
Abstract
With the sequencing and annotation of genomes and transcriptomes of several eukaryotes, the importance of noncoding RNA (ncRNA)—RNA molecules that are not translated to protein products—has become more evident. A subclass of ncRNA transcripts are encoded by highly regulated, multi-exon, transcriptional units, are processed like typical protein-coding mRNAs and are increasingly implicated in regulation of many cellular functions in eukaryotes. This study describes the identification of candidate functional ncRNAs from among the RIKEN mouse full-length cDNA collection, which contains 60,770 sequences, by using a systematic computational filtering approach. We initially searched for previously reported ncRNAs and found nine murine ncRNAs and homologs of several previously described nonmouse ncRNAs. Through our computational approach to filter artifact-free clones that lack protein coding potential, we extracted 4280 transcripts as the largest-candidate set. Many clones in the set had EST hits, potential CpG islands surrounding the transcription start sites, and homologies with the human genome. This implies that many candidates are indeed transcribed in a regulated manner. Our results demonstrate that ncRNAs are a major functional subclass of processed transcripts in mammals.
Noncoding RNA (ncRNA) is a global term for transcripts that lack an apparent open reading frame (ORF) and do not encode a protein product. Until recently, the only known functional ncRNAs were ribosomal RNA, transfer RNA, and several small nucleolar RNAs. Other classes of small ncRNAs—such as microRNAs (miRNAs), C/D box snoRNAs, small interfering RNAs (siRNAs), and small temporal RNAs (stRNAs)—have been identified and characterized in all three domains of life (bacteria, archaea, and eukarya) based on experimental expression analysis and computational screening (Sharp 2001; Eddy 2002; Grosshans and Slack 2002; Tang et al. 2002; Wassarman 2002). Lau et al. (2001) had reported that abundantly expressed 21- to 24-nt-long miRNAs are found in Caenorhabditis elegans, and the pattern of their expression varies with developmental stages (Lau et al. 2001). These classes of small ncRNAs are believed to contribute to processes such as transcriptional regulation, translational repression, and mRNA degradation (Storz 2002).
Longer ncRNAs, sometimes referred to as mRNA-like ncRNAs, form a quite distinct class. Unlike the classes noted above, they are processed like mRNA; that is, they are transcribed by RNA polymerase II, spliced, polyadenylated, and conceivably capped (Erdmann et al. 2000, 2001). For example, Xist, which acts as an X-chromosome inactivator to achieve dosage compensation in mammalian females, encodes a 17-kb-long RNA molecule with no significant ORFs, even though it is comprised of seven exons and polyadenylated (Hong et al. 1999; Nesterova et al. 2001). Such mRNA-like processed ncRNAs have been identified in plants and animals and are expressed in a tissue-specific manner (Erdmann et al. 2001). The diversity of such transcripts in the mammalian transcriptome has not been evident from genome sequencing, because exon boundaries are difficult to define, and ncRNAs tend to be less conserved between mammalian species. In this situation, the RIKEN mouse full-length cDNA collection, called FANTOM2 clone set (Okazaki et al. 2002), provides the largest available resource in any mammal for the discovery of candidate functional ncRNAs. The set contains many sequences that do not show apparent protein coding regions according to human-curated annotation. Some of these are likely to be the result of incomplete cDNA synthesis or incompletely processed transcripts (e.g., 3′ untranslated regions [UTRs]), or perhaps transcriptional “noise,” so the identification of strong candidate functional ncRNAs requires additional annotation criteria.
There are two ways to identify ncRNAs computationally. One is the “genome based” approach, which detects ncRNAs from genomic sequence. More than 200 candidate ncRNA genes are predicted in Escherichia coli by computational comparative genomics using “intergenic” sequence data from four related bacteria (Rivas et al. 2001). Two similar approaches are reported by other groups, and at least 20 ncRNA genes have been experimentally confirmed in E. coli (Argaman et al. 2001; Wassarman et al. 2001). This approach is not feasible in the more complex mammalian genomes.
The other approach is “transcripts-based.” MacIntosh et al. (2001) attempted to identify and characterize new ncRNAs by using Arabidopsis thaliana EST sequences. Through systematic computational screening, they extracted dozens of ncRNA candidates and putative RNAs encoding small peptides. The investigators concluded that there are numerous functional ncRNAs in A. thaliana.)
This study describes the initial effort at comprehensive identification of mammalian mRNA-like processed ncRNAs based on the comprehensive mouse transcriptome survey provided by the RIKEN Mouse Gene Encyclopedia project.
RESULTS AND DISCUSSION
Characterization of Previously Reported ncRNAs in FANTOM2 Clone Set
To assess the representation of ncRNAs in the FANTOM2 clone set (see below), we initially identified previously described ncRNAs. The set of previously reported ncRNAs was based on the Noncoding RNAs Database (http://biobases.ibch.poznan.pl/ncRNA/), which was constructed by Erdmann et al (2000, 2001). The query sequence set from the database contains ncRNAs from both mammalian and nonmammalian origins, including plants and flies.
We used a homology search based on BLASTN and Ssearch (Pearson 1991), to search for 18 of the murine known ncRNAs, and nine of them were identified in the FANTOM2 clone set. Likewise, putative homologs of several ncRNAs previously described in other mammalian organisms were also found (Table 1). We have newly identified homologs of rat NTAB (French et al. 2001), 7H4 (Velleca et al. 1994), human NTT (Liu et al. 1997), NCRMS (Chan et al. 2002), U19 snoRNA host gene (Bortolin and Kiss 1998), and hamster adapt33 (Wang et al. 1996) in the FANTOM2 clone set. These results indicate that ncRNAs are well represented in the FANTOM2 clone set, and sequence conservation across species is a useful criterion in annotation and validation.
Summary of Previously Identified ncRNAs or Their Possible Homologs Found in FANTOM2 Clone Set
FANTOM2 clone ID | Homologous noncoding RNA | GenBank/EMBL/DDBJ accession no. | Status of identity | Organism | References |
|---|---|---|---|---|---|
| C130002M05 | KvLQT-as | AF119385 | 97.8% in 2467 nt | Mm | Smilinich et al. 1999 |
| 3830421G02 | U17 snoRNA host gene | AJ006836 | 99.7% in 383 nt | Mm | Pelczar and Filipowicz 1998 |
| A730062M15 | U22 snoRNA host gene | U40654 | 100% in 476 nt | Mm | Tycowski et al. 1996 |
| E130201N16 | G90 | AJ132433 | 98.4% in 833 nt | Mm | Krause et al. 1999 |
| A430022B11 | XIST | L04961 | 99.3% in 1497 nt | Mm | Hong et al. 1999 |
| B230105C16 | IPW | U69888 | 97.9% in 617 nt | Mm | Wevrick and Francke 1997 |
| 1100001A04 | H19 | NM_023123 | 99.2% in 870 nt | Mm | Hurst and Smith 1999 |
| 9630004F23 | CIOR | AF140607 | 99.9% in 2118 nt | Mm | Inoue et al. 2002 |
| 6430597C21 | Rian | AB063319 | 100% in 1496 nt | Mm | Hatada et al. 2001 |
| 2900019G14 | NTAB | AY035551 | 89.7% in 955 nt | Rn | French et al. 2001 |
| 5930439P04 | Synapse-specific 7H4 | L33722 | 79.1% in 3668 nt | Rn | Velleca et al. 1994 |
| A530032L19 | NTT | U54776 | 61.0% in 2635 nt | Hs | Liu et al. 1997 |
| D630034O16 | DGCR5 | X91348 | 73.7% in 243 nt | Hs | Sutherland et al. 1996 |
| D930049J19 | NCRMS | XR_000104 | 72.6% in 446 nt | Hs | Chan et al. 2002 |
| E430001E02 | U19 snoRNA host gene | AJ224166 | 80.7% in 451 nt | Hs | Bortolin and Kiss 1998 |
|
5430416N02 |
adapt33 |
U29660 |
63.6% in 676 nt |
Cg |
Wang et al. 1996 |
[i] Homology search was initially performed by BLASTN. Then the best hit sequence having its e value <1.0e - 10 in same orientation were picked up. For this sequence, Ssearch (Pearson 1991) was carried out for stricter alignment. Hs indicates Homo sapiens; Mm, Mus musculus; Rn, Rattus norvegicus; and Cg, Cricetulus griseus
Computational Screening of the Novel ncRNA Candidates
The FANTOM2 clone set contains 60,770 cDNA clones selected from >260 normalized, subtracted, and full-length enriched cDNA libraries of C57BL/6J strain of mouse. The 60,770 clones of FANTOM2-set were clustered into 33,409 transcriptional units (TUs), and approximately half of the TUs contained a deduced protein sequence (Representative Protein Set [RPS]) based on ORF prediction and/or homology with known proteins. The remaining set of cDNAs (15,815 sequences) that are defined as non–protein-coding TUs (Okazaki et al. 2002) represent the starting set for identification of ncRNAs. To eliminate other possible sources of transcripts that lack an apparent functional ORF, such as UTR-only sequences (incomplete cDNA synthesis), unprocessed mRNAs with retained introns, and chimeric cDNA clones, we applied the following strategy of computational filtering. In addition to removing the RPS, we eliminated sequences that showed any homology with known protein sequences, even if they did not contain any evidence of an ORF. We then mapped the remaining sequences to the mouse genome (MGSCv3). Comparison between the alignments with the genome and exon predictions by GENSCAN (Burge and Karlin 1997) was also considered. Because GENSCAN may fail to identify untranslated regions of protein-coding transcripts, we eliminated any sequences that mapped within 10 kb of any predicted exon on the grounds that they may be part of the same TU, for example, an alternative 3′UTR or splice variant (see Methods).
Consequently, we extracted 4280 transcripts as the candidate set of ncRNAs. The procedures for computational screening and number of remaining sequences in each filtering step are shown in Table 2. The average length of the candidate clones was 1778.9 nt, and that of predicted protein-coding transcripts in RTS was 2131.8 nt. Likewise, the average length of the longest ORF of resulting candidates was 200.6 nt, whereas that of transcripts in RPS was 1088.7 nt. A full set of accession numbers of the candidate set is provided in the Supplementary tables, and the annotation of each individual candidate can be assessed at the FANTOM2 Web interface as described in the overview of the RIKEN project (Okazaki et al. 2002).
Summary of Computational Screening for Novel ncRNA Candidates
Procedures for computational screening | No. of remaining clones |
|---|---|
| Defined as Non-protein-coding TU | 15,815 |
| No homologies to other known protein sequences (BLASTX) | 12,382 |
| Could be aligned to mouse genome sequence (identity >90%, length >90%) | 11,652 |
|
CDS prediction could not be made by GENSCAN (10 kb around mapped region) |
4,280 |
Characterization of the Candidate Set
The strategy used here is conservative, and we know that not all known ncRNAs would meet these criteria. Several previously reported ncRNAs were eliminated at each filtering step of the strategy described above, but as we would hope, the frequency within the remaining set increased (albeit the number of known ncRNAs is too small to assess the statistical validity of this assessment). To characterize the candidate set further, we used several additional criteria. First, we conducted a homology search against publicly available EST sequences of mouse, human, and rat, as well as against the human genome. We additionally searched CpG islands in 5′ boundary regions and polyadenylation signals in 3′ ends (see more details in Methods). CpG islands and polyadenylation signal are observed not only in a large number of protein-coding genes but also in several known ncRNAs. This can be taken as supporting evidence that they are actually transcribed by RNA polymerase II. We also determined the intron–exon boundaries of the loci encoding the transcripts, and we identified the subset of candidates that is produced by splicing of a primary transcript. These results are summarized in Table 3.
Summary of Further Characterization of the ncRNA Candidates
Evidence | Hit no. (%) |
|---|---|
| Mouse EST hit | 1,200 (28.0) |
| Human EST hit | 111 (2.6) |
| Rat EST hit | 252 (5.8) |
| Human genome homology | 454 (10.6) |
| Potential CpG islands | 919 (21.5) |
| Potential PolyA signal | 1,395 (32.6) |
|
Spliced sequences (no. of exons ≥2) |
1,150 (26.9) |
There were 1200 (28.0%) ncRNA candidates, which were homologous with mouse EST sequences (BLASTN with e value lower than 1.0e - 100). This indicates that approximately one fourth of the candidates have independent evidence of reproducible expression. This is not an especially stringent criterion, because the RIKEN Project is itself the largest mouse EST project, and library construction involves strong selection to avoid redundancy. Furthermore, functional ncRNAs may not be abundant transcripts. More interestingly, 111 (2.6%) clones showed strong homology with human ESTs, and 252 (5.8%) clones showed strong homology to rat ESTs. Furthermore, 454 (10.6%) clones could be aligned with the human genome sequence at ≥50% homology and ≥70% of length. Sequence conservation is also not a strict criterion for exclusion or functional significance, because several known ncRNAs would fail to meet this criterion.
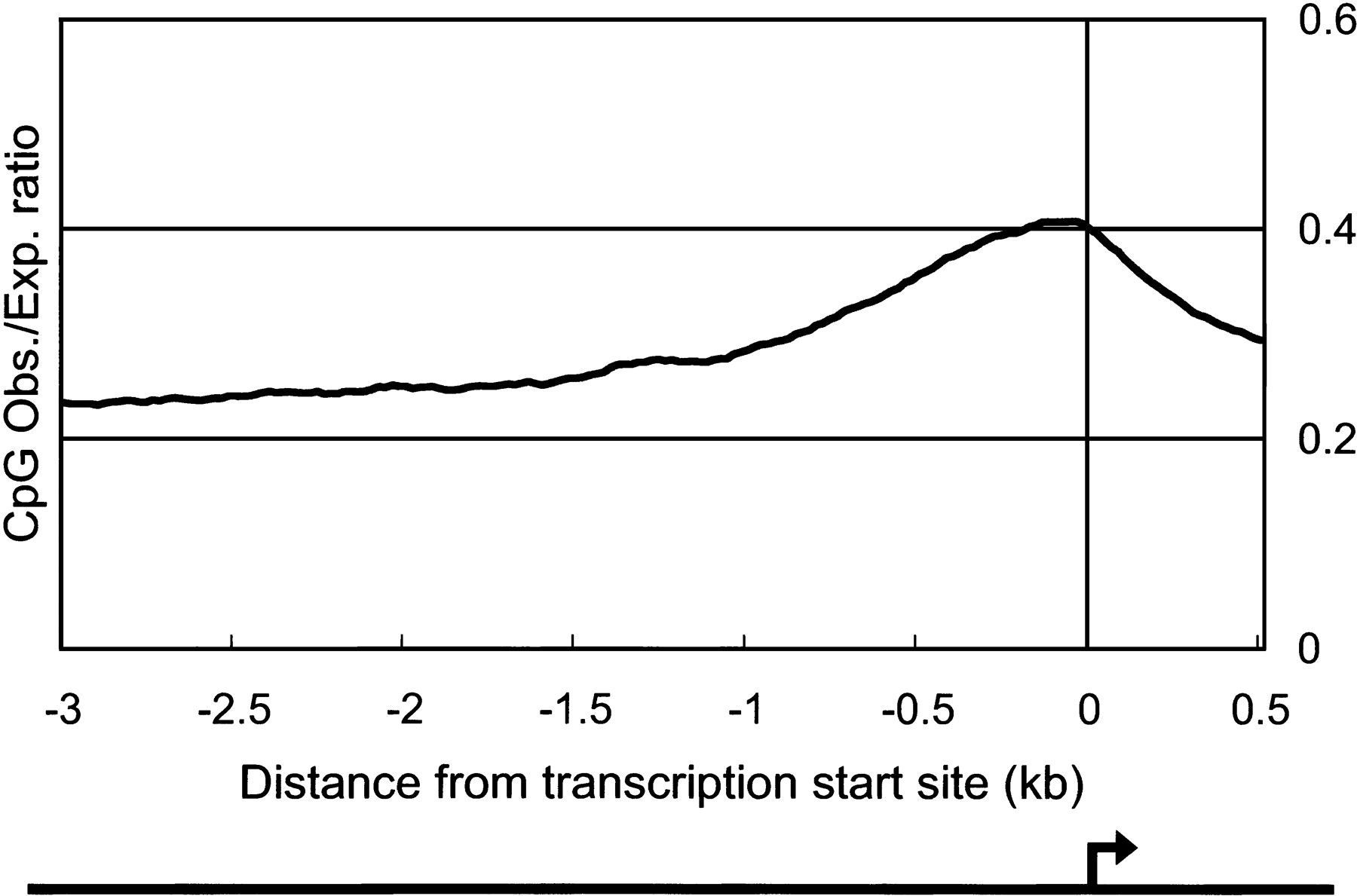
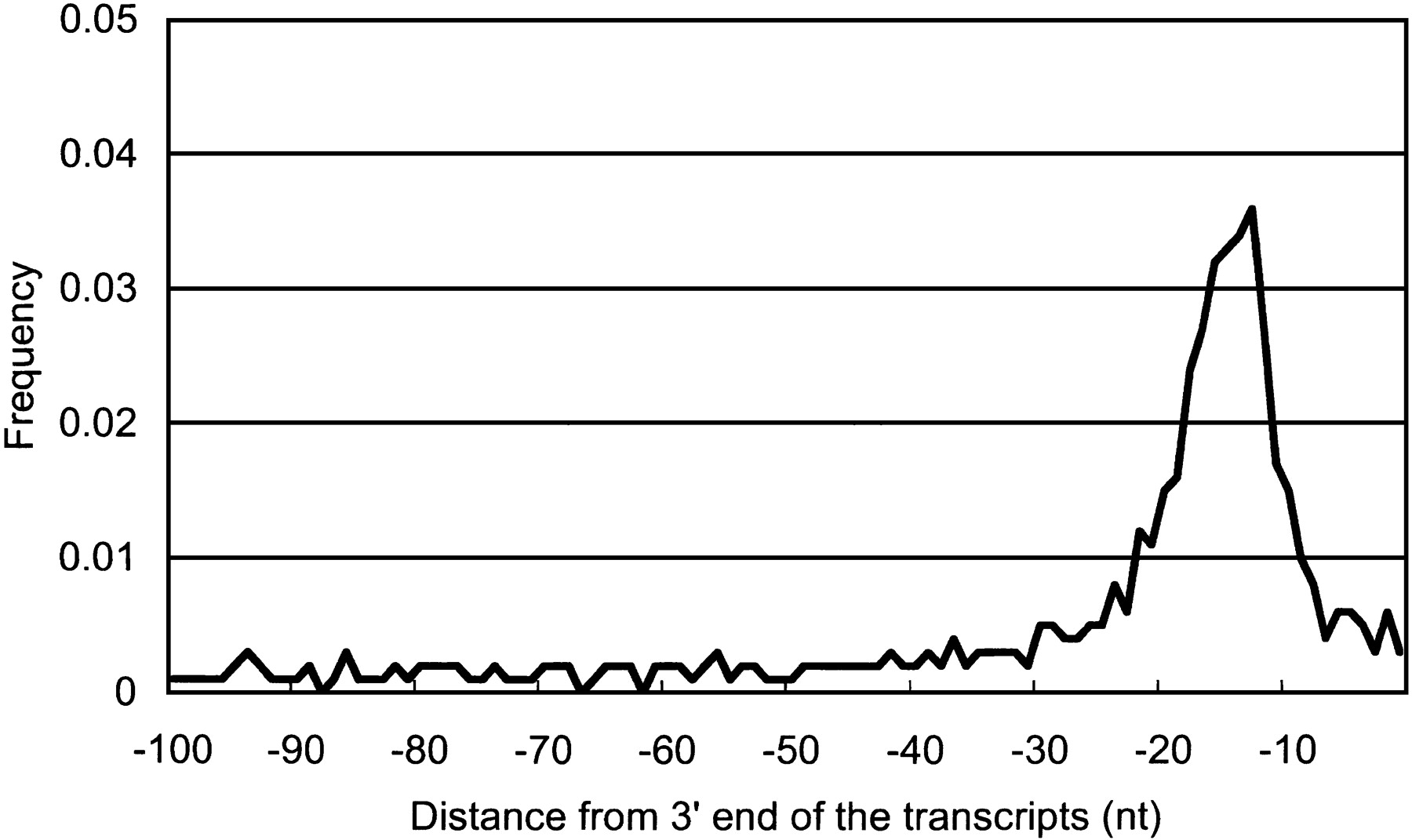
We identified CpG islands in the 5′ boundary genomic region for 919 (21.5%) of the clones. CpG islands are associated with ∼40% of promoters for mammalian genes, most commonly those of housekeeping genes that are widely expressed (Takai and Jones 2002). A polyadenylation signal was found at the 3′ end of 1395 (32.6%) of the clones. This is a rather more stringent criterion, implying that the transcript is a genuine polyadenylated mRNA-like molecule. However, cDNAs that lack this signal might have arisen by internal oligo-dT priming but might still be bona fide ncRNAs. To clarify the robustness of these criteria, we plotted the average of CpG observed/expected (O/E) ratio and frequency of poly-A–like signals around the 5′ boundaries of mapped genomic regions, and the 3′ end of cDNA sequences, respectively. As shown in Figures 1 and 2, there is a clear peak of the CpG O/E ratio and poly-A signals that delineates the subsets of transcripts in the candidate set into separate classes.
Average of CpG observed/expected (O/E) value around putative transcription start site. The average of CpG O/E ratio for each transcription start site (from 3 kb upstream to 0.5 kb downstream) of the 4280 largest-candidate set was plotted. The set contains 919 sequences, which have potential CpG islands surrounding transcription start site, as referred in Table 3. Putative transcription start sites (TSSs) were defined by 5′ boundaries of mapped genomic regions as indicated by an arrow. CpG O/E ratio was calculated every 200-bp window with sliding 20 bp. The formula for producing CpG O/E ratio is described in Methods.
Frequencies of polyadenylation signal-like sequences upstream of 3′ end. Frequencies of polyadenylation signal-like sequences located upstream of 3′ end sites were plotted. The sequence pattern AATAAA/ATTAAA was searched for every position from the 3′ end of the 4280 largest-candidate set. The set contains 1395 sequences, which contain polyA-signal like sequence in the 3′ end, as mentioned in Table 3.
Among the candidate set that could be mapped to the genome, 1150 (26.9%) revealed multiple exons. Several known ncRNAs such as Xist, Gas5, and BIC (Smith and Steitz 1998; Hong et al. 1999; Tam 2001) are known to undergo splicing. Again, this criterion provides a strong indication that the transcript is a genuine product of RNAPol II–mediated transcription and is likely to be functional.
In a separate analysis, the FANTOM2 cDNA set was found to contain >2400 sense–antisense pairs (Okazaki et al. 2002; Kiyosawa et al. 2003). Three hundred twenty-three (7.5%) members of the candidate set are also included in the antisense transcript candidates. Antisense transcripts have been implicated in transcription control, especially in genomic imprinting. For example, AIR, which is an antisense transcript from Igf2r locus, silences three kinds of paternal imprinting genes (Sleutels et al. 2002). In another study (Holmes et al. 2003), new and novel antisense transcripts from among the FANTOM2 set were mapped to the imprinted GNAS locus.
Among the candidate set, 68.0% of the clones fitted at least one of the criteria, and 54.8% of these fitted clones satisfied more than two criteria, as shown in Table 3. Therefore, we believe that the set contains many potential ncRNAs and that a substantial subset will be shown to be functional in some aspect of mammalian biology. Twenty-five strong candidates extracted by our filtering are listed as examples for putative ncRNAs in Table 4. The average length of the longest ORFs of them is 195.2 nt. As an additional index that the transcripts are unlikely to code for even a small peptide, in all of the clones except TF14562 and TF9816, the longest ORFs are not started from the first ATG of each clone, which is normally used as the initiation codon in >90% of mammalian protein coding transcripts. It remains possible that a small subset does, indeed, encode small proteins. Detailed annotation of candidate CDS encoding proteins between 50 and 99 amino acids is described elsewhere (Grimmond et al. 2003), and candidates that arise from this analysis will be eliminated from the candidate set.
Twenty-Five Examples of ncRNA Candidates
EST hit | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
ID |
Chromosome no. |
Length (nt) |
Tissues library |
Mm |
Hs |
Rn |
Hs genome homolog | ||
| TF14562 | Chr1 (4) | 408 | 18-day embryo | 37 | 0 | 0 | None | ||
| TF15290 | Chr10 (5) | 1511 | Testis | 0 | 0 | 0 | None | ||
| TF16305 | Chr10 (4) | 1742 | Testis | 0 | 0 | 0 | 1390 (0.625) | ||
| TF8960 | Chr11 (3) | 1658 | Stomach/C. striatam/cecum | 23 | 12 | 3 | None | ||
| TF8434 | Chr11 (2) | 891 | Cerebellum/embryo10 + embryo11/pancreas | 21 | 0 | 0 | None | ||
| TF15141 | Chr11 (5) | 1878 | Embryo/testis/thymus | 10 | 0 | 0 | None | ||
| TF23639 | Chr12 (1) | 1584 | Testis (embryo15) | 12 | 0 | 2 | 1551 (0.613) | ||
| TF19580 | Chr13 (2) | 2816 | Cerebellum | 1 | 0 | 20 | 2585 (0.663) | ||
| TF9816 | Chr13 (2) | 1152 | C. striatum/embryo 13-head | 15 | 1 | 0 | None | ||
| TF9905 | Chr17 (2) | 350 | Testis | 1 | 0 | 0 | 273 (0.675) | ||
| TF12671 | Chr18 (6) | 1901 | Cecum | 0 | 0 | 0 | None | ||
| TF31326 | Chr18 (1) | 1636 | ES cells/E12 upper body | 7 | 0 | 1 | 1226 (0.659) | ||
| TF28373 | Chr19 (4) | 2349 | N7 Cerebellum/embryo 9/bone | 64 | 1 | 0 | None | ||
| TF14099 | Chr2 (8) | 1174 | Testis | 0 | 0 | 0 | None | ||
| TE70920 | Chr2 (2) | 1318 | U. bladder/N0 thymus | 7 | 0 | 2 | 1160 (0.681) | ||
| TE14398 | Chr3 (1) | 317 | Testis | 1 | 0 | 3 | 223 (0.562) | ||
| TF11833 | Chr2 (3) | 454 | Embryo 8 | 0 | 0 | 0 | 387 (0.872) | ||
| TF27297 | Chr3 (2) | 1321 | Aorta and vein | 1 | 0 | 0 | 1247 (0.752) | ||
| TF12549 | Chr5 (5) | 604 | Head (neonate 6 day)/E12 upper body | 10 | 0 | 0 | None | ||
| TF22931 | Chr5 (6) | 2408 | Forelimb (embryo13)/head | 4 | 0 | 0 | None | ||
| TF33090 | Chr5 (3) | 2703 | N0 eyeball | 0 | 0 | 0 | 2337 (0.715) | ||
| TF13219 | Chr6 (3) | 479 | Testis | 0 | 0 | 0 | 343 (0.724) | ||
| TF13544 | Chr9 (5) | 999 | Embryo10 + embryo11 | 25 | 0 | 0 | None | ||
| TF21967 | Chr9 (2) | 573 | Embryo10 + embryo11 | 3 | 0 | 2 | 571 (0.806) | ||
|
TF8931 |
ChrX (7) |
3433 |
Embryo10 + embryo11/tongue/hippocampus/skin |
12 |
0 |
1 |
None | ||
[i] Examples of ncRNA candidates were listed with their length, mapped chromosome (the number in parenthesis indicates mapped number of exons), tissue library, and number of EST hit (Mm indicates Mus musculus; Hs, Homo sapiens; and Rn, Rattus norvegicus). Status of human genome homology with aligned length and identity (in parenthesis) was also noted. None indicates that no significant homology was observed under the threshold of >70% length, >50% identity. All of these candidates were selected based on visual inspection through FANTOM2 Web interface (http://fantom2.gsc.riken.go.jp/db/), and observed both of CpG islands in their 5′ upstream region of transcription start site and polyadenylation signal-like sequence in the 3′ end
The next stage in further validation of these candidate ncRNAs is documentation of their regulation and expression profiles. A number of candidates are isolated from tissue-specific and stage-specific libraries (Table 4). The ncRNAs that show tissue-restricted expression would clearly be candidates for functions in differentiation and development. Examples in this class would include BC1 and NTAB (French et al. 2001; Muddashetty et al. 2002), which are specifically transcribed in the brain. They make a complex with certain ribonucleoproteins (RNPs) and regulate the RNA translation, transport, and turnover. The FANTOM2 Web interface for all candidate transcripts provides some indication of expression pattern based on the profile of libraries from which ESTs have been identified. The RIKEN project includes systematic cDNA microarray analysis of all of the clones in the FANTOM2 set, and some of this information is already in the public domain (Bono et al. 2003).
In conclusion, the FANTOM2 cDNA collection clearly contains thousands of candidate ncRNAs, a significant subset of which has all the characteristics of an mRNA other than protein-coding function. The era of the central dogma, DNA-RNA-protein, as the major conduit for expression of genome-encoded biological information, is clearly at an end.
METHODS
Computational Screening of the Candidates
The Mouse Representative Transcripts Set (RTS) was used as the nonredundant sequence set for this transcriptome screening. First, the RTS sequences that are defined as non–protein coding TU by FANTOM consortium, that is, RTS that cannot produce proteins in the Representative Protein Set (RPS), were extracted (Okazaki et al. 2002). Homology search with known amino-acid sequences (ftp://us.expasy.org/databases/sp_tr_nrdb/) according to BLASTX (http://www.ncbi.nlm.nih.gov/blast) was performed to eliminate any likelihood of homology with a protein-coding transcript. If the result of BLASTX was produced with e value <1.0e - .05, the clone was eliminated. It should be noted that this criterion would eliminate transcripts from expressed pseudogenes. As the next filtering step, cDNA sequences were aligned to genomic sequence by using BLAST and SIM4 (http://globin.cse.psu.edu/dist/sim4/). If they were aligned at >90% identity over >90% of their length, cDNA clones were kept, otherwise they were discarded. In addition, the prediction of protein-coding regions in genomic sequences by GENSCAN (http://genes.mit.edu/GENSCANinfo.html) was also carried out. The remaining sequences were kept as the candidates, if the entire 10-kb region of genome sequence around the mapped region—the size was determined according to previous work (MacIntosh et al. 2001)—did not overlap with protein-coding regions predicted by GENSCAN.
Further Characterization of the Largest Candidate Set
All of the homology searches with publicly available EST sequences were performed by BLASTN. Only EST sequences with e value <1.0e - 100 were regarded as corresponding homologous mouse ESTs (ftp://ftp.ncbi.nih.gov/blast/db/est_mouse.Z), and sequences with E-values lower than 1.0e - 50 were regarded as the likely human (ftp://ftp.ncbi.nih.gov/blast/db/est_human.Z) and rat (ftp://ftp.ncbi.nih.gov/genomes/R_norvegicus/rn_est.gz) orthologous ESTs. Reverse hits were not considered.
A homology search with human genomic sequences was performed according to BLAST and SIM4 in the identical way as gene mapping to the mouse genome sequence (see Computational Screening of the Candidates). Because there is less selection pressure on contiguous homology of noncoding sequences, hits of 70% of the full-length, and at least 50% of nucleotide identity, were considered significant.
The CpG island analysis was performed according to calculation of CpG O/E ratio and (G+C) content for every 200-bp window with moving 1-bp intervals around 5′ boundaries of aligned region of cDNAs. If the region had (G+C) content ≥50% and CpG O/E ratio ≥ 0.6, it was considered as a CpG island. CpG O/E ratio was calculated by using the Gardiner-Garden and Frommer formula (Gardiner-Garden and Frommer 1987), ([number of CG × N]/[number of C × number of G]), where N denotes the total number of nucleotides in the analyzed sequence. The search of polyadenylation signal is based on statistic pattern search. Two hexamer sequences, AATAAA or ATTAAA, were searched for each 30 nucleotides of the 3′ end.
Notes
[4] Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1011603.
[5] Takahiro Arakawa, Piero Carninci, and Jun Kawai.
[6] Corresponding author. E-MAIL [email protected]; FAX 81 (466) 47-5099.
We acknowledge Dr. I. Yamanaka, Dr. H. Bono, and I. Nikaido for technical support; S. Fujimori, A. Sakurai, and H. Kochiwa for helpful discussions; and M. Chishima for surveys of previously identified ncRNAs.
References
- ↵Argaman, L., Hershberg, R., Vogel, J., Bejerano, G., Wagner, E.G., Margalit, H., and Altuvia, S. 2001. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 11: 941 -950.
- ↵Bono, H., Yagi, K., Kasukawa, T., Nikaido, I., Tominaga, N., Miki, R., Mizuno, Y., Tomaru, Y., Goto, H., Nitanda, H., et al. 2003. Systematic expression profiling of the mouse transcriptome using RIKEN cDNA microarrays. Genome Res. (this issue).
- ↵Bortolin, M.L. and Kiss. T. 1998. Human U19 intron-encoded snoRNA is processed from a long primary transcript that possesses little potential for protein coding. RNA 4: 445-454.
- ↵Burge, C. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78-94.
- ↵Chan, A.S., Thorner, P.S., Squire, J.A., and Zielenska, M. 2002. Identification of a novel gene NCRMS on chromosome 12q21 with differential expression between rhabdomyosarcoma subtypes. Oncogene 21:3029 -3037.
- ↵Eddy, S.R. 2002. Computational genomics of noncoding RNA genes. Cell 109: 137 -140.
- ↵Erdmann, V.A., Szymanski, M., Hochberg, A., Groot, N., and Barciszewski, J. 2000. Non-coding, mRNA-like RNAs database Y2K. Nucleic Acids Res. 28: 197 -200.
- ↵Erdmann, V.A., Barciszewska, M.Z., Hochberg, A., de Groot, N., and Barciszewski, J. 2001. Regulatory RNAs. Cell. Mol. Life Sci. 58: 960 -977.
- ↵French, P.J., Bliss, T.V., and O'Connor, V. 2001. Ntab, a novel non-coding RNA abundantly expressed in rat brain. Neuroscience 108: 207 -215.
- ↵Gardiner-Garden, M. and Frommer, M. 1987. CpG islands in vertebrate genomes. J. Mol. Biol. 196: 261 -282.
- ↵Grimmond, S.M., Miranda, K.C., Yuan, Z., Davis, M.J., Hume, D.A., Yagi, K., Tominaga, N., Bono, H., Hayashizaki, Y., Okazaki, Y., et al. 2003. The mouse secretome: Functional classification of the proteins secreted into the extracellular environment. Genome Res. (this issue).
- ↵Grosshans, H. and Slack, F.J. 2002. Micro-RNAs: Small is plentiful. J. Cell. Biol. 156: 17-21.
- ↵Hatada, I., Morita, S., Obata, Y., Sotomaru, Y., Shimoda, M., and Kono, T. 2001. Identification of a new imprinted gene, Rian, on mouse chromosome 12 by fluorescent differential display screening. J. Biochem. (Tokyo) 130:187 -190.
- ↵Holmes, R., Williamson, C., Peters, J., Denny, P., RIKEN GER Group and GSL Members, and Wells, C. 2003. A comprehensive transcript map of the mouse Gnas imprinted complex. Genome Res. 13: (this issue).
- ↵Hong, Y.K., Ontiveros, S.D., Chen, C., and Strauss, W.M. 1999. A new structure for the murine Xist gene and its relationship to chromosome choice/counting during X-chromosome inactivation. Proc. Natl. Acad. Sci. 96: 6829 -6834.
- ↵Hurst, L.D. and Smith, N.G. 1999. Molecular evolutionary evidence that H19 mRNA is functional. Trends Genet. 15: 134 -135.
- ↵Inoue, A., Kobayashi, Y., Ishizuka, M., Hirose, S., and Hagiwara, H. 2002. Identification of a novel osteoblastic gene, inducible by C-type natriuretic peptide, whose transcript might function in mineralization as a noncoding RNA. Calcif. Tissue Int. 70: 111 -116.
- ↵Kiyosawa, H., Yamanaka, I., Osato, N., RIKEN GER Group and GSL Members, and Hayashizaki, Y. 2003. Antisense transcripts with FANTOM2 clone set and their implications for gene regulation. Genome Res. 13: (this issue).
- ↵Krause, R., Hemberger, M., Himmelbauer, H., Kalscheuer, V., and Fundele, R.H. 1999. Identification and characterization of G90, a novel mouse RNA that lacks an extensive open reading frame. Gene 232: 35 -42.
- ↵Lau, N.C., Lim, L.P., Weinstein, E.G., and Bartel, D.P. 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858 -862.
- ↵Liu, A.Y., Torchia, B.S., Migeon, B.R., and Siliciano, R.F. 1997. The human NTT gene: Identification of a novel 17-kb noncoding nuclear RNA expressed in activated CD4+ T cells. Genomics 39: 171 -184.
- ↵MacIntosh, G.C., Wilkerson, C., and Green, P.J. 2001. Identification and analysis of Arabidopsis expressed sequence tags characteristic of non-coding RNAs. Plant Physiol. 127: 765 -776.
- ↵Muddashetty, R., Khanam, T., Kondrashov, A., Bundman, M., Iacoangeli, A., Kremerskothen, J., Duning, K., Barnekow, A., Huttenhofer, A., Tiedge, H., et al. 2002. Poly(A)-binding protein is associated with neuronal BC1 and BC200 ribonucleoprotein particles. J. Mol. Biol. 321: 433 -445.
- ↵Nesterova, T.B., Slobodyanyuk, S.Y., Elisaphenko, E.A., Shevchenko, A.I., Johnston, C., Pavlova, M.E., Rogozin, I.B., Kolesnikov, N.N., Brockdorff, N., and Zakian, S.M. 2001. Characterization of the genomic Xist locus in rodents reveals conservation of overall gene structure and tandem repeats but rapid evolution of unique sequence. Genome Res. 11: 833 -849.
- ↵Okazaki, Y., Furuno M., Kasukawa T., Adachi J., Bono H., Kondo S., Nikaido I., Osato N., Saito R., Suzuki, H., et al. 2002. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420: 563 -573.
- ↵Pearson, W.R. 1991. Searching protein sequence libraries: Comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics 11: 635 -650.
- ↵Pelczar, P. and Filipowicz, W. 1998. The host gene for intronic U17 small nucleolar RNAs in mammals has no protein-coding potential and is a member of the 5′-terminal oligopyrimidine gene family. Mol. Cell. Biol. 18: 4509 -4518.
- ↵Rivas, E., Klein, R.J., Jones, T.A., and Eddy, S.R. 2001. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr. Biol. 11: 1369 -1373.
- ↵Sharp, P.A. 2001. RNA interference: 2001. Genes & Dev. 15: 485 -490.
- ↵Sleutels, F., Zwart, R., and Barlow, D.P. 2002. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415: 810 -813.
- ↵Smilinich, N.J., Day, C.D., Fitzpatrick, G.V., Caldwell, G.M., Lossie, A.C., Cooper, P.R., Smallwood, A.C., Joyce, J.A., Schofield, P.N., Reik, W., et al. 1999. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc. Natl. Acad. Sci. 96: 8064 -8069.
- ↵Smith, C.M. and Steitz, J.A. 1998. Classification of gas5 as a multi-small-nucleolar-RNA (snoRNA) host gene and a member of the 5′-terminal oligopyrimidine gene family reveals common features of snoRNA host genes. Mol. Cell. Biol. 18: 6897 -6909.
- ↵Storz, G. 2002. An expanding universe of noncoding RNAs. Science 296: 1260 -1263.
- ↵Sutherland, H.F., Wadey, R., McKie, J.M., Taylor, C., Atif, U., Johnstone, K.A., Halford, S., Kim, U.J., Goodship, J., Baldini, A., et al. 1996. Identification of a novel transcript disrupted by a balanced translocation associated with DiGeorge syndrome. Am. J. Hum. Genet. 59: 23 -31.
- ↵Takai, D. and Jones, P.A. 2002. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. 99: 3740 -3745.
- ↵Tam, W. 2001. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene 274: 157 -167.
- ↵Tang, T.H., Bachellerie, J.P., Rozhdestvensky, T., Bortolin, M.L., Huber, H., Drungowski, M., Elge, T., Brosius, J., and Huttenhofer, A. 2002. Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus. Proc. Natl. Acad. Sci. 99: 7536 -7541.
- ↵Tycowski, K.T., Shu, M.D., and Steitz, J.A. 1996. A mammalian gene with introns instead of exons generating stable RNA products. Nature 379: 464 -466.
- ↵Velleca, M.A., Wallace, M.C., and Merlie, J.P. 1994. A novel synapse-associated noncoding RNA. Mol. Cell. Biol. 14: 7095 -7104.
- ↵Wang, Y., Crawford, D.R., and Davies, K.J. 1996. adapt33, a novel oxidant-inducible RNA from hamster HA-1 cells. Arch Biochem. Biophys. 332: 255 -260.
- ↵Wassarman, K.M. 2002. Small RNAs in bacteria: Diverse regulators of gene expression in response to environmental changes. Cell 109: 141 -144.
- ↵Wassarman, K.M., Repoila, F., Rosenow, C., Storz, G., and Gottesman, S. 2001. Identification of novel small RNAs using comparative genomics and microarrays. Genes & Dev. 15: 1637 -1651.
- ↵Wevrick, R. and Francke, U. 1997. An imprinted mouse transcript homologous to the human imprinted in Prader-Willi syndrome (IPW) gene. Hum. Mol. Genet. 6: 325-332.
WEB SITE REFERENCES
- ↵http://biobases.ibch.poznan.pl/ncRNA/; Noncoding RNAs database.
- ↵ftp://us.expasy.org/databases/sp_tr_nrdb/; data set for known protein sequences.
- ftp://ftp.ncbi.nih.gov/blast/db/; database of mouse EST sequences and human EST sequences.
- ftp://ftp.ncbi.nih.gov/genomes/R_norvegicus/; database of rat EST sequences.
- ↵http://www.ncbi.nlm.nih.gov/blast; executable files of BLASTN and BLASTX.
- ↵http://globin.cse.psu.edu/dist/sim4/; SIM4, a program for gene mapping to the genomic sequences.
- ↵http://genes.mit.edu/GENSCANinfo.html; GENSCAN, a program for ORF prediction from genomic sequences.
- ↵http://fantom2.gsc.riken.go.jp/db/; Web interface for FANTOM2 database.