a new source for refining the pipeline; (2) a system for determining whether or not an annotation needs to be checked manually; and (3) a system for detecting annotation that might be changed by the database update.
Conclusion
The development of a well engineered and structured annotation system is essential for the success of large-scale sequence analysis projects such as FANTOM2. Two key elements of such a system are the creation of an automated pipeline for doing preliminary annotation of the sequences, and development of an intuitive graphic interface that will allow curators and users of the database to have ready access to the annotation and the underlying evidence. The CAS, which provided the interface we used in our MATRICS annotation, is available for community access to the FAMTON2 data at http://fantom2.gsc.riken.go.jp/.
An analysis of the pattern of human curation suggests that a well designed preliminary annotation pipeline can obviate the need for human intervention in many cases. If we exclude the uninformative annotation, the hypothetical proteins, unknown ESTs, and unclassifiable sequences, automated annotation produced by our pipeline was accepted in more than 85% of the cases. In the instances where the preliminary annotation was not accepted, the curator was not always correct and commonly breached the annotation rules. With further refinement of the “uninformative rule” and other elements of this system, based on an understanding of how human annotation works, we believe that complete computational annotation, and continuous updating of that sequence, will be possible and that this can lead to a much more dynamic resource for functional genomics studies.
METHODS
Sequence Set and Curated Annotation Set
We ran our automated annotation pipeline with the 60,770 FANTOM2 sequences derived from the RIKEN mouse cDNA clone libraries (Carninci et al. 2003). These sequences were clustered into 33,409 transcriptional units (TUs), and one sequence was selected as a representative sequence from each TU. We used human-curated annotation of the 33,409 representative sequences for evaluating results from our pipeline.
Sequence Analysis and Database Searches
Assembled full-length cDNA sequences were first masked using RepeatMasker (A.F.A. Smit and P. Green, unpubl.) to exclude regions containing known repetitive sequences. DNA searches were performed using BLASTN (Altschul et al. 1990) using the “-F” option, which turns off filtering of the query sequences. FANTOM2 query sequences were searched against mouse DNA sequences in LocusLink (Pruitt and Maglott 2001; http://www.ncbi.nih.gov/LocusLink/), RefSeq (Pruitt and Maglott 2001; http://www.ncbi.nih.gov/LocusLink/refseq.html), and the MGI (Mouse Genome Informatics) database (Blake et al. 2002; http://www.informatics.jax.org/), and in separate searches against the mouse sequences in dbEST (Boguski et al. 1993; http://www.ncbi.nih.gov/dbEST/). Non-EST database searches were filtered to exclude hits with less than 98% identity or less than 100 bp in length (excluding repetitive regions); EST database searches required a minimum of 95% identity and 100 bp of matched sequence for inclusion in the analysis. Protein databases were searched using the FASTY program (Pearson et al. 1997) in the FASTA3 package. FASTY uses possible frameshifts to extend potential DNA query sequence matches to protein sequences in the database. This is useful because cDNA sequences may contain frameshifts, including insertions and deletions, and these can cause the protein-coding amino acid sequence to be incorrectly deduced. The FANTOM2 sequences were searched against SPTR-NRDB (Bairoch and Apweiler 2000; ftp://ftp.ebi.ac.uk/pub/databases/sptr_nrdb/) and PIR (Wu et al. 2002; http://pir.georgetown.edu/). SPTR-NRDB is composed of SWISS-PROT, TrEMBL, TrEMBL-new, and splice variants. Open reading frames in the cDNA sequences were predicted using ProCrest, and those with predicted CDS regions greater than or equal to 100 amino acids in length were subjected to three separate motif-prediction analyses. InterProScan was used to search the InterPro database (Apweiler et al. 2001; http://www.ebi.ac.uk/interpro/); HMMER (http://hmmer.wustl.edu/) was used to search the MDS database (Kawaji et al. 2002), which contains novel motifs and their hidden Markov models (http://motif.ics.es.osaka-u.ac.jp/MDS/), and SCOP analysis was preformed to search the SuperFamily database (Gough et al. 2001; http://www.supfam.org/). Finally, sequences were analyzed using UniGene (Boguski and Schuler 1995; http://www.ncbi.nih.gov/UniGene/) and the TIGR Gene Indices (Quackenbush et al. 2001; http://www.tigr.org/tdb/tgi/), which use different approaches to group ESTs and gene sequences into clusters and to provide various annotation for the sequences represented by each cluster.
Gene Name Nomenclature and Controlled Vocabulary Terms
Query sequences falling into categories 1–3, were assigned the gene name of the matched target sequence DNA entry in MGI/LocusLink; gene symbols and synonyms were also transferred to our annotation database. Queries falling into categories 4–10 were assigned a gene name corresponding to the matched protein name. For query sequences falling into category 5 or 6, the keyword “homolog” was appended to the matching protein name. Sequences assigned to category 7 or 8 were denoted with the prefix “similar to” attached to the target sequence name. The prefix “weakly similar to” was used to identify sequences assigned to category 9 or 10. For all sequences in categories 5–10, the name of the organism corresponding to the matched protein was appended to the assigned gene name. Sequences falling into categories 11, 12, or 13 were assigned the annotation “inferred (cluster name)”, where the cluster name was “UniGene Cluster Name/TIGR cluster name”, or the “UniGene cluster name”, or “TIGR cluster name” as appropriate. If a query was assigned to category 14 or 15, its gene name was “hypothetical (InterPro/MDS domain/motif name)” containing protein. Those queries falling into category 16 were assigned the name “hypothetical (SCOP domain names concatenated with “/”) structure containing protein.” Query sequences assigned to category 17, 18, or 19 were annotated as “hypothetical protein,” “unknown EST,” or “unclassifiable,” respectively.
Comparing Automated Annotation and Human Curation
To assess the relative performance of our annotation pipeline and human curation, we compared the assigned annotations for the 33,409 representative transcript sequences in the FANTOM2 data set. The two annotations were considered equivalent if any of the following conditions were satisfied:
The two gene name assignments were identical.
Both gene names were derived from the same database entry. This means that a curator modified the assigned gene name by editing the assigned name for clarity.
Both gene names were derived from InterPro domains, but the entries were not the same. In the FANTOM2 annotation system, only a single InterPro domain annotation is allowed, although proteins may contain multiple domains. Consequently we regard this case as “matched,” although we are considering a means of including multiple domain assignments in our next revision of the pipeline and database.
Both gene names were derived from SCOP entries, but the entries were not the same.
System Architecture
The FANTOM annotation pipeline was implemented as a Perl script that evaluated the evidence at each stage in the process and made a decision at each stage, writing the appropriate annotation to the database using the appropriate controlled vocabulary terms. The regular expressions used in the “uninformative filter” and the filtering program are available at http://fantom2.gsc.riken.go.jp/.
The CAS was implemented as a Web-based application using mod_perl and the gd graphics library on a Linux system running an Apache 1.3 server. All curated annotations and annotation histories were stored in a custom database implemented in a Sybase relational database management system; the database schema is presented as Supplemental Information 2. Other data such as similarity search alignments and clone sequences were stored in indexed flat files.
Access to the Annotation System During MATRICS
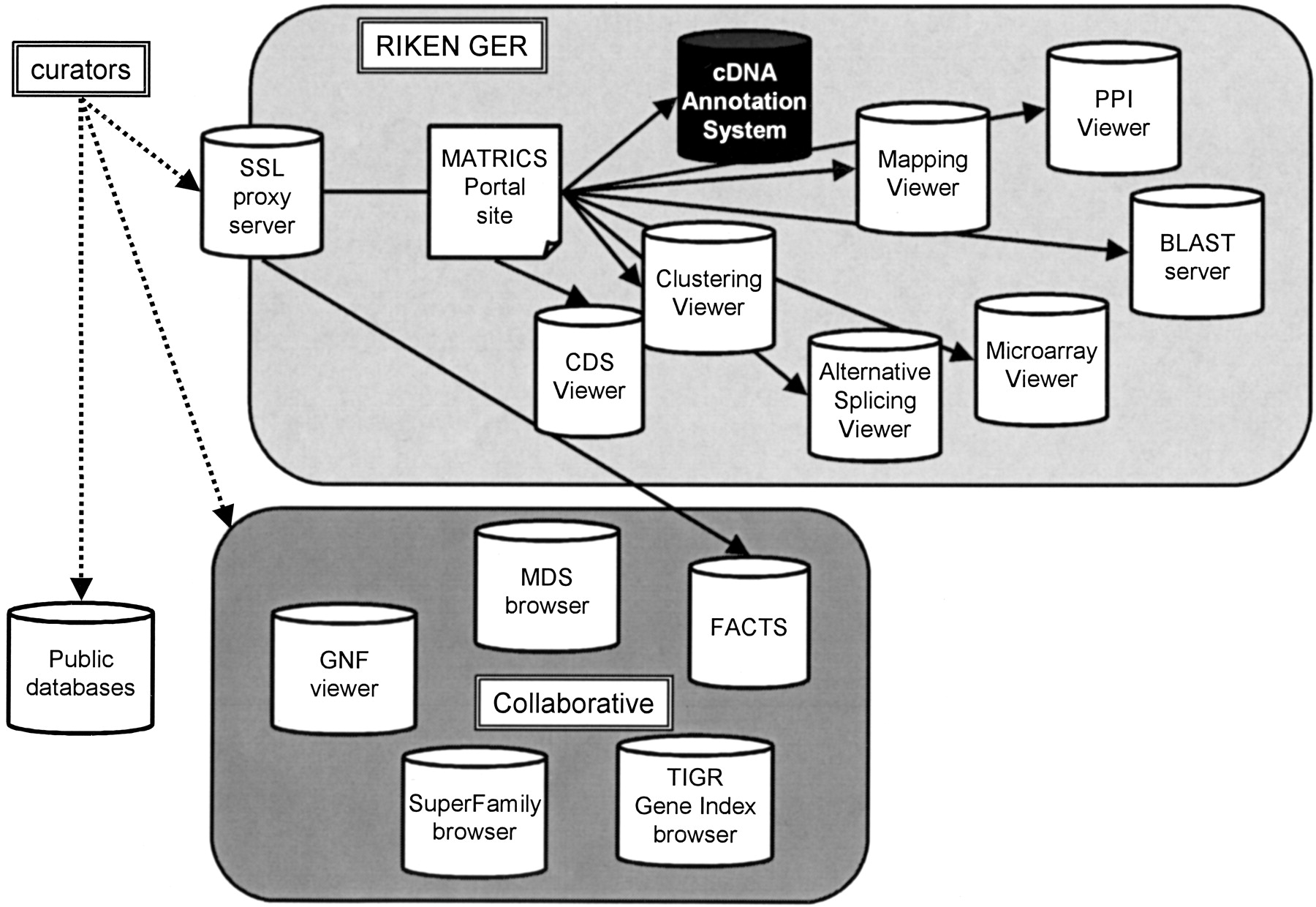
The CAS was published through the main RIKEN Web site, and curators accessed the system through an SSL proxy server (Policy director). Figure 4 shows an overview of the system architecture in MATRICS.
The MATRICS annotation phase used an SSL proxy server at the RIKEN Genome Exploration Laboratory in Japan to provide a gateway to a wide range of annotation databases and tools available at RIKEN, as well as additional external databases maintained by our collaborators on this project.
Notes
[2] Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.992803.
[3] Corresponding authors. E-MAIL [email protected]; FAX 81-45-503-9216. E-MAIL [email protected]; FAX +1-301-838-0208.
This work was supported by a Research Grant to the RIKEN Genome Exploration Research Project from the Ministry of Education, Culture, Sports, Science and Technology of Japan; by ACT-JST (Research and Development for Applying Advanced Computational Science and Technology) of the Japan Science and Technology Corporation to Y.H. J.Q. was supported by grants from the U.S. DOE, the NSF, and the National Heart, Lung, and Blood Institute of the NIH.J.Q. thanks the TIGR Gene Index team, and particularly Geo Pertea and Razvan Sultana for assistance with this project. The Mouse Genome Sequencing (MGS) Project, The Mouse Genome Database (MGD), the Gene Expression Database (GXD), and the Gene Ontology (GO) project are components of the MGI database system. The MGS is supported by DOE grant FG02-99ER62850, the MGD by National Human Genome Research Institute (NHGRI) grant HG-00330, the GXD by National Institute of Child Health and Human Development grant HD-33745, and the GO project by NHGRI grant HG-002273.
References
- ↵Adams, M.D., Celniker, S.E., Holt, R.A., Evans, C.A., Gocayne, J.D., Amanatides, P.G., Scherer, S.E., Li, P.W., Hoskins, R.A., Galle, R.F., et al. 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185 -2195.
- ↵Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403 -410.
- ↵Apweiler, R., Attwood, T.K., Bairoch, A., Bateman, A., Birney, E., Biswas, M., Bucher, P., Cerutti, L., Corpet, F., Croning, M.D., et al. 2001. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 29: 37 -40.
- ↵Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28: 45 -48.
- ↵Blake, J.A., Richardson, J.E., Bult, C.J., Kadin, J.A., and Eppig, J.T. 2002. The Mouse Genome Database (MGD): The model organism database for the laboratory mouse. Nucleic Acids Res. 30: 113 -115.
- ↵Boguski, M.S. and Schuler, G.D. 1995. ESTablishing a human transcript map. Nat. Genet. 10: 369 -371.
- ↵Boguski, M.S., Lowe, T.M., and Tolstoshev, C.M. 1993. dbEST—Database for “expressed sequence tags”. Nat. Genet. 4: 332 -333.
- ↵Bono, H., Kasukawa, T., Hayashizaki, Y., and Okazaki, Y. 2002. READ: RIKEN Expression Array Database. Nucleic Acids Res. 30: 211 -213.
- ↵Bono, H., Yagi, K., Kasukawa, T., Nikaido, I., Tominaga, N., Miki, R., Mizuno, Y., Tomaru, Y., Goto, H., Nitanda, H., et al. 2003. Systematic expression profiling of the mouse transcriptome using RIKEN cDNA microarrays. Genome Res. (this issue).
- ↵Carninci, P., Waki, K., Shiraki, T., Konno, H., Shibata, K., Itoh, M., Aizawa, K., Arakawa, T., Ishii, Y., Sasaki, D., et al. 2003. Targeting a complex transcriptome: The construction of the mouse full-length cDNA encyclopedia. Genome Res. (this issue).
- ↵The FANTOM Consortium and the RIKEN Genome Exploration Research Group Phase I and II Team. 2002. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420: 563 -573.
- ↵The Gene Ontology Consortium. 2001. Creating the gene ontology resource: Design and implementation. Genome Res. 11: 1425 -1433.
- ↵Gordon, D., Abajian, C., and Green, P. 1998. Consed: A graphical tool for sequence finishing. Genome Res. 8: 195-202.
- ↵Gough, J., Karplus, K., Hughey, R., and Chothia, C. 2001. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol. 313:903 -919.
- ↵Kawaji, H., Schonbach, C., Matsuo, Y., Kawai, J., Okazaki, Y., Hayashizaki, Y., and Matsuda, H. 2002. Exploration of novel motifs derived from mouse cDNA sequences. Genome Res. 12: 367 -378.
- ↵Pearson, W.R., Wood, T., Zhang, Z., and Miller, W. 1997. Comparison of DNA sequences with protein sequences. Genomics 46: 24 -36.
- ↵Pruitt, K.D. and Maglott, D.R. 2001. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 29: 137 -140.
- ↵Quackenbush, J. 2000. Viva la revolution! A report from the FANTOM meeting. Nat. Genet. 26: 255 -256.
- ↵Quackenbush, J., Cho, J., Lee, D., Liang, F., Holt, I., Karamycheva, S., Parvizi, B., Pertea, G., Sultana, R., and White, J. 2001. The TIGR Gene Indices: Analysis of gene transcript sequences in highly sampled eukaryotic species. Nucleic Acids Res. 29: 159 -164.
- ↵Wu, C.H., Huang, H., Arminski, L., Castro-Alvear, J., Chen, Y., Hu, Z.Z., Ledley, R.S., Lewis, K.C., Mewes, H.W., Orcutt, B.C., et al. 2002. The Protein Information Resource: An integrated public resource of functional annotation of proteins. Nucleic Acids Res. 30: 35 -37.
WEB SITE REFERENCES
- ↵http://fantom2.gsc.riken.go.jp/; FANTOM2 Web site.
- ↵http://fantom2.gsc.riken.go.jp/ITOP/; ITOP Web site.
- ↵ftp://ftp.ebi.ac.uk/pub/databases/sptr_nrdb/; SPTR-NRDB FTP site.
- ↵http://hmmer.wustl.edu/; S.R. Eddy. HMMER: Profile hidden Markov models for biological sequence analysis.
- ↵http://motif.ics.es.osaka-u.ac.jp/MDS/; MDS Web site.
- ↵http://pir.georgetown.edu/; PIR Web site.
- ↵http://www.ebi.ac.uk/interpro/; InterPro Web site.
- ↵http://www.informatics.jax.org/; Mouse Genome Informatics (MGI) Web site.
- ↵http://www.ncbi.nih.gov/dbEST/; dbEST Web site.
- ↵http://www.ncbi.nih.gov/LocusLink/; LocusLink Web site.
- ↵http://www.ncbi.nih.gov/LocusLink/refseq.html; RefSeq Web site.
- ↵http://www.ncbi.nih.gov/UniGene/; UniGene Web site.
- ↵http://www.supfam.org/; SuperFamily Web site.
- ↵http://www.tigr.org/tdb/tgi/; TIGR Gene Indices Web site.