
Fly Factory
It is a great time to be a biologist. The raw information encoded in the genomes of humans and a host of model organisms is being extracted, but we are still far from understanding what all this raw data means. Browsing a genome and looking at all of the sequences known only by gene prediction ID numbers is both humbling and exciting. Ultimately, the biggest revolution ushered in by ongoing genome projects may well be large-scale biology, not novel techniques. After all, sequencing has been around for decades and creative scientists exploited sequencing for cottage industry gene-by-gene analysis from the beginning. The genomic era is handing us the books of life, but the genomic era is also a story of building the infrastructure and management skills necessary to bring big science to biology. We will need new strategies and more collaborative interactions to systematically explore sequenced genomes.
Simin et al. (2002) generated a collection of expressed sequence tags (ESTs) from Drosophila embryos and then determined the spatial pattern of expression of the corresponding transcripts by in situ hybridization. This effectively points out how a big dataset can help us map functions onto sequenced genomes. The manuscript is a harbinger of the future for the development of a flexible robotic process that can be applied not only to genomic problems, but for a range of techniques that fall under the banner of biology.
Drosophilists are justifiably proud of how much we have learned about patterning during development through the analysis of mutant phenotypes. The classic screens for embryonic lethality showed that genes are required in restricted domains in the embryo and act to progressively subdivide the embryo into not only tissue layers, but also into distinct structures along the anterior-posterior and dorsal-ventral planes (Jürgens et al. 1984;Nüsslein-Volhard et al. 1984; Wieschaus et al. 1984). These genes, which are all regulators of information flow through genetic networks, also show spatially restricted expression by in situ hybridization. Understanding the network of regulators is terrific, but a longstanding question has been the identity of the genes that do the work of development. Simin et al. (2002) and related studies report a host of restricted patterns of transcripts, such as ribosomal protein-encoding mRNAs, which one would not expect to find in a genetic screen for a particular pattern defect, because the wild-type genes are either required for all cells (as in the case of the ribosome) or have subtle phenotypes obscured by the action of other genes (Kopczynski et al. 1998; Liang and Biggin 1998; Simin et al. 2002). These patterned transcripts identify good candidates for the terminal differentiation (or effector) genes that are likely to tweak cells in the subtle ways required for their function as part of the organism. Systematic analysis of spatial mRNA patterns is likely to be an important tool in parsing the nucleic acid-binding proteins that decode the genome from those that ultimately build cells with slightly differing roles.
This study also holds interest for those who are curious about the current status of genome annotation. ESTs in combination with in situ hybridization experiments provide high-quality biological evidence for predicted genes and unique evidence for genes missed in the early rounds of genome annotation. Interestingly, 11% of the EST sequences characterized in this study failed to align with pre-existingDrosophila ESTs (Andrews et al. 2000; Rubin et al. 2000) or predicted genes (Adams et al. 2000). Mounting evidence from this and other manuscripts clearly indicates that genome annotation is difficult (Ashburner 2000; Gaasterland and Oprea 2001). Whereas none of us would willingly go back to the pre-annotated genome, it is quite clear that genome annotation should be taken with a grain of salt. It is also quite clear that in the rush to proteomics, we should not forget to expend efforts to finish mapping biological evidence (such as full-length cDNAs, array data, spatial patterns of expression, and mutant phenotypes) onto the finished genome sequence. Genome annotation should be seen as a process, not an event.
There is a serious and unresolved issue of how to track and make sense of large sets of biological information, such as spatial and temporal gene expression profiles. The authors have joined the growing list of laboratories that list Gene Ontology terms (Ashburner et al. 2000) in addition to the standard DNA ID numbers. This is important, but there is room for improvement. FlyBase (1999), the outstanding database of the Drosophila genome will need to develop ways to readily incorporate large data sets such as seen in Simin et al. (2002). Authors will need to help too, by including FlyBase ID numbers in their manuscripts and associated supplemental tables. However, none of these IDs are ultimately stable. As we dig deeper into issues such as alternative splicing and overlapping transcripts in metazoan genomes, not to mention evolving gene models for genes, using IDs for genes is increasingly problematic. Perhaps the sequence itself is the only real tag. It would be good practice to routinely associate any feature, such as an in situ or gene-expression profile, with the corresponding genomic sequence. The result will be a sequenced genome that will be a book and also a card catalog for a library. Finally, although lists of genes and associated patterns will be of interest to most Drosophilists working on the development of the embryo, photos of all of the in situ patterns would be valuable. Experts on a particular tissue or pattern would be able to extract additional information from the primary data. Developing databases for storing and especially querying in situ hybridization patterns, not just the descriptions, will be important.
Of key interest these days too, given the current large (and growing) size of datasets, is the automation of experimental procedures required for extracting such data. My view of laboratory automation consists of machine-aided manual labor, that is, a standard laboratory populated with a few PCR machines, a liquid handling station, and a microarrayer to handle the most onerous repetitive steps. With the exception of the microscopic analysis (a major bottleneck), robotics was used throughout the Simin et al. (2002) project. The range of steps that were automated is really quite impressive. The authors used a robotic laboratory that can seamlessly handle many of the tasks performed by a seasoned laboratory worker (Fig. 1). The laboratory web site lists developed protocols for micro- and macro-array generation, plasmid preps, PCR reactions, RFLP analysis, agarose gel analyses, filter and bead-binding assays, ELISAs, density gradient fractionation analyses, in vitro transcription and translation, and in situ hybridization. Additionally, the robotic laboratory is not constrained by a linear hand off of materials between one machine and another. Because the tasks are broken down into modules, the system can be programmed to perform new protocols by daisy chaining standard tasks. The layout of the machines and the tracking system also improves flexibility and throughput. Multiple protocols can be intertwined, such that while one process is in a long incubation step (for 16 h in the case of in situ hybridization), another unrelated job can be run.
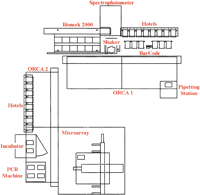
Schematic of work station located in the Eccles Institute of Human Genetics. It occupies approximately one-half of a 600 sq. ft. laboratory bay. Two ORCA robotic arms address a work area of ∼50 sq. ft. Key pieces of equipment are shown in red. (Fig. taken with permission from http://metherall.genetics.utah.edu/HTR.html. A layout video is also available at this web site).
The exploration of new ways to maximize throughput is an exciting part of biology today. Time will tell how this plays out, but this new era will almost certainly involve more extensive collaborations among specialized groups of biologists, computer scientists, mathematicians, and engineers who concentrate on maximizing speed. Having a robotics laboratory that is limited more by the imagination and commitment of the staff and collaborators, and less by the list of protocols or the process pipeline, is clearly moving automation in the right direction.
Footnotes
-
E-MAIL oliver{at}helix.nih.gov; FAX (301) 496-5239.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.295602.
- Cold Spring Harbor Laboratory Press








