
Interrelating Different Types of Genomic Data, from Proteome to Secretome: 'Oming in on Function
Abstract
With the completion of genome sequences, the current challenge for biology is to determine the functions of all gene products and to understand how they contribute in making an organism viable. For the first time, biological systems can be viewed as being finite, with a limited set of molecular parts. However, the full range of biological processes controlled by these parts is extremely complex. Thus, a key approach in genomic research is to divide the cellular contents into distinct sub-populations, which are often given an “-omic” term. For example, the proteome is the full complement of proteins encoded by the genome, and the secretome is the part of it secreted from the cell. Carrying this further, we suggest the term “translatome” to describe the members of the proteome weighted by their abundance, and the “functome” to describe all the functions carried out by these. Once the individual sub-populations are defined and analyzed, we can then try to reconstruct the full organism by interrelating them, eventually allowing for a full and dynamic view of the cell. All this is, of course, made possible because of the increasing amount of large-scale data resulting from functional genomics experiments. However, there are still many difficulties resulting from the noisiness and complexity of the information. To some degree, these can be overcome through averaging with broad proteomic categories such as those implicit in functional and structural classifications. For illustration, we discuss one example in detail, interrelating transcript and cellular protein populations (transcriptome and translatome). Further information is available athttp://bioinfo.mbb.yale.edu/what-is-it.
“[It] does not consist of individuals, but expresses the sum of interrelations, the relations within which these individuals
stand.” —adapted from Karl Marx,Grundrisse (1857)
The raw data produced by genome sequencing projects currently provides little insight into the precise workings of an organism at the molecular level (Luscombe et al., in press). Therefore, the goal of functional genomics is to complement the genomic sequence by assigning useful biological information to every gene. Through this, we aim to improve our understanding of how the different biological molecules contained within the cell (i.e., DNA, RNA, proteins, and metabolites) combine to make the organism viable. Clearly, the main challenge is the elucidation of all molecular, cellular, and physiological functions of each gene product. However, there are many subsidiary goals as part of this challenge, such as defining the three-dimensional structures of these macromolecules, their subcellular localizations, intermolecular interactions, and expression levels. Although gathering and classifying the necessary information is central to this process, it is impractical to rely on individual experiments for the potentially thousands of genes in each organism. Furthermore, with large-scale proteomic experiments still yet to be used widely, computational techniques — while sometimes based on less than ideal information — provide a crucial resource for assigning biological data.
The paper by Antelmann et al. in this issue of Genome Research(Antelmann et al. 2001) evaluates their earlier attempts to assign protein functions through computational means. Previously, the group used computational methods to predict all exported proteins(or members of the secretome) in Bacillus subtilis by searching for signal peptides and cell retention signals in the protein sequences. A better understanding of how and why a protein is secreted is valuable as the bacterium's ability to export numerous enzymes enables it to degrade extracellular substrates and survive in a continuously changing environment. Moreover, it will eventually allow these bacteria to be employed as “cellular factories” for secreting commercially valuable proteins in large quantities (Tjalsma et al. 2000).
Antelmann et al.'s present paper aims to verify their previous predictions by experimentally characterising the entire population of secreted proteins using 2D gel electrophoresis and mass spectrometry. They showed that the original predictions correctly identified about 50% of all secreted proteins. Most of the disagreements were due to the inability to predict the secretion of proteins lacking the appropriate signal, or those containing seemingly inappropriate signals (cell retention signals). In summary, Antelmann et al.'s work highlights both the encouraging aspects of computational assignments of biological data, and reveals some of the shortcomings in the current methods.
The Path to Function is Filled with 'omes
To describe their studies, Antelmann et al. coined the term “secretome”. This 'omic term is an example of the new lexicon that has appeared recently to define the varied populations and sub-populations in the cell (Fig. 1). These terms are generally suffixed with “-ome”, with an associated research topic of “-omics”.
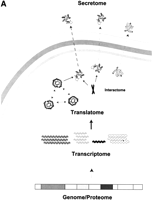
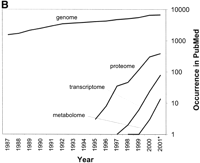
An overview of the current ‘omic terminology. (A) A schematic of the main ’omes in the process of gene expression. (B) The literature citations of four of the most widely used 'omes over time.
Broadly, the existing 'omes can be divided into those that represent a population of molecules, and those that define their actions (Fig. 1). For the first category, populations provide an inventory or “parts list” of molecules contained within an organism (Gerstein and Hegyi 1998; Skolnick and Fetrow 2000; Vukmirovic and Tilghman 2000; Qian et al. 2001). The genome, the entire DNA sequence of an organism, presents a basis for defining the proteome, a list of coding DNA regions that result in protein products. Transcription of these coding sequences produces the transcriptome (Velculescu et al. 1997), which is the cellular complement of all mRNA under a variety of cellular conditions. Note, this population is weighted by the expression level of each molecule and, ideally, should incorporate the results of alternative splicing. Following translation of the transcriptome, we suggest the term “translatome” to describe the cellular population of proteins expressed in the organism at a given time, explicitly weighted by their abundance. It is important to note that, whereas the membership of the genome and proteome are virtually static, the transcriptome and translatome are dynamic and continually change in response to internal and external events. Additional 'omes describe the presence of molecules that are not encoded by the genome, but are nonetheless essential, for instance, the metabolome (Tweeddale et al. 1998). Because of the newness of most 'omic terms, a few still have competing definitions. This is most evident for the proteome (see Table1).
A Table of 'omes, Together with their Occurrence in the Literature and on the World Wide Web
The second group of 'omes are fewer in number and describe the actions of the protein products. For example, the secretome is a subset of the proteome that is defined by its action, that is, it is actively exported from the cell. The interactome (Sanchez et al. 1999) lists all of the specific interactions that are made between macromolecules in the cell. More abstractly, the regulome (Web references only; see Table1) defines the genome-wide regulatory network of the cell and most notably includes transcription regulation pathways.
The elucidation of each of these 'omes contributes to the ultimate goal of functional genomics, defining the functome,which describes all of the functions that are assigned to each gene in the genome (theRison et al. 2000, http://www.biochem.ucl.ac.uk/∼rison). The functions of a gene can be described at many levels, including their biochemical, cellular and physiological roles (Ashburner et al. 2000), and also depend on additional factors that are not immediately associated with their basic functions, such as subcellular localization and intermolecular interactions. Therefore, aspects of the functome may be expressed in terms of other 'omes, for example those that group similar biochemical functions, for example the immunome (Pederson 1999); similar localizations, for example the secretome; and similar interactions, for example, the interactome. For the record, we coin our own term here; at present, a large proportion of genes can only be described as members of the “unknome”: those with currently no functional information!
Computational Methods for Defining 'omes
There are a variety of computational approaches for defining 'omes (Gerstein and Honig 2001):
- (1)
- Algorithmic methods for predicting genes, protein structure, interactions, or localization based on patterns in individual sequences or structures; for example, defining the proteome or orfeome using a gene-finding algorithm on the genome (Claverie 1997; Guigo et al. 2000;Harrison et al. 2001; Yeh et al. 2001), determining the foldome from structure prediction of the proteome (Simons et al. 2001), determining the interactome from the foldome, using known binding sites (Teichmann et al. 2001), and determining the secretome through identifying signal sequences in the proteome (Tjalsma et al. 2000).
- (2)
- Annotation transfer through homology, that is, inferring structure or function based on sequence and structural information of homologous proteins (Genstein 1997, 1998; Brenner 1999; Hegyi and Gerstein 1999;Wilson et al. 2000; Thornton 2001; Hegyi and Gerstein, in press).
- (3)
- Using a “guilt-by-association” method based on clustering where functions or interactions are inferred from clusters of functional genomic data, such as expression information. For example, similar functions can sometimes be inferred through interactions with other proteins or similar expression profiles (Eisen et al. 1998; Marcotte et al. 1999; Gerstein and Jansen 2000; Ito et al. 2001).
Experimental Methods for Defining 'omes
Although still in their infancy, several large-scale experimental techniques are designed to assess the nature of different 'omes. Gene expression studies are now well established and microarray or GeneChip technologies can be used to measure mRNA abundance in the cell and hence define the transcriptome (Epstein and Butow 2000). Detection of protein concentration and definition of the translatome is more difficult, however, as evidenced by the dearth of such data. At present, the most prominent method employs two-dimensional electrophoresis to isolate proteins followed by mass spectrometry for their identification (Futcher et al. 1999; Gygi et al. 1999;Naaby-Hansen et al. 2001) followed by quantification (Appel et al. 1997; Aebersold et al. 2000; Gygi et al. 2000). The two-hybrid system enables detection of specific protein–protein associations to build the interactome (Uetz et al. 2000; Ito et al. 2001; Walhout and Vidal 2001). Antelmann et al. (2001) used two-dimensional electrophoresis to determine the membership of the secretome.
Given the goal of determining the functome, perhaps the most exciting technology is the protein chip system, which is capable of high-throughput screening of protein biochemical activity. (Zhu et al. 2000; Zhu 2001, in press). Other methods for obtaining large-scale protein functional characterization include a transposon insertion methodology (Ross-Macdonald et al. 1999).
Although we discuss the computational and experimental methods separately, there is, in fact, an inseparable relationship between the two. On the one hand, data resulting from high-throughput experimentation require intensive computational interpretation and evaluation (Carson et al. 2001). On the other hand, computational methods use empirical data to build a knowledge base for predictions. Furthermore, they sometimes produce questionable predictions that should be reviewed and confirmed through experiments, as Antelmann et al. point out. In addition to these high-throughput techniques, another interesting tactic is to aggregate the results of individual experiments through comprehensive literature searches. Although there clearly are difficulties with differing experimental conditions and varying interpretations, preliminary results have shown this to be an effective method (Jenssen et al. 2001; Marcotte et al. 2001; Ono et al. 2001).
Interrelating Different 'omes
Having categorized the organism into different sub-populations, a fundamental approach in genomics is to establish relationships between the different 'omes. In other words, by piecing the individual 'omes together, we hope to build a full and dynamic view of the complex processes that support the organism. For example, how do the proteome and regulome combine to produce the translatome?
As with defining the 'omes, these relationships can be explored in different ways:
- (1)
- Defining or assigning one 'ome based on another, as described above.
- (2)
- Comparing one 'ome with another to better understand the processes that shift one population into its successor. For instance, this could be done by correlating expression measurements for the transcriptome and translatome (see below).
- (3)
- Calculating “missing” (experimentally unattainable) information in one 'ome based on information in another one – for example, using the known relationships between gene expression level and subcellular location to help predict the destination of proteins of unknown localization (Drawid and Gerstein 2000; Drawid et al. 2000).
- (4)
- Describing the intersection between multiple populations. For example, combining data from the transcriptome and the functome could describe the array of biochemical, and potentially, physiological functions that are available to the cell at any given time (Hegyi and Gerstein 1999).
The Use of Broad Categories to Interpret Noisy Data
Functional genomics experiments generally give rise to very complicated data that are inherently hard to interpret. Furthermore, these data are often plagued with noise (Kerr et al. 2000). Both factors can lead to inaccuracies and conflicting interpretations.
A good example is gene expression measurements, which are known to fluctuate between experiments even if the conditions are apparently identical (Baldi and Long 2001). These fluctuations are often due to measurement errors, but there are also inherent biological variations of expression levels, relating to the stochastic nature of gene expression (Szallasi 1999). One cause is the very low cellular concentrations of many transcription factors, meaning, that they bind promoters very rarely. Such events approximate to a Poisson process, and in fact, macroscopic chemical kinetics would fail to describe the resulting expression level of the gene (McAdams and Arkin 1999; Thattai and van Oudenaarden 2001). In another example, the interactome, when determined using the yeast two-hybrid technique, is notorious for false positives and negatives (Ito et al. 2000; Serebriiskii et al. 2000; Ito et al. 2001; Legrain et al. 2001).
A useful way to tackle noise and complexity of functional genomics information is to average the data from many different genes into broad 'omic categories (Jansen and Gerstein 2000). For instance, instead of looking at how the level of expression of an individual gene changes over a timecourse, we can average all the genes in a functional category (e.g., glycolysis) together. This gives a more robust answer about the degree to which a functional system changes over the timecourse. Likewise, if one wants to investigate the relationship between a gene's essentiality — whether or not it is essential (Winzeler et al. 1999) — and its subcellular localization, it might be useful to combine the results for all proteins in the same compartment. This would give the average degree of essentiality of all nuclear proteins, cytoplasmic proteins, and so forth. In an actual study for predicting protein subcellular localization, we obtained more accurate predictions for the overall populations (96% accuracy) of a given subcellular compartment than for individual genes (75% accuracy) (Drawid et al. 2000).
Thus, the strength of genomic studies lies in the global comparisons between biological systems rather than detailed examination of single genes or proteins. Genomic information is often misused when applied exclusively to individual genes. If one is interested only in one particular gene, there are many more conclusive experiments that should be consulted before using the results from genomics datasets. Therefore, genomic data should not be used in lieu of traditional biochemistry, but as an initial guideline to identify areas for deeper investigation and to see how those results fit in with the rest of the genome.
Moreover, most genomics datasets give relative rather than absolute information, which means that information about a single gene has little meaning in isolation. For example, they are best used to identify “outlier” genes that are particularly highly-expressed, or have especially many interactions, rather than to focus on the individual measurements for a particular gene. A gene that makes a particularly large number of interactions may indicate that it is a key component of the cell. One numerical technique that is particularly useful with regard to dealing with this information is expressing results through ranks (i.e., not giving the number of interactions of a particular gene product, but how it ranks when compared with others). Furthermore, it provides a powerful way to combine many different heterogeneous sources of information into a common and statistically robust numerical framework (Gerstein and Levitt 1997; Gerstein and Hegyi 1998; Qian et al. 2001).
These observations should be kept in mind when interacting with genomics tools and databases. Many websites focus on providing a lot of information for a single gene sequence or protein, in a “non-genomic” fashion. Rather, such sites should be designed to simultaneously display and manipulate large populations of genes. In the absence of such an 'omic interface, it is important that information resources at least accommodate bulk downloading of standardized data.
A Case Study: Interrelating the Transcriptome and the Translatome
A specific example of comparing the transcriptome and translatome will illustrate the points we made about interrelating 'omes and using categories to interpret noisy data. Here the question is to what degree do highly expressed genes (transcriptome) correspond to highly expressed proteins (translatome)? We can get very different answers depending on the perspective we take:
Theoretical View
Turning to the entire mRNA and protein populations, the change in protein concentration over time is equal to the rate of translation minus the rate of degradation. Borrowing from chemical kinetics, this is approximately expressed by the equation dP(i,t)/dt = SE(i,t) -DP(i,t), where P is the abundance of protein i at time t, E is the corresponding expression level of this protein, S is a general rate of protein synthesis per mRNA, and D is a general rate of protein degradation per protein. Obviously, this is highly simplified and in a more general context one would expect that the rates of synthesis and degradation to be different for each gene and dependent on the regulatory effects of other genes over time. In addition, the equation does not take into account the stochastic nature of gene expression (see above) (Chen et al. 1999).
Direct Comparison of Individual mRNA and Protein Data
At the moment, we do not have good enough data to apply models such as the equation above. However, there is an intuitive sense that highly expressed genes correspond to highly abundant proteins. (One can see this by imagining the situation at steady-state, when the lefthand side of the equation is zero and a positive correlation between Eand P results.) Figure 2A shows the direct comparison between raw measurements of mRNA expression and protein abundance data for 181 genes in yeast drawn from two recent studies (Futcher et al. 1999; Gygi et al. 1999). The two variables show a high degree of variation for individual data pairs and investigators have come to different conclusions about the general correlation between them. This is, to some degree, dependent on the subjective way of analyzing the data.
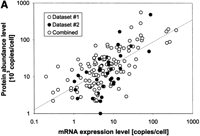
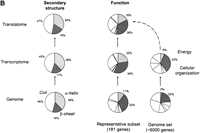
Interrelating the transcriptome and the translatome.(A) A direct comparison of protein abundance and mRNA expression. The abundance data is from two recent studies (datasets 1 and 2) of a global comparison of protein and mRNA expression levels in yeast (Futcher et al. 1999; Gygi et al. 1999). The combined protein abundance dataset is an average of the data points from the two studies if the given gene product appears in both studies. The mRNA expression data is mainly derived from Holstege (1998). Although there is a general trend for protein concentration to rise with mRNA levels, the actual correlation is weak and protein concentrations can sometimes vary by more than two orders of magnitude for a given mRNA level. Similar observations were reported by a study in human liver cells (Anderson and Seilhamer 1997). The mRNA expression data was scaled and the process is described on our Web site (http://bioinfo.mbb.yale.edu/expression). (B) The composition of the genome (proteome), transcriptome and translatome in terms of broad categories: protein secondary structures and functions. This is based on the analysis in Jansen and Gerstein (2000) with updates to include protein abundance data. The bottom piecharts give the composition in the genome, the middle charts in the transcriptome and the top charts in the translatome. The compositions for the transcriptome and the translatome are calculated by weighting each mRNA/protein with its respective expression level. The secondary structure composition does not vary significantly between the different 'omes, mainly because transcription and translation are independent of secondary structure. The right five piecharts analyse the functional composition. We highlight the Energy and Cellular Organization categories determined from MIPS (Mewes et al. 2000). A problem in comparing the different 'omes is that each represents a different set of genes. For instance, protein levels have been measured only for a fraction of genes whereas mRNA levels are known for almost all genes. The piecharts show the compositions for the whole genome in the right column and a representative subset of genes with known protein levels in the left column. Comparing the left to the right immediately shows the experimental bias of two-dimensional electrophoresis (the method for measuring protein abundance) with respect to certain functional categories. There is good agreement between the composition in the translatome and the transcriptome, despite the low correlation of protein and mRNA levels for individual genes. In comparison, the compositions in the genome are much lower.
Analysis of the Data in Terms of Categories
Although the relationship between mRNA and protein levels is vague for individual genes, some of the statistics for broad categories of protein properties are much more robust. Figure 2B shows the protein secondary structure and functional composition in the genome, the transcriptome (i.e., weighted by mRNA abundance), and in the translatome (i.e., weighted by protein abundance). In contrast to the differences between mRNA and protein data for individual genes, the broad categories show that the transcriptome and translatome populations are remarkably similar; both contain roughly the same proportions of secondary structure and functional categories. Moreover, this contrasts with the genome, which appears to have a distinctly different composition of functional categories. This illustrates that we get a more consistent picture when we average across the population; that is, there is broad similarity between the characteristics of highly expressed mRNA and highly abundant proteins.
Conclusion
The ultimate goal of genomics is the elucidation of the functome, but there are many intermediate steps. By viewing the cell in terms of a list of distinct parts, we can define, part by part, each 'ome in an effort to determine and categorize functional information for each gene. High-throughput experimentation and computational techniques are valuable and complementary; that is, conclusive results often cannot be made based on a single methodology. It must be noted that this data is only valuable with regard to large populations, and as such, should only be used as a secondary source for single gene queries. Moreover, genomic approaches result in inaccurate and noisy data. This noise, while deafening on the single gene level, can be tolerated through the use of broad categories to analyze the data.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL mark.gerstein{at}yale.edu
-
Article and publication are athttp://www.genome.org/cgi/doi/10.1101/gr.207401.
- Cold Spring Harbor Laboratory Press








