
Physical Maps for Genome Analysis of Serotype A and D Strains of the Fungal Pathogen Cryptococcus neoformans
- Jacqueline E. Schein1,4,
- Kristin L. Tangen2,4,
- Readman Chiu1,
- Heesun Shin1,
- Klaus B. Lengeler3,
- William Kim MacDonald2,
- Ian Bosdet1,
- Joseph Heitman3,
- Steven J.M. Jones1,
- Marco A. Marra1, and
- James W. Kronstad2,5
- 1Genome Sciences Centre, BC Cancer Agency, Vancouver, British Columbia V5Z 4E6, Canada; 2Biotechnology Laboratory, Department of Microbiology and Immunology, and Faculty of Agricultural Sciences, The University of British Columbia, Vancouver, British Columbia V6T 1Z3, Canada; 3Departments of Genetics, Pharmacology and Cancer Biology, and Microbiology and Medicine, Howard Hughes Medical Institute, Duke University Medical Center, Durham, North Carolina 27710, USA
Abstract
The basidiomycete fungus Cryptococcus neoformans is an important opportunistic pathogen of humans that poses a significant threat to immunocompromised individuals. Isolates of C. neoformans are classified into serotypes (A, B, C, D, and AD) based on antigenic differences in the polysaccharide capsule that surrounds the fungal cells. Genomic and EST sequencing projects are underway for the serotype D strain JEC21 and the serotype A strain H99. As part of a genomics program for C. neoformans, we have constructed fingerprinted bacterial artificial chromosome (BAC) clone physical maps for strains H99 and JEC21 to support the genomic sequencing efforts and to provide an initial comparison of the two genomes. The BAC clones represented an estimated 10-fold redundant coverage of the genomes of each serotype and allowed the assembly of 20 contigs each for H99 and JEC21. We found that the genomes of the two strains are sufficiently distinct to prevent coassembly of the two maps when combined fingerprint data are used to construct contigs. Hybridization experiments placed 82 markers on the JEC21 map and 102 markers on the H99 map, enabling contigs to be linked with specific chromosomes identified by electrophoretic karyotyping. These markers revealed both extensive similarity in gene order (conservation of synteny) between JEC21 and H99 as well as examples of chromosomal rearrangements including inversions and translocations. Sequencing reads were generated from the ends of the BAC clones to allow correlation of genomic shotgun sequence data with physical map contigs. The BAC maps therefore represent a valuable resource for the generation, assembly, and finishing of the genomic sequence of both JEC21 and H99. The physical maps also serve as a link between map-based and sequence-based data, providing a powerful resource for continued genomic studies.
[This paper is dedicated to the memory of Michael Smith, Founding Director of the Biotechnology Laboratory and the BC Cancer Agency Genome Sciences Centre. Supplemental material is available online at http://www.genome.org.]
The basidiomycete fungus Cryptococcus neoformans is capable of causing serious infections in immunocompromised and immunocompetent people. Infection is initiated upon inhalation of fungal spores or yeast cells, and dissemination can occur to numerous sites including the skin, bones, and the central nervous system. C. neoformans frequently causes meningioencephalitis, and this manifestation of cryptococcosis occurs in ∼10% of AIDS patients. The ability of C. neoformans to cause disease has been associated with a number of virulence factors including the ability to grow at 37°C, the elaboration of a polysaccharide capsule, melanin production, and the MATα mating-type locus (Mitchell and Perfect 1995; Casadevall and Perfect 1998).
Isolates of C. neoformans have been divided into three varieties known as grubii (serotype A), neoformans(serotype D), and gattii (serotypes B and C). The serological separations for these groups are defined primarily on the basis of antigenic differences in the capsular polysaccharide. Molecular phylogenetic work revealed that the grubii andneoformans varieties are separated by ∼18.5 million years of evolution, and these varieties differ from the gattii variety by ∼37 million years (Xu et al. 2000). Serotypes A and D and a hybrid AD serotype are found worldwide; in contrast, serotypes B and C are mainly restricted to tropical and subtropical regions, although isolates with these serotypes can also be obtained from temperate regions (Mitchell and Perfect 1995; Sorrell 2001). The majority of clinical isolates in North America are serotype A strains; serotype D strains are more prevalent in specific European countries, for example, Denmark, France, and Italy (Bennett et al. 1984; Dromer et al. 1996;Franzot et al. 1998). Several studies have attempted to characterize the differences between serotype A and D strains. Thus, isolates have been characterized with respect to 26S rRNA sequences, PCR fingerprinting, enzyme electrophoretic profiles, and electrophoretic karyotypes (Perfect et al. 1989, 1993; Brandt et al. 1993; Guehó et al. 1993; Meyer et al. 1993; Wickes et al. 1994; Boekhout and van Belkum 1997; Bertout et al. 1999). In a recent study, strains of serotype A and D were distinguished by the RFLP patterns obtained upon hybridization with a repeated element (CNRE-1) and by the nucleotide sequence analysis of specific genes (e.g., URA5; Franzot et al. 1998; Xu et al. 2000). Xu et al. (2000) have performed a more detailed phylogenetic analysis of strains from the different serotypes using sequence analysis of the mitochondrial large ribosomal subunit, the internal transcribed spacer region of nuclear rRNA, and the genes encoding orotidine monophosphate pyrophosphorylase (URA5) and diphenol oxidase (CnLAC). This work supports the current separation of strains into the three varieties and provides a phylogenetic framework for understanding the evolution and geographic distribution of C. neoformans. The population structure ofC. neoformans has also been examined in detail using AFLP genotyping (Boekhout et al. 2001) and PCR fingerprinting (Meyer et al. 1999; Ellis et al. 2000). Finally, although strains of serotype A and D have been shown to be genetically distinct, it is possible to obtain mating between isolates from the two different serotypes (Kwon-Chung 1975).
Several groups have performed electrophoretic karyotyping to determine the size and number of chromosomes in strains representing the different varieties of C. neoformans (Perfect et al. 1989,1993; Polacheck and Lebens 1989; Wickes et al. 1994; Boekhout and van Belkum 1997; Boekhout et al. 1997; Forche et al. 2000). In addition,Wickes et al. (1994) and Spitzer and Spitzer (1997) assigned several markers to electrophoretically separated chromosomes by hybridization with known genes or ESTs. Overall, the current view of the karyotype inC. neoformans indicates a genome size in the range of 15 to 27 Mb with an average chromosome number of 12 for varietyneoformans and 13 for variety gattii. Forche et al. (2000) recently described the construction of a meiotic linkage map forC. neoformans serotype D. A mapping population of 100 progeny was used with a total of 181 AFLP, RAPD, and gene markers to identify 14 major linkage groups. Six of the linkage groups were assigned to specific chromosomes.
We initiated a physical characterization of the C. neoformansgenome as part of an international effort to obtain the complete genomic sequence of two strains representing the A and D serotypes (Heitman et al. 1999). Genomic shotgun sequencing for C. neoformans is presently underway at the Stanford Genome Technology Center, The Institute for Genome Research (TIGR), and the Duke University Center for Genome Technology. As described in this report, we used the bacterial artificial chromosome (BAC) fingerprinting technology first described by Marra et al. (1997) to generate large contigs that will form the framework for assembly and finishing of the genomic sequence for the serotype A and D strains. We also sequenced the ends of the fingerprinted BAC clones and contributed the traces to the shotgun sequence databases for both strains. Our mapping approach has been used previously for whole-genome, random BAC clone fingerprinting projects that supported sequencing of theArabidopsis thaliana (Marra et al. 1999; Mozo et al. 1999) and human (McPherson et al. 2001) genomes. Finally, we placed markers on the BAC maps and used these markers both to compare the conservation of synteny between the serotype A and D strains and to attempt to correlate BAC clone contigs with specific chromosomes.
RESULTS AND DISCUSSION
Construction of Fingerprinted BAC Clone Physical Maps
Genomic DNA was isolated from H99 and JEC21, and a BAC library was constructed for each strain (see Methods). For each library, a total of 3072 bacterial clone glycerol stocks arrayed randomly into eight 384-well plates were processed for fingerprint map construction. Each BAC clone was fingerprinted to determine the number and size ofHindIII restriction fragments contained in the insert. Fingerprints were successfully obtained for 2642 JEC21 clones and 2612 H99 clones. The average insert size for fingerprinted clones in the JEC21 library was 108,560 bp and in the H99 library 107,648 bp, as determined by the fingerprint analysis. A fingerprint database for each library was created and analyzed using the program FPC(Soderlund et al. 1997, 2000; Ness et al. 2002;http://www.genome.clemson.edu/fpc/). A high-stringency automated assembly was first performed in FPC to bin together clones with substantial overlap based on shared restriction fragments. To maximize the likelihood that each bin represented a region of contiguous DNA, or contig, a minimum of 85%–90% shared restriction fragments was required for clones to be binned together. The automated fingerprint assembly resulted in the creation of 276 contigs in the JEC21 database and 261 contigs in the H99 database. Additional contig integrity was achieved by manual interrogation and editing of each contig via tools within the FPC software, using fingerprint similarities to refine clone order and clone overlaps. Clones with fingerprints that appeared to be contaminated (comprised of DNA from more than one clone) and partially digested clones were removed from the database during the editing process. Following the refined positioning of clones within all contigs, clones at the ends of each contig were compared with all other clones within theFPC database at a reduced minimum required fingerprint overlap (∼50% shared restriction fragments) to identify potential joins between contigs. Potential joins between contig ends were manually examined and permitted only where the joins did not result in inconsistencies in the fingerprint data. Upon completion of these manual edits, the JEC21 map contained 2322 clones, the H99 map contained 2529 clones, and each map had been assembled into 20 sequence-ready contigs. An example of the FPC display for the contig that carries the mating-type locus (MATα) for JEC21 is provided as supplementary data in Supplementary Figure 1 (available online at http://www.genome.org).
For the JEC21 map, the contigs range in size from 184,760 bp (6 clones) to 1,748,127 bp (321 clones). For H99, the smallest contig (84,272 bp) contains two clones, and the largest of 1,356,533 bp contains 246 clones. Summaries of the 20 assembled contigs for each strain are provided as Supplementary Tables A and B (available online athttp://www.genome.org). These tables also list the number of markers that mapped to each contig by hybridization (see below) and the estimated total amount of DNA represented in each of the assembled contigs for both strains. With regard to genome size, our estimates of 15.79 Mb for JEC21 and 15.55 Mb for H99 are at the lower end of the range (15–27 Mb) estimated from electrophoretic karyotyping of different C. neoformans strains (Perfect et al. 1989; Wickes et al. 1994; Boekhout et al. 1997).
The genome sizes estimated from the maps are likely to be underestimates because there may be areas of the genomes that are not represented in the BAC libraries. These may include areas that are difficult to clone or maintain in Escherichia coli such as telomere or centromere sequences or areas with an unusual distribution of HindIII sites. Of course, estimates of genome size from electrophoretic karyotyping experiments can also be confounded by problems with chromosome size determination and the comigration of different chromosomes. These issues will be resolved as the shotgun sequence data are combined with the physical mapping information for JEC21. This approach is presently underway at the Institute for Genome Research (TIGR), and the estimated genome size is in the range of 19 to 23 Mb (http://www.tigr.org/tdb/edb2/crypt/htmls/index.shtml).
The availability of fingerprinted BAC data from two closely related strains allowed a test of whether contigs could be assembled with orthologous clones from each genome. Specifically, FPC was used to analyze the combined set of fingerprints from both strains. No contigs could be generated that were composed of clones from both strains. Thus, the genomes of the serotype A strain H99 and the serotype D strain JEC21 are sufficiently divergent to preclude analysis of synteny based on HindIII restriction digestion patterns.
Finally, BAC clones comprising a minimally overlapping tiling set were manually selected for each contig in both databases. Great care was taken to ensure that shared restriction fragments could be identified in the fingerprints of overlapping clone pairs. The selected tiling path clones represent a collection of overlapping clones covering the genomes of JEC21 (165 tiling path clones) and H99 (163 tiling path clones). These tiling sets will therefore be useful for assembling and finishing the genomic sequences of these strains.
Comparison of a Genome Shotgun Sequence Assembly to the Fingerprint Map of JEC21
The BAC fingerprint contigs for strains JEC21 and H99 represent the first genome-wide physical maps for C. neoformans, and provide a minimum tiling path of BAC clones for systematic sequencing of the genomes of these strains. As mentioned above, shotgun sequencing projects for JEC21 are in progress at Stanford University and TIGR, and a limited shotgun-sequencing project is underway for H99 at Duke University. Correlation of shotgun sequence data with fingerprinted BAC clones would allow the contigs to provide a framework on which to assemble the existing shotgun sequence data for both strains. To facilitate the alignment of the physical maps with the emerging genomic sequence data, we sequenced the ends of the fingerprinted BAC clones and contributed the traces to the shotgun sequence databases for both strains.
BAC-end sequence reactions were performed for both ends of all 3072 clones from each fingerprinted BAC library, for a total of 6144 attempted BAC-end reads per strain. A total of 4772 (78%) successful BAC-end sequences were obtained for JEC21 BACs with an average read length of 540 bp (see Methods). Of the successful reads, 4186 were derived from clones that had fingerprints in the map. Of the fingerprinted JEC21 BAC clones with successful BAC-end sequences, 1939 had both ends represented in the data set (3878 total end reads, or 93%), and 308 clones had a single associated end read. For H99 clones, 4908 (80%) successful BAC-end sequences were obtained with an average read length of 560 bp (see Methods). Of these successful reads, 4390 were derived from clones that had fingerprints in the H99 map. For the fingerprinted H99 BACs with end sequences, 1957 had sequences represented from both ends (3914 total reads, or 89%), and 476 had a single associated end read. The BAC-end sequences are available athttp://www.bcgsc.bc.ca and have been incorporated into the shotgun sequence assemblies at the Stanford Genome Technology Center (http://www-sequence.stanford.edu/group/C.neoformans), TIGR (http://www.tigr.org/tdb/edb2/crypt/htmls/index.shtml), and the Duke University Center for Genome Technology (http://cgt.genetics.duke.edu/data/index.html).
We undertook the correlation of JEC21 BAC-end sequences derived from mapped BACs with the JEC21 whole-genome shotgun sequence assembly generated at TIGR, representing nominally 3.5-fold coverage of theC. neoformans genome and including the BAC-end sequence data. Each of the BAC-end sequences was compared with the complete set of genomic sequence assembly contigs using the BLASTalgorithm (Altschul et al. 1990). Only those alignments that satisfied the criteria of a minimum 95% sequence identity across 90% of the high-quality portion of the BAC-end sequence were selected for further analysis. The subset of TIGR assembly contigs remaining for analysis could be classified into one of four groups. The first group contains unique matches between a TIGR sequence assembly contig and a BAC-end sequence, that is, each sequence assembly contig in this group aligned to a single BAC-end sequence and vice versa. The second category contains TIGR sequence assembly contigs that had alignments with multiple BAC-end sequences, where the corresponding BAC clones were all in the same physical map contig. The TIGR sequence assembly contigs in this second category therefore have good evidence supporting their correlation to a specific fingerprint contig. Alignments classified into the third category were those where more than one TIGR sequence assembly contig aligned with the same BAC-end sequence. This situation could potentially result from duplicated regions of the genome or possibly from misassembled shotgun sequence contigs. The fourth category contained TIGR sequence assembly contigs that aligned with multiple BAC-end sequences derived from BACs in many different fingerprint contigs. This category most likely contains sequences derived from regions of the genome containing repeat sequences. The alignments in the first and second categories represent unambiguous correlations between physical map contigs and whole-genome shotgun assemblies (the results of the data from these two categories are summarized in Supplementary Table C, available online athttp://www.genome.org). The sum of the TIGR shotgun sequence assemblies correlated to each fingerprint contig is calculated, as is the overall coverage of the contig based on the estimated contig size. Using this methodology, 7,643,886 bases of TIGR shotgun sequence were unambiguously correlated with the fingerprint contigs, or 48% coverage of the physical map.
Comparison of the H99 and JEC21 Fingerprint Maps
Hybridization experiments were performed to place markers for known genes, ESTs, and BAC ends onto the maps to identify corresponding contigs between the maps and to examine the conservation of synteny between the strains. Hybridization data from probes derived from BAC-end sequences were used as additional evidence for the identification and evaluation of potential contig merges. A summary of the marker data from the hybridization experiments is presented in Table 1. Three sets of probes were used to identify hybridizing clones arrayed as a set of 9216 BACs from the JEC21 library and 6528 BACs from the H99 library on a high-density filter. First, a set of 96 Overgo (Ross et al. 1999; Methods) probes (40-mers) was used in a pooled format to rapidly match genes and ESTs with contigs. Second, Overgo probes to BAC-end sequences were used to fill in missing data in the cross-reference analysis of the contigs. Finally, Overgo and plasmid-derived probes for specific markers linked to the electrophoretic karyotype (Spitzer and Spitzer 1997) were also used in an attempt to match contigs with specific chromosome-sized bands. Note that more BAC clones were available on the high-density filter than were fingerprinted. Overall, 82 and 102 markers were placed on contigs for JEC21 and H99, respectively. Shared markers were found for 17 and 18 of the 20 contigs for JEC21 and H99, respectively. Note that no markers were found for contigs 2 and 17 of JEC21, but markers were found for all 20 contigs in strain H99. Hybridization with two different probes for C. neoformans rDNA sequences revealed that BAC clones carrying rDNA genes were found in contig 7 for JEC21, but were not present in the 6528 clones from the H99 BAC library. This result may indicate that the rDNA sequences from H99 have a different organization of HindIII restriction sites that precluded cloning of the region. DNA blot analysis of complete HindIII digests of H99 and JEC21 genomic DNA revealed that the H99 rDNA contains fewer HindIII restriction sites (data not shown), which may explain its inability to be cloned in pBeloBAC11 using our protocols. Lists of the sequences that were used as probes for each marker are provided in Supplementary Table D (available online athttp://www.genome.org).
Summary of the Hybridization of Selected Markers to BAC Clones of H99 and JEC21
As shown in Figure 1, the hybridization data and the fingerprint information allowed us to match 18 of the 20 H99 contigs to 17 of the 20 JEC21 contigs. Markers were used for the comparison only in situations where they could confidently be mapped to a single contig. The comparisons of marker positions between the maps revealed considerable conservation of synteny between the strains, with several clusters of markers showing identical order. In addition to the overall similarities between the two maps, the conservation of marker order was particularly striking for contigs 13 (JEC21) and 3 (H99), for contigs 19 (JEC21) and 9 (H99), and for contigs 11 (JEC21) and 5 (H99). The hybridization probes for genes known to be in the MATαlocus identified contig 11 in JEC21 and contig 5 in H99 as arising from the mating-type chromosome (Fig. 1; see below). The MATαregion has been the focus of targeted sequencing (Karos et al. 2000;Lengeler et al. 2002) in part because of the association of the α mating type with virulence (Kwon-Chung et al. 1992). The combined FPC and hybridization data also revealed several examples of rearrangements between the two genomes. Specifically, we identified eight markers whose positions did not agree between the two maps relative to flanking markers (Fig. 1; Spi25, Spi35, a1b07cn.r1, a1e04cn.f1, H003C03.F, URA5, and H003M21.R). The positions of some of these markers indicate the presence of an inverted region between the genomes (e.g., Spi25 and Spi35), whereas the positions of others indicate translocations (e.g., a1b07cn.r1 and a1e04cn.f1).
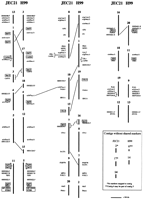
Conservation of synteny between the genomes of JEC21 and H99. The 20 contigs for each map are shown with the markers found on each contig. Note that only markers that mapped to contigs in both strains are shown. Contigs are represented as vertical lines with the contig numbers above. The relative positions of the markers within the contigs are approximated based on hybridization results. Where the relative positions of adjacent markers in a contig could not be reliably interpreted they are grouped by a square bracket. Corresponding contigs from JEC21 and H99, based on marker content and order, are placed adjacent to each other for comparison. Differences in marker locations between the two strains are also indicated. No markers were found for the JEC21 contigs 2 and 17; these contigs, along with contig 14 and the H99 contigs 6 and 14, did not have shared markers between the genomes. The boxed and shaded markers also were used to relate the contigs to electrophoretically separated chromosomes (Fig. 2).
The locations of specific sets of markers between the two maps also implies that certain contigs may be regions from the same chromosome. For example, the comparisons of shared markers between the maps indicate that contigs 1, 3, and 13 could be regions on the same chromosome for JEC21, and contigs 3 and 17 could be joined in H99. Similar connections are indicated for contigs 10 and 15, and contigs 7 and 9 for JEC21. For H99, contigs 4 and 18, 12 and 15, 1 and 19 and 7, and 8 and 16 could be joined; the mapping data also indicate that contig 6 could be joined to contig 5, which contains theMATα locus. Overall, these joins would reduce the number of contigs to ∼15 for the H99 map and ∼17 for the JEC21 map. For comparison, the reported range for chromosome number for the majority of strains of C. neoformans is between 11 and 14 (Boekhout et al. 1997).
Relationship of Specific Contigs to Chromosome-Sized Bands From the C. neoformans Electrophoretic Karyotype
The positions of specific markers on the contigs were also compared with the published locations of the same markers on electrophoretically separated chromosomes and the meiotic map. For this analysis, the electrophoretic karyotypes of two progenitors of JEC21, B3501 and NIH12, were used to represent the chromosomes because these strains were used for previous hybridization experiments (Fig.2A; Wickes et al. 1994; Spitzer and Spitzer 1997). The patterns of chromosome-sized bands for these strains appear to be similar or identical to the pattern of JEC21, as determined byLengeler et al. (2000). The hybridization probes also included the genes URA5, CAP64, CnLAC, andSTE20α that have been placed on the meiotic map by Forche et al. (2000). The JEC21 contigs that hybridized with markers previously assigned to specific chromosomes are shown in Figure 2A. Our results indicate that the three largest contigs (1, 7, and 11) from JEC21 contain the same markers that map to the three largest chromosome-sized bands in the two other serotype D strains. The chromosome represented by the third largest band in these strains contains the MATlocus, and this chromosomal location has also been established in JEC21 (Lengeler et al. 2000).
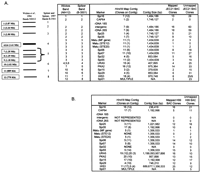
Relationship between electrophoretically separated chromosomes and the contigs of the JEC21 and H99 maps. (A) Diagrammatic representation of the electrophoretically separated chromosomes from the serotype D strains B-3501 and NIH12 (Wickes et al. 1994; Spitzer and Spitzer 1997). The panel on the right shows the contigs of JEC21 that hybridized to the same markers used by Spitzer and Spitzer (1997)and Wickes et al. (1994) to identify specific chromosomes or groups of chromosomes. (B) Locations of chromosome-specific markers on mapped and unmapped clones and specific contigs of the serotype A strain H99. The number of bacterial artificial chromosome (BAC) clones that hybridized with each marker is also presented for both the mapped BAC clones and the additional clones (unmapped) that were present on the high-density filter.
The rDNA markers are present on the second largest chromosome-sized band in strains B3501 and NIH12 (Wickes et al. 1994). Similarly, we found that the rDNA probes hybridized to the second largest band on a blot of separated chromosomes (data not shown) and to contig 7 in JEC21 (Fig. 2A). These results are in agreement with the hybridization data obtained by Wang et al. (2001) for the CPA1 and CPA2genes; these genes hybridize to the second largest band in JEC21 and are found with the rDNA on contig 7 (Fig. 2A). However, we found that the HIS3 probe hybridized to a band equivalent to chromosome 7 (data not shown), in contrast to the reported location of this gene on band 11 (Wickes et al. 1994). This result indicates that there are differences in the locations of some markers for JEC21 when compared with progenitor strains B-3501 and NIH12. We also found that the Spi01 probe hybridized to multiple contigs, whereas Spitzer and Spitzer (1997) found that this marker is located on the smallest chromosome in B-3501. These observations indicate that caution should be exercised for some of the comparisons because of possible differences in the karyotypes between JEC21 and the progenitor strains. In this regard, several reports have described the variability of the karyotype inC. neoformans (Perfect et al. 1989; Wickes et al. 1994;Boekhout and van Belkum 1997). However, in addition to correlating contigs with the largest chromosomes, the hybridization data may provide insight into the contigs that represent the smallest chromosomes; this information may have utility for the analysis of chromosome structure in C. neoformans. For example, both contig 4 and the 10th chromosome-size band of JEC21 hybridize with the Spi29 marker and have similar sizes (0.853 Mb vs. 1.02 Mb, respectively). Thus, contig 4 may represent most of chromosome 10 if the karyotypes are equivalent for B-3501 and JEC21 for this band (Fig.2A).
The markers for rDNA and the MAT locus are found on the largest chromosome-size band in the serotype A strain NIH371 (Wickes et al. 1994). The MAT locus is known to be on the second largest karyotype band in H99 (Lengeler et al. 2000), and our hybridization data link this chromosome with contig 5 in the BAC map (Fig. 2B). Results with the CAP64 and Spi35 markers also indicate that contig 17 represents part of one of the largest chromosomes in H99; this conclusion is supported by the comparison of the conservation of synteny (Fig. 1) because contig 17 of H99 shares two other markers with contig 1 of JEC21. We noted earlier that the rDNA markers did not hybridize to any of the clones in the H99 BAC library; the rDNA cluster is therefore not represented on the contig map. However, hybridization of the rDNA probes to electrophoretically separated chromosomes did locate the sequences on the largest band (data not shown). Furthermore, the location of the rDNA region between contigs 8 and 16 in H99 is indicated by the comparisons of the shared markers; that is, these contigs carry the markers Spi16/H002K09.F and CPA1 that flank the rDNA on contig 7 in JEC21 (Fig. 1). For H99, the URA5 andHIS3 probes each hybridized to two contigs, although clones from one of the two contigs (19 for URA5 and 11 forHIS3) were more frequently detected. These results indicate that cross-hybridizing sequences may be present for these markers.
Summary
The BAC fingerprint maps described here for strains JEC21 and H99, along with the sequences of the ends of the mapped clones, provide a partial framework for the completion of the genomic sequences of these strains. The maps with the minimum tiling set of BAC clones for the genome and the end sequences of the BAC clones have been contributed to the genomic sequencing effort already underway for JEC21. The maps also provide the first comparison of the conservation of synteny between the genomes of C. neoformans strains from the A and D capsular serotypes that represent varieties grubii andneoformans, respectively. Furthermore, the maps provide the opportunity to use arrays of the minimum tiling sets of BAC clones to make comparisons between genomes from different isolates from the same or different varieties. This approach has been used successfully to explore genome variability in the Mycobacterium complex (Gordon et al. 1999).
METHODS
BAC Clone Fingerprinting
C. neoformans DNA for the construction of the BAC libraries was isolated as previously described (Lengeler et al. 2000), and the libraries were prepared at ResGen in the BAC vector pBeloBAC11 (Wang et al. 1997). For the BAC clones that were fingerprinted, the average insert size was reported to be 114.54 kb for the H99 library and 110.74 kb for the JEC21 library (based on a sample of clones). High-throughput, agarose gel-based BAC fingerprinting, fingerprint map assembly, and manual editing were performed as described previously (Marra et al. 1997, 1999; McPherson et al. 2001; J. Schein et al. 2002) with the exception that restriction fragment identification, fragment mobility, and size determination were performed using recently developed automated analysis software (D. Furhmann, S. Jones, J. Schein, and M. Marra, unpubl.).
Contig Size Estimation
An automated algorithm was used to compare the restriction fragments of overlapping clone pairs in the tiling clone sets selected for each contig. The unique fragments for each tiling path clone were identified, and their sizes were summed to estimate the overall size of the contigs. Specifically, the algorithm performed the following for each contig: (1) add the sizes of all the fragments in the left-most tiling path clone in the contig to create a cumulative size estimate; (2) identify the next left-most tiling path clone and identify its unique fragments (any fragments not shared with the previous clone), then add those sizes to the cumulative size estimate; (3) repeat step 2 until all unique fragments in the tiling path clones have been identified and summed to give a total size estimate. Shared fragments are as defined by the FPC parameters used such that two fragments are considered the same if their calculated mobilities are within 7 mobility units of each other.
BAC-End Sequencing
The BAC DNA isolated for fingerprinting was of sufficient quality for the generation of end sequence data. The protocol for BAC-end sequencing reactions was provided by Shaying Zhao (TIGR) and is available at the Web sitehttp://www.tigr.org/tdb/bac_ends/mouse/bac_end_intro.html. The data were collected on ABI Prism 3700 DNA Analyzer sequencing instruments. The trace data were processed by the program phred (Ewing and Green 1998a,b) using default parameters and the sequence trimmed for quality and vector. Reads that contained <15 bp of sequence following processing were removed from the data set. Average read lengths were calculated from the quality length reported byphred for each read.
BAC-End Sequence Alignments to TIGR Shotgun Sequence
Nucleotide sequence comparisons of BAC-end sequences to the whole-genome shotgun assembly contigs at TIGR (July 25, 2001 assembly; 3.5× coverage) were performed using BLAST (Altschul et al. 1990). Default parameters were used with the exceptions that the repeat masking function was turned off to include polynucleotide runs in the alignment length score, and the word size was set to 32.
Hybridization to BAC Clones
The Overgo method as developed by J.D. McPherson (Ross et al. 1999; Vollrath 1999) and the software program Overgo Maker(http://www.genome.wustl.edu/gsc/overgo/overgo.html) were used to design 123 probes for hybridization to the fingerprinted BAC clones. The sequences for the hybridization probes originated from knownC. neoformans genes in GenBank (http://www.ncbi.nlm.nih.gov), putative genes identified in the JEC21 genomic database at the Stanford Genome Technology Center (SGTC;http://baggage.stanford.edu/group/C.neoformans/), expressed sequence tags (ESTs; http://www.genome.ou.edu/cneo.html), karyotype markers (Spitzer and Spitzer 1997), and BAC-end sequences (http://www.bcgsc.bc.ca). The 40-mer Overgo probes were checked for redundancy by searching against the JEC21 genomic database (http://baggage.stanford.edu/group/C.neoformans/) and the H99 EST database (http://www.genome.ou.edu/cneo.html) with theBLASTn algorithm. Oligonucleotides were purchased from GIBCO BRL and from the Nucleic Acid and Protein Service Facility (NAPS) at the Biotechnology Laboratory, University of British Columbia. Sequences for the PKA2 and HIS3 genes were on a 4.5-kb fragment (PKA2; pCD49) and a 604-bp cDNA in a TOPO TA vector (HIS3; pMJB54). Overgo probe labeling was performed using the Overgo protocol (Ross et al. 1999), and plasmid-derived DNA fragments were labeled with an Oligonucleotide Labeling Kit (Amersham Pharmacia Biotech.). Detailed information about the hybridization probes can be found in the Supplementary Material (available online athttp://www.genome.org).
High-density filters containing C. neoformans BAC clones were purchased from ResGen. The Overgo protocol was used for hybridization (Ross et al. 1999) except that free nucleotides were removed with a nucleotide removal kit (QIAGEN) and filter washes were performed in 50 mL of 4× SSC/0.1% SDS, 1.5× SSC/0.1% SDS, and 0.75% SSC/0.1% SDS at 55°C. Filters were exposed to film for 3 d at −80°C.
Data Availability
The fingerprint maps are available in FPC format at the Web site of the BC Genome Sciences Centre (http://www.bcgsc.bc.ca). There the maps can be viewed with Internet Contig Explorer (iCE), and the BAC-end sequences are also available.
WEB SITE REFERENCES
http://baggage.stanford.edu/group/C.neoformans/; JEC21 genomic database at the Stanford Genome Technology Center.
http://cgt.genetics.duke.edu/data/index.html; sequence information for strain H99 at the Duke University Center for Genome Technology.
http://www.bcgsc.bc.ca; fingerprint maps and BAC-end sequences at the British Columbia Genome Sciences Centre.
http://www.genome.clemson.edu/fpc/; FPC program to create fingerprint databases.
http://www.genome.ou.edu/cneo.html; C. neoformans EST data at the University of Oklahoma Advanced Center for Genome Technology.
http://www.genome.wustl.edu/gsc/overgo/overgo.html; Overgo Maker to design hybridization probes.
http://www.ncbi.nlm.nih.gov; National Center for Biotechnology Information, GenBank.
http://www-sequence.stanford.edu/group/C.neoformans/; sequence data for strain JEC21 at the Stanford Genome Technology Center.
http://www.tigr.org/tdb/bac_ends/mouse/bac_end_intro.html; protocols for BAC DNA sequencing reactions.
http://www.tigr.org/tdb/edb2/crypt/htmls/index.shtml; sequence data for strain JEC21 at the The Institute for Genome Research (TIGR) Web site.
Acknowledgments
The authors thank Steven Ness for the automated algorithms for contig size calculations. We also thank members of the mapping and sequencing groups of the BC Cancer Agency Genome Sequence Centre (J. Asano, Y. Butterfield, S. Chan, S. Chittaranjan, C. Fjell, N. Girn, C. Gray, R. Guin, M. Krzywinski, R. Kutsche, S. Leach, D. Lee, S. Lee, C. Mathewson, C. McLeavy, S. Ness, T. Olson, P. Pandoh, A. Prabhu, P. Saeedi, D. Smailus, L. Spence, J. Stott, S. Taylor, M. Tsai, N. Wye, and G. Yang) for their contributions to this work. The authors gratefully acknowledge Richard Hyman, Eula Fung, Don Rowley, and Ron Davis at the Stanford Genome Technology Center (funded by the cooperative agreement U01 AI47087); Brendan Loftus and Claire Fraser at The Institute for Genomic Research (funded by the NIAID/NIH under cooperative agreement U01 AI48594); and Fred Dietrich at the Duke Center for Genome Technology for access to the CryptococcusGenome Project data. We also thank Bruce A. Roe, Doris Kupfer, Jennifer Lewis, Sola Yu, Kent Buchanan, Dave Dyer, and Juneann Murphy at the University of Oklahoma for access to data from the Cryptococcus neoformans cDNA Sequencing Project (strains JEC21 and H99; NIH-NIAID grant number AI147079). This work was supported by a Genomics Program grant from the Natural Sciences and Engineering Research Council of Canada (to S.J., J.K., and M.M.) and by scholar awards from the Burroughs Wellcome Fund to J.H. and J.K. M.M. is a Michael Smith Foundation for Health Research Biomedical Scholar.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 These authors contributed equally to this work.
-
↵5 Corresponding author.
-
E-MAIL kronstad{at}interchange.ubc.ca; FAX (604) 822-2114.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.81002. Article published online before print in August 2002.
-
- Received January 13, 2002.
- Accepted July 3, 2002.
- Cold Spring Harbor Laboratory Press








