
A Bioinformatics-Based Strategy Identifies c-Myc and Cdc25A as Candidates for the Apmt Mammary Tumor Latency Modifiers
Abstract
The epistatically interacting modifier loci (Apmt1 and Apmt2) accelerate the polyoma Middle-T (PyVT)-induced mammary tumor. To identify potential candidate genes loci, a combined bioinformatics and genomics strategy was used. On the basis of the assumption that the loci were functioning in the same or intersecting pathways, a search of the literature databases was performed to identify molecular pathways containing genes from both candidate intervals. Among the genes identified by this method were the cell cycle-associated genes Cdc25A and c-Myc, both of which have been implicated in breast cancer. Genomic sequencing revealed noncoding polymorphism in both genes, in the promoter region of Cdc25A, and in the 3′ UTR of c-Myc. Molecular and in vitro analysis showed that the polymorphisms were functionally significant. In vivo analysis was performed by generating compound PyVT/Myc double-transgenic animals to mimic the hypothetical model, and was found to recapitulate the age-of-onset phenotype. These data suggest that c-Myc and Cdc25A are Apmt1and Apmt2, and suggest that, at least in certain instances, bioinformatics can be utilized to bypass congenic construction and subsequent mapping in conventional QTL studies.
Women carrying mutations in the breast cancer susceptibility genesBRCA1 and BRCA2 are highly predisposed to developing cancer compared with the general population. However, women carrying the same mutation, even within families, can exhibit significantly different clinical expression (Goldgar et al., 1994; Friedman et al., 1995; Langston et al., 1996; Easton et al., 1997), with some women developing cancer early in life, whereas others remain unaffected until >70 years of age (Narod et al., 1995). Although environmental exposures are likely to account for some of the variability, there is evidence that there may be additional genetic elements that contribute to the differential expressivity of the phenotype (Krontiris et al., 1993; Ford and Easton, 1995; Phelan et al., 1996; Kristensen et al., 1998). Identification and characterization of these additional genetic factors will hopefully lead to a greater understanding of the etiology of breast cancer and potentially novel ways to prevent or treat it.
To study these genetic factors, our laboratory uses a transgenic mouse mammary tumor model, the FVB/N-TgN(MMTV-PyVT)634Mul mouse (Guy et al., 1992). These animals express the mouse polyoma Middle-T antigen (PyVT) from a mouse mammary tumor virus enhancer and promoter, resulting in the development of synchronous, multifocal tumors by 57 days of age on average (Guy et al., 1992; Lifsted et al., 1998). Previously, we have shown by outcrossing to the I/LnJ inbred strain of mice, the presence of epistatically interacting latency modifiers in the I/LnJ genome that significantly accelerate tumor appearance (Lifsted et al., 1998). Backcross analysis showed the presence of two epistatically interacting loci on Chrs 9 and 15 (LeVoyer et al., 2000) and generation of congenic animals for high-resolution conventional quantitative trait analysis initiated (J. Rouse and K. Hunter, unpubl.).
The conventional strategy for identification of modifier or QTL candidate genes requires the creation of congenic animals, followed by mapping the trait in question in a series of subcongenic intervals. This method, although effective, is slow and laborious, entailing significant time, animal costs, and effort. In an attempt to circumvent this process, we therefore developed a combined genetics, genomics, and bioinformatics approach to identify interesting candidate genes for analysis and testing. In this study, we show the feasibility of this approach and provide evidence that the genes identified by the bioinformatics method, c-Myc and Cdc25A, are strong candidates for the tumor latency modifiers Apmt1 andApmt2.
RESULTS
The latency modifier genes Apmt1 and Apmt2function in a conditional epistatic interaction (Le Voyer et al., 2000). The FVB/NJ allele of Apmt1 on Chr 15 acts additively to accelerate tumor latency, but only in the presence of an I/LnJ allele of Apmt2 on Chr 9. This interaction suggested that the two loci might be members of a common pathway. On the basis of this assumption, a bioinformatics search was performed to identify potential biochemical pathways that might be the basis of the tumor acceleration phenotype. A PubMed search was performed to look for articles that contained at least one gene from each of the 25-cM long QTL candidate regions (see Fig. 1). The search query consisted of 44 genes and 85 genes from the Chr 15 and Chr 9 candidate intervals, respectively. The resulting list of abstracts 588 was then hand curated to identify gene pairs that were known to interact or to be in a common pathway, and were considered interesting cancer-related genes. The most interesting gene pair to be identified by this screen was c-Myc and Cdc25A, which were observed in 37 abstracts, both of which have been implicated in breast cancer (Cangi et al., 2000; Chrzan et al., 2001). Due to the role of these genes in cell cycle control, they were considered the primary candidates, therefore, no other gene pair was analyzed.

Graphical representation of PubMed pathway query. Abstracts were searched for the appearance of at least one member of each chromosomal candidate region list of genes. The set of articles in common with both lists was then hand curated, and select genes were subjected to genomic analysis.
A polymorphism screen of c-Myc was therefore performed. Primers were designed in the genomic DNA flanking the three c-Myc exons and the PCR products from FVB/NJ and I/LnJ were sequenced. No coding polymorphisms were observed. However, a 2-bp deletion in the 3′ UTR of I/LnJ was observed in a region associated with mRNA stability (Cole and Mango, 1990) and might therefore affect c-Myc mRNA and protein levels. cDNA expression chip data was examined to determine whether there were differences in the amounts of c-Myc mRNA present in FVB/NJ (n = 4) and [I/LnJ x FVB]F1 (n = 5) mammary tumors. Tumor RNA was assayed on the NCI Oncochip cDNA array, the intensities measured, and then compared by the nonparametric Mann-Whitney test. c-Mycwas overexpressed ∼1.3-fold in FVB/NJ tumors compared with the [I/LnJ x FVB]F1 tumors (P <0.007; see Fig.2) consistent with the possibility that the 2-bp deletion in the I/LnJ 3′ UTR might be destabilizing the message. To confirm these results, c-Myc mRNA levels were also assayed by quantitative PCR. Spleen RNAs were isolated from FVB/NJ and I/LnJ animals. Two independent primer sets were assayed, and the results consistently showed, across all of the experiments, that FVB/NJ c-Myc mRNA levels were higher relative to the I/LnJ animals, consistent with the chip analysis (data not shown).
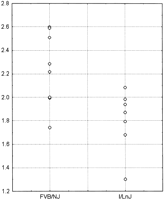
Scatterplot of the Expression Chip c-Myc results. The FVB/NJ homozygous results are displayed at left and the [I/LnJ x FVB/NJ]F1 results at right. The data is presented as the ratio of the c-Myc expression of the samples versus the reference RNA.
Because Cdc25A is thought to be a direct transcriptional target of c-Myc (Galaktionov et al., 1996; Amati et al., 1998), sequencing of the promoter region and the ORF was performed. Sequence analysis of the Cdc25A gene revealed a single-coding polymorphism present in I/LnJ mice, resulting in a Q127H change from the consensus in FVB/NJ. Approximately 1 kb of region 5′ of the transcriptional start site (Paskind et al., 2000) was sequenced in FVB/NJ and I/LnJ genomic DNA to identify potential promoter polymorphisms. Multiple polymorphisms were observed, including two single basepair polymorphisms, two single basepair deletions in I/LnJ compared with FVB/NJ, and the presence of a variable poly(A) element (see Fig. 3a). Because c-Myc is known to transcriptionally activate Cdc25A, and as it had been shown in yeast that poly(A) elements in promoters can effect transcriptional regulation (Iyer and Struhl, 1995; Suter et al., 2000), further analysis focused on the promoter polymorphism. To assess their effect, the I/LnJ and FVB/NJ Cdc25A promoters were subject to in vitro transcription assays. The promoter elements were cloned into reporter plasmids, transfected into NIH-3T3 cells, and the relative transcriptional activity assessed. As can be seen in Figure 3b, the I/LnJ promoter activity was ∼1.6-fold that of FVB/NJ (P <0.0002), showing that promoter region polymorphisms observed are functionally significant.

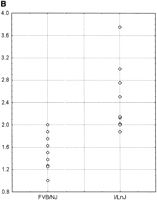
(A) Cdc25A promoter polymorphisms. The site of single base pair polymorphisms between FVB/NJ and I/LnJ are boxed. Bases deleted in the I/LnJ promoter are shaded in gray. The ATG translational start site of Cdc25A is underlined in bold. (B) In vitro Cdc25A promoter expression assay. The I/LnJ and FVB/NJCdc25A promoters were cloned into a luciferase reporter plasmid, transfected into NIH-3T3 cells and the promoter efficiency measured. The data is presented as relative expression compared with the internal cotransfection marker.
The following model can be derived from these data to fit the epistatic interaction observed between the tumor latency accelerating lociApmt1 and Apmt2. In the mammary glands of homozygous I/LnJ animals, the more efficient Cdc25A promoter would be compensated for by the lower levels of the direct transcriptional activator c-Myc. In the accelerated [I/LnJ x FVB/NJ]F1 tumors, the Cdc25A mRNA levels would be hyperinduced by the higher levels of the c-Myc protein, mediated by the more stable FVB/NJ c-Myc allele. The up-regulation of Cdc25A would relax the G1/S checkpoint, permitting earlier or more rapid entry into the cell cycle, resulting in faster tumor development. In the backcross, introduction of a second FVB/NJ allele would further induce the I/LnJCdc25A promoter, resulting in an additive effect at theApmt1 locus.
This model predicts that overexpression of the c-Myc allele in the PyVT animal above normal FVB/NJ levels would result in overexpression of Cdc25A, leading to the acceleration of tumor kinetics. To test this double transgenic, animals were generated by breeding the MMTV-PyVT mouse with the MMTV-Myc mouse, resulting in overexpression of both transgenes in the mammary epithelium. Both transgenes are carried on the FVB/N background, thereby eliminating the concerns of confounding genetic background effects. As predicted by the model, all of the double transgenic animals (n = 8) developed palpable mammary tumors between 24 and 30 days of age compared with between 50 and 60 days for the polyoma middle-T littermates (P <10−6; see Figs. 4 and5). The more rapid appearance of the tumors in the double transgenics compared with the [I/LnJ x FVB/NJ] F1 animals is likely due to the higher levels of c-Myc expression in the double-transgenic animals compared with the heterozygous animals. Western blots showed that bothCdc25A and c-Myc were overexpressed in the double-transgenic animals as predicted (data not shown).
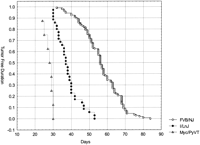
Kaplan-Meier Tumor free duration curve of PyVT/Myc double transgenic animals compared with the FVB/NJ homozyogous and [I/LnJ x FVB/NJ]F1 heterozygous animals.
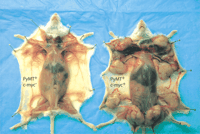
Example of the c-Myc/PyVT double-transgenic animals. The animals are littermates, killed at 57 days of age. The animal atleft is a PyVT transgenic. The mammary glands are just beginning to develop palpable tumors. The animal at right is a c-Myc/PyVT double transgenic.
DISCUSSION
To date, the identification of the underlying genetic basis of quantitative trait loci has been a significant hurdle for researchers to overcome. The conventional strategy requires the sequential generation and analysis of congenic and subcongenic animals to obtain higher resolution mapping and limit the number of potential candidate genes. This strategy, although having a relatively high chance of success, is long and laborious, requiring years to complete and large numbers of animals. As a result, only a handful of modifier genes have been identified (MacPhee et al., 1995; Zhang et al., 1998). Recently, however, it has been suggested that this process would likely become somewhat less difficult as the ever-expanding genomic, proteomic, and expression array data became publicly available (Davenport et al., 1988). Belknap et al. (200l) suggested that using these data would enable investigators to rapidly narrow down the potential candidate genes to a manageable number that could be analyzed in parallel with generation of the high-resolution mapping congenics and subcongenics, potentially accelerating QTL discovery by orders of magnitude.
In accord with this approach, in this study we have applied a pathway-based bioinformatics search to limit the number of candidate genes requiring further analysis. By use of the known genetic interaction, and by making the assumption that the interacting genes lie within the same pathway, a simple PubMed search permitted the identification of potentially interesting gene pairs for analysis. The secondary screen, in this case hand curating, was critical to limiting the number of genes that needed to be examined. Understanding the possible mechanism underlying the phenotypes being examined will play an important role in being able to identify the appropriate genes to examine. In this case, we clearly benefited from the extensive pathway analysis that has been performed on oncogenes and cell cycle regulation. Pathways or phenotypes that are less well understood would be less likely to successfully identify appropriate gene pairs. However, many high-throughput protein–protein interactions studies are being performed to identify many of the molecular interactions, as well as a number of other proteomic-based analyses. As these datasets become increasingly available, it should be progressively easier to identify interesting gene pairs for QTL analysis on the basis of pathway interactions.
In this study, the application of the bioinformatics pathway analysis led to strong evidence that c-Myc is the Apmt1 tumor latency modifier locus. The fact that c-Myc can modulate mammary tumorigenesis is not surprising. c-Myc has been known to synergize with other oncogenes in cellular transformation in a variety of studies and is known to be involved in human breast cancer (Davenport et al., 1988; Belknap et al., 2001). What is more interesting, is the level of c-Myc that appears to have such a functional significance in this system. Unlike the cell culture experiments, significant overexpression of c-Myc was not observed in the [I/LnJ x FVB/NJ]F1 tissues. Instead, only a ∼30%–40% mRNA expression level difference was observed. This suggests that modest expression level differences of some genes, in the presence of other strongly tumor-promoting genes like PyVT, might play a major role in the disparity in age-of-onset observed in the human population. It also should be pointed out that this important and significantly different expression level difference would not normally be considered in most expression array analysis, in which the cutoff for consideration is usually a twofold difference.
The evidence that Cdc25A is Apmt2, although compelling, is less definitive than for c-Myc. Like c-Myc, the functional polymorphism is noncoding and presumably affects the steady-state levels of the G1/S checkpoint protein. Like c-Myc, we believe that the combined evidence suggests that Cdc25A is a credible candidate for one of the tumor latency modifiers. Cdc25A is a known target of c-Myc (Iyer and Struhl, 1995; Suter et al., 2000), therefore, altering expression of c-Myc would be expected to result in different levels of the Cdc25A protein. The role ofCdc25A overexpression in cancer has been implicated in a number of studies. Overexpression has been observed in small cell lung cancer (Wu et al., 1998), as well as associated with papillomavirus E7 expression (Katich et al., 2001; Nguyen et al., 2002). In addition, overexpression of Cdc25A has been shown to cooperate with oncogenes or loss of tumor suppressors in the formation of high-grade tumors (Galaktionov et al., 1995). Cdc25A has also been implicated recently as an important component of theAtm checkpoint against radioresistant DNA synthesis (Falck et al., 2001). Defects in Atm or other upstream radiation-responsive elements are thought to lead to an overexpression of Cdc25A. Overexpression of Cdc25A would be predicted to give a cell a proliferative advantage by increasing the probability of passing through the G1/S checkpoint, resulting in more rapid tumor development. The in vitro promoter assays are in concordance with this possibility. Attempts to directly measureCdc25A mRNA levels between I/LnJ and FVB/NJ by expression chip and quantitative PCR assays in tumors or spleen were inconclusive, due to variability between samples (data not shown). Regardless, the most compelling evidence of Cdc25A being Apmt2 would be the generation of a Cdc25A overexpressing transgenic and the determination of tumor latency in Cdc25A/PyVT double transgenics. We have therefore chosen to focus on this strategy rather than collecting and analyzing the large number of samples required to obtain statistically significant Cdc25A expression results. These experiments are currently underway in our laboratory.
METHODS
Bioinformatics Search
The bioinformatics search was performed as follows. The list of genes in a 25-cM region centered on either Apmt1 orApmt2 was identified by searching MGI (Bult et al., 2000). On the basis of the assumption that the epistatically interacting genes operated in the same pathway, a PubMed search was performed to identify abstracts in the last 5 yr that had one or more genes from each list present. The query was designed as follows: (gene A OR gene B OR gene C…) AND (gene α OR gene β OR gene χ. . . . .). The resulting list was then hand curated to identify interesting candidate gene pairs.
Expression Chip Analysis
Five mammary tumor samples from FVB/N-Tg (MMTV-PyVT)634Mul and [I/LnJ x FVB/NJ]F1, were collected, snap frozen, and stored at −80°C for RNA isolation. Total RNA was extracted from spleen tissue by using TRIzol Reagent (Life Technologies) according to the standard protocol. Reference RNA was extracted and pooled from 10- to 11-week-old FVB/NJ virgin mammary glands. A total of 3 μg of total RNA from reference and tumor samples was amplified using the modified Eberwine (Eberwine et al., 1992) method. Briefly, first-strand synthesis of cDNA was performed using Superscript II reverse transcriptase (Life Technologies) and a T7 oligo-dT primer, and the second strand of cDNA was synthesized using DNA polymerase I. RNAase H was then used to cleave the RNA from the RNA–DNA hybrid and generate RNA primers for DNA polymerase I-mediated chain extension. The cDNA was cleaned up and used as a template for amplification of RNA. In vitro transcription to amplify RNA was performed using the T7-Megascript kit (Ambion) following the manufacturer's instructions.
Ten micrograms of linearly amplified RNA was used to generate Cy3-dUTP- or Cy5-dUTP-labeled first-strand cDNA by reverse transcription using random primers. The cDNA products synthesized from sample and reference were hydrolyzed with NaOH, and then purified in microcon YM-30 columns (Amicon). Each tumor sample was labeled reciprocally by Cy3-dUTP or Cy5-dUTP fluors and hybridized on a microarray. cDNA clones (GEM1 set, ∼8700 elements) were purchased from Incyte Genetics. The cDNA microarray was fabricated by the National Cancer Institute microarray facility and used to analyze the gene expression profiles in mouse mammary tumor tissues initiated by MMTV-PyVT transgene in different genomic backgrounds.
Microarrays were prehybridized for at least 1 h in 5× SSC, 0.1% SDS, 0.1% BSA at 42°C. The chips were then washed in distilled water and isopropanol before application of the probes. The fluor-tagged cDNA probes synthesized from tumor sample and reference were mixed, denatured at 100°C, and subsequently cohybridized to an array slide in hybridization buffer (25% formamide, 5× SSC, and 0.1% SDS) at 42°C overnight. The microarrays were subsequently washed sequentially in 2× SSC, 0.1% SDS in 1× SSC, 0.1% SDS in 0.2× SSC, and 0.5× SSC. The arrays were air dried and scanned using the Axon GenePix400A scanner and images were processed using GenePix-Pro3.0 program. Both image and signal intensity data were stored in a database supported by the Center for Information Technology at the National Institutes of Health.
Sequencing
All sequencing was performed with Perkin Elmer BigDye Dye Terminator sequence kits and analyzed on a Perkin Elmer 3100 Automated Fluorescent Sequencer. Sequences were compiled and analyzed with the computer software packages PHRED and PHRAP(Gordon et al., 1998). The primers used are shown in Table1.
Sequencing Primers
Construction of Cdc25A Promoter Luciferase Plasmids
Promoter regions from FVB/NJ and I/LnJ (−109 to −1057 bp upstream of the ATG translational start site) were amplified with the Cdc25AP1F and Cdc25AP2R primers under the standard conditions. The ∼950-bp product was cloned into the pCR2.1 vector, following the manufacturer's protocol (Invitrogen) and the clones sequence verified. The KpnI/XbaI fragment from pCR 2.1 containing the promoter region of Cdc25A was then subcloned intoKpnI/NheI sites of pGL2-Enhancer vector (Promega).
Cdc25A Promoter Luciferase Assays
NIH/3T3 were grown in DMEM supplemented with 10% FBS, penicillin (100 U/mL) and streptomycin (100 μg/mL) (DMEM; GIBCO BRL). Plasmids were prepared using the Qiagen-EndoFree prep kit (QIAGEN) and stored in Endotoxin-free TE buffer. Concentration and purity were determined by UV absorbtion. To measure promoter efficiency, Cdc25Apromoter-pGL2-Enhancer Firefly luciferase constructs were cotransfected with the Renilla luciferase control plasmid pRL-SV40 (Promega). The Dual-Luciferase TM Reporter Assay system (Promega) was used to determine activity of firefly luciferase (Photinus pyralisus) and sea pansy Renilla luciferase (R. reniformis) according to the manufacturer's instructions. The luciferase activity was determined on a 96 Well Microtiter Plate Luminometer (Dynex Technologies). Light output was measured for 10 sec, and the results integrated to yield of activity. All pGL2 firefly luciferase measurements were standardized to the Renilla luciferase (control) activity.
Animals
FVB/N-TgN(MMTVPyVT)634Mul mice were obtained from The Jackson Laboratory. MMTV-myc mice were purchased from Charles River Laboratory. Inheritance of the transgenes was determined by PCR amplification of weanling tail biopsy DNA with the following primers: (1) MMTV-PyVT transgene: 5′-AACGGCGGAGCGAGGAACTG-3′, 5′-ATCGGGCTCAGC AACACAAG-3′, and (2) MMTV-c-myc transgene: 5′-GGT GATAGTCCCTTCACATC-3′, 5′-GTGCCACCTGACGTC TAAGA-3′ (Bearss et al., 2000). Diagnosis of mammary tumors was performed by palpation. Animals were checked for tumors every other day. After the initial identification of the primary tumor, animals were further aged to confirm the diagnosis.
Taqman Assays
Total RNA was extracted from spleen tissue by using TRIzol Reagent (Life Technologies) according to the standard protocol. The quantity and quality of RNA samples was determined by the Agilent Technologies 2100 Bioanalyer (Bio Sizing Software version A.02.01., Agilent Technologies). Total RNA was processed directly to cDNA by reverse transcriptase with ThermoScript RT–PCR System (Invitorgen), according to the manufacture's protocol in a total volume of 20 μL. The target message was quantified by measuring Ct, then applying a standard curve to determine the quantity of starting target message. A standard curve was produced by quantifying transcripts of the housekeeping gene 18S using the Pre-Developed TaqMan Assay Reagent Control Kit (Applied Biosystems) as the endogenous RNA control. Each target sample was normalized on the basis of its 18S content. One primer and probe set was designed using Primer Express Oligo Design software (Applied Biosystems version 1.5). A second probe (cmyc1) was selected on the basis of previous work from Decallonne et al. (2000). All primers were purchased from Applied Biosystems Custom Oligo Synthesis Service. The standard curve was constructed by making twofold serial dilutions of mouse spleen cDNA produced (as described above) from total RNA isolated with TRIzol by reverse transcriptase with ThermoScript RT–PCR System (Invitrogen), according to the manufacture's protocol in a total volume of 20 μL. Amplification reactions contained equal amounts of cDNA (as determined by analyzing a 1:100 diluted sample of individual RT–PCR reaction on the Spectramax Plus (SoftmaxPro software version 3.1.1), 25 μL of TaqMan Universal PCR Mastermix (Applied Biosystems), 45 pM of each of the specific primers, and 200 nM of the fluorescent probe. All reactions were performed in triplicate in an Applied Biosystems Prism 7700 Sequence Detection System and the thermal cycling conditions were as follows: 2 min at 50.0°C, 10 min at 95.0°C, followed by 40 cycles of 95.0°C for 15 sec, and 60.0°C for 1 min. The primers used are as follows: cmyc1-F: 5′-TGCCCCTCAACGTGAACTTC-3′, cmyc1-R: 5′-CAGATATCCTCACTGGGCGC-3′, cmyc1 probe: 5′-ACGA GGAAGAGAATTTCTATCACCAGCAACAGC-3′, cmyc2-F: 5′-TGAGCCCCTAGTGCTGCAT-3′, cmyc2-R: 5′-ACGCCGACTC CGACCTCTT-3′, cmyc2-probe: 5′-CTTCTTGCTCTTCTTCAG AGTCGCTGCTG-3′.
Acknowledgments
We thank the members of the Laboratory of Population Genetics for their assistance and helpful discussions and Drs. J. Jen and J. Struewing for critical review of this manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Present address: National University of Singapore, Singapore 117604.
-
↵4 Corresponding author.
-
E-MAIL Hunterk{at}mail.nih.gov; FAX (301) 435-8963.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.210502.
-
- Received February 26, 2002.
- Accepted April 3, 2002.
- Cold Spring Harbor Laboratory Press








