
High-Performance Multiplex SNP Analysis of Three Hemochromatosis-Related Mutations With Capillary Array Electrophoresis Microplates
Abstract
An assay is described for high-throughput single nucleotide polymorphism (SNP) genotyping on a microfabricated capillary array electrophoresis (CAE) microchip. The assay targets the three common variants at the HFE locus associated with the genetic disease hereditary hemochromatosis (HHC). The assay employs allele-specific PCR (ASPCR) for the C282Y (845g->a), H63D (187c->g), and S65C (193a->t) variants using fluorescently-labeled energy-transfer (ET) allele-specific primers. Using a 96-channel radial CAE microplate, the labeled ASPCR products generated from 96 samples in a reference Caucasian population are simultaneously separated with single-base-pair resolution and genotyped in under 10 min. Detection is accomplished with a laser-excited rotary four-color fluorescence scanner. The allele-specific amplicons are differentiated on the basis of both their size and the color of the label emission. This study is the first demonstration of the combined use of ASPCR with ET primers and microfabricated radial CAE microplates to perform multiplex SNP analyses in a clinically relevant population.
With the completion of the draft human genome sequence, the next major molecular biological challenge involves determining and understanding the range of sequence variation found not only in humans but also in other genomes. The largest amount of genomic variability is found as single nucleotide polymorphisms, or SNPs (Landegren et al. 1998; Cargill et al. 1999; Zubritsky 1999). This SNP mapping task is of increasing importance due to the growing link between particular SNP-based genotypes and the predisposition/susceptibility to many diseases (Cargill et al. 1999). Current SNP typing formats have significant drawbacks, including cost, complexity, the requirement of specialized equipment or reagents, difficulty of multiplexing, and inability to be easily miniaturized and integrated. For example, realtime PCR analysis with TaqMan has a very rigid format that limits the number of samples that can be run concurrently (Livak et al. 1995). Heteroduplex and sequence-specific conformational polymorphism (SSCP) analyses are limited by the number of amplicons that can be co-generated, or mixed together, and by the complexity of analyzing the resulting electropherograms (Jackson et al. 1997; Bosserhoff et al. 1999; Simonsen et al. 1999; Wenz et al. 1999). Additionally, typing by hybridization with allele-specific oligonucleotides (Beutler et al. 1996), as well as the aforementioned methods, is complicated by adjacent mutations. Finally, conventional gel-based allele-specific PCR (ASPCR) and restriction analysis of PCR products can be extremely time-consuming (Smillie 1997, 1998; Takeuchi et al. 1997).
New generations of genetic analysis devices, along with supporting methodology, are needed to exploit the SNP genotyping opportunities. These new approaches must have the ability to perform complex, sensitive, specific, high-speed, and high-throughput analyses. Capillary array electrophoresis (CAE) is one important step in this direction (Kheterpal and Mathies 1999). CAE analysis has already been utilized for rapid microsatellite genotyping as well as sequencing of mitochondrial DNA and the human genome (Kheterpal et al. 1996;Kheterpal and Mathies 1999; Fox 1999).
Microfabricated capillary electrophoresis (CE) devices offer additional advantages for genetic analysis including smaller sample volumes, higher speed and sensitivity, and the ability to densely pack separation channels into smaller monolithic platforms (Kheterpal and Mathies 1999). High-speed sizing of DNA restriction fragments, PCR products, ASPCR products, and short tandem repeats as well as multiplex short tandem repeat (STR) typing have already been demonstrated on prototype microfabricated CE systems (Woolley and Mathies 1994;Schmalzing et al. 1997; Woolley et al. 1997; Simpson et al. 1998a;Medintz et al. 2000a). The rapid analysis capabilities of these devices have also been utilized for diagnosis of lymphoproliferative disorders and herpes simplex encephalitis (Hofgartner et al. 1999; Munro et al. 1999), and the high-performance genotyping of 96 MTHFR alleles in <90 sec has recently been demonstrated on systems that couple CAE microplates to laser-excited scanning confocal detection (Shi et al. 1999).
The SNP variants at the locus associated with hereditary hemochromatosis (HHC) provide an excellent target for critically evaluating high-speed SNP typing on CAE microplates. This autosomal recessive disease of iron metabolism affects 1 in 300 individuals of northern European ancestry and may be the most common genetic disease in the United States because ∼1.5 million Americans are affected (Mura et al. 1999). Given these considerations, a comprehensive genetic test for the HHC variants is desirable. The effects of the C282Y [845g->a substitution] and H63D [187c->g] mutations in the HFE gene have been well described (Jazwinska et al. 1996; Mura et al. 1997, 1999). An S65C [193a->t] variant has also been recently implicated in HHC, and its frequency was found to be 1.5% in Danish blood donors and 2.4% in a Swiss population (Mura et al. 1999;Simonsen et al. 1999; Medintz et al. 2000a). The close proximity of this S65C variant to the H63D mutation (6 bp) would obviously complicate most analysis methods.
A preliminary evaluation of ASPCR methodology for typing the S65C variant has been presented (Medintz et al. 2000a). Differentially labeled and sized S65C allele-specific (AS) amplicons were rapidly genotyped (<120 sec) on a radial CAE microplate (Medintz et al. 2000a). ASPCR is ideally suited for specific SNP typing because the 3′ terminal nucleotide of the AS primer is complementary to and determines whether that allele is present in the template DNA. Additional advantages are that the sample generation is reduced to a single step, and the labeled primer eliminates the need for intercalating dyes (Woolley and Mathies 1994; Shi et al. 1999).
Here we use energy-transfer (ET) covalent labeling to enhance the specificity of HFE genotyping to facilitate high-throughput multiplex SNP typing on CAE microplates. ET-labeled AS primers were used to generate ASPCR products for the C282Y (845g->a), H63D (187c->g), and S65C (193a->t) mutations in a reference population of >100 samples. The mixed ASPCR products were then rapidly separated and genotyped in <10 min on a 96-channel radial CAE microchip coupled to a laser-excited rotary scanning confocal fluorescence scanner.
The resulting data were processed using the Genetic Profiler analysis software. This is the first study demonstrating the practical utility of CAE microchips for high-performance SNP analysis.
RESULTS
CAE Microplate Multiplex SNP Analysis
In our assay, between three and six differentially labeled AS amplicons define the HHC-related alleles present in each sample, and six control amplicons confirm the efficacy of each ASPCR. All amplicons are of different sizes and are thus separated and identified against a standard during electrophoresis. Figure1A shows the processed electropherogram resulting from the analysis of a wild-type (WT) control sample along with the raw data collected for that same sample. The ASPCR and PCR control amplicons appear as red peaks (ET-ROX). The tetrachlorofluorescein (TET)-labeled standards appear as green peaks. Note the ASPCR amplicons eluting at 174, 211, and 223 bp for the H63(187c), S65(193a), and C282(845g) WT alleles, respectively. The PCR control amplicons elute at 244, 268, 281, 293, 363, and 528 bp. The 280 bp TET control and the 281 bp ET-ROX control peaks demonstrate that single-base-pair resolution is achieved. Figure 1B presents analyzed data from an S65C heterozygote sample along with the raw data. The mutant allele appears as a blue peak (ET-R110) at 201 bp corresponding to the 65C(193t) allele. The PCR control peaks in this sample appear in black corresponding to their ET-TAM labeling. Figure 1C presents the processed data from an H63D(187c->g)/C282Y(845g->a) compound heterozygote along with the raw data. The mutant peaks appear in blue, eluting at 160 and 232 bp, corresponding to the 63D(187c) and 282y(845a) alleles, respectively. The mutant alleles demonstrated in Figure 1B and 1C along with the WT alleles illustrate all the alleles screened for in this assay.
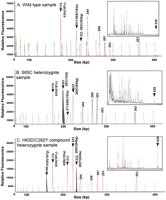
CAE microplate multiplex SNP genotyping analysis of three HFE variants. (A) Separation of amplicons generated from a wild-type sample. The three wild-type ET-ROX (red) labeled AS amplicons are found at 174 bp (63H-187c), 211 bp (65S-193a), and 223 bp (282C-845g). The other ET-ROX (red) PCR controls are found at 244, 268, 281, 293, and 363 bp. The inset presents the raw unprocessed data. The 528 bp control is marked in this raw data but is not displayed in the analyzed data because it is outside the sizing standard range. The 20 TET-labeled sizing standards are detected in the green channel. (B) Separation of the amplicons generated from an S65C heterozygote sample with PCR controls labeled with ET-TAM (black). The presence of the 65C-193t mutant allele in this sample appears as an extra peak labeled with ET-R110 (blue) at 201 bp. (C) Separation of the amplicons generated from an H63D/C282Y compound heterozygote sample. Note the presence of the extra ET-R110 (blue) peaks at 160 and 232 bp, generated by the presence of the 63D-187g and 282Y-845a mutant alleles, respectively.
High-Throughput CAE Analysis
To demonstrate that this assay is easily applied in a high-throughput format, we performed 96 simultaneous separations on the radial CAE microplate. Figure 2 shows the electropherograms from the separation of 96 of the 100-sample CEPH Caucasian panel. Each electropherogram in this image was individually aligned to match the 70 bp and 400 bp standard peaks. Note that all separations were completed in <10 min and all the amplicons and standards were resolved. The small amount of channel-to-channel variation in migration times and patterns observed is due to the nonuniform positioning of the electrodes in the injector reservoirs and does not affect each channel's internally referenced migration (Shi et al. 1999). Each of the samples in this study was genotyped on the CAE microplate, as described, between four and eight times to verify separation reproducability.
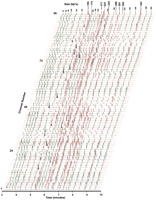
Ninety-six CEPH samples genotyped for three HHC-associated variants simultaneously on a 96-channel radial CAE microplate. The numbers at the top of the electropherograms indicate the standard and amplicon sizes in bp. Electropherograms were aligned to match the 70 bp and 400 bp flanking standards. Samples with mutant alleles are easily identified by the presence of the extra R110 (blue) peaks at 160, 201, and 232 bp (63D-187g, 65C-193t, and 282Y-845a alleles, respectively).
Sizing of PCR Amplicons
Table 1 presents an evaluation of the amplicon sizes as determined by Genetic Profiler. The largest amount of variation from the predicted amplicon size is seen in the 160–232-bp amplicon range, whereas the variation of the larger amplicons decreases. For example, the predicted 160 bp amplicon had a mean value of 161.9 bp, median of 162.1 bp, with a standard deviation of 0.8 bp and a standard error of 0.3 bp for the 25 160-bp amplicons that were analyzed. This compares to the predicted 363-bp amplicon, which had a mean value of 362.8 bp, median of 362.1 bp, standard deviation of 0.3 bp and standard error 0.04 bp (N = 100). These variations in size correspond to the electrophoretic migration differences caused by sizing ET-labeled amplicons against a mono-labeled DNA sizing standard. Another factor contributing to variation might be the presence of the unlabeled complement of all the amplicons, which although we electrophoresed under denaturing conditions (see Methods), might still affect migration. The small variation from predicted size in no way hinders the unambiguous genotyping of all samples.
Analysis of Amplicon Sizes
Frequencies of Alleles
The frequencies of the C282Y (845g->a), H63D (187c->g), and S65C (193a->t) variants in the 100 reference Caucasian samples were found to be 3%, 11%, and 2%, respectively. The CEPH family of 18 samples and the 'universal' CEPH donor did not contain any mutant alleles. Three samples with H63D homozygote mutant genotypes were detected in the 100 Caucasian samples. No other homozygote mutant or compound heterozygote genotypes were detected. All variant alleles found were confirmed with restriction digestion analysis and the results matched in all cases.
DISCUSSION
This study was performed to demonstrate the capabilities of radial CAE microplates with four-color fluorescence detection for high-performance multiplex SNP analysis. To accomplish this we developed and applied a clinically relevant three-site 'multiplex' assay for the most common HHC-associated mutations. The ASPCR format applied here has numerous advantages. By utilizing joint coamplification of ASPCR amplicons and PCR controls, the efficacy of each reaction is easily verified. The use of differentially sized and labeled amplicons for genotyping allows for the unambiguous identification of all alleles present. The actual volumes of final product analyzed are so small (1%–5% of the total generated product) that scaling down reactant volumes and the subsequent cost savings are easily feasible. Lastly, the assay can be modified to add/substitute additional SNP sites at the HFE locus as they are developed, without having to redo any of the existing PCR schemes.
To verify the high-performance capabilities of this assay, we simultaneously separated and genotyped 96 samples at the three HFE variant sites in <10 min. This demonstrates the feasibility of collecting large amounts of data on a very rapid time scale using radial CAE chip analysis. Some of these samples had been analyzed previously on the Molecular Dynamics 96-capillary MegaBACE 1000 instrument (Medintz et al. 2000b); the MegaBACE analysis took 75 min as opposed to the 9.5 min demonstrated here. Each sample analyzed on the CAE microplates consisted of a complex mixture of ladder and multiple differentially labeled amplicons, and yet single-base-pair resolution was achieved. This is due to the use of denaturing conditions and LPA as the separation matrix. This 1-bp resolution is a substantial improvement over previous microplate CAE analyses that utilized hydroxyethylcellulose (HEC) as the matrix resulting in 5–10-bp resolution (Shi et al. 1999; Medintz et al. 2000a). The low resolution in the earlier studies was due to the poorer separation qualities of HEC and the shorter (33 mm) capillary separation lengths on the 100 mm diameter microplate. The increased resolution achieved here is illustrated by the wide single-bp resolution range (70–528 bp) compared to the 136–400 bp range with 5–10-bp resolution demonstrated previously (Medintz et al. 2000a).
By coupling ASPCR of multiple loci to high-performance radial CAE microplate analysis we were able to easily analyze three HHC-associated variants. The rapid simultaneous analysis of three variant sites could not be undertaken with current SSCP or heteroduplex technology due to the inherent complexity of the electrophoregrams generated therein. Additionally, two of the three sites analyzed here are very closely located within the HFE gene (6 bp). This would be a severe problem for hybridization-based assays that utilize allele-specific oligonucleotide (ASO) probes. Closely located polymorphisms substantially reduce the discrimination of the ASO probes in genotyping assays. (Keller and Manak 1993; Landegren et al. 1998).
This report also demonstrates the use of four-color DNA analysis for genotyping using ET labels. Previous microplate CAE genotyping utilized two-color analysis with bis-intercalating dyes (Shi et al. 1999). These dyes are effective but they do require the development and execution of careful DNA quantitation and incubation prior to analysis (Shi et al. 1999). Additionally, even when utilizing covalent single-dye fluorescent-labeling of ASPCR amplicons, it was found that a large concentration of PCR product was needed for detecting ROX-labeled amplicons due to its poor fluorescent excitation at 488 nm (Medintz et al. 2000a). The use of ET primers overcomes these problems due to the efficient excitation of all four labels with a single laser line and its higher sensitivity (Ju et al. 1996a). We are currently developing convenient ET cassette labeling technologies that will make the conversion to ET formats even more facile (Ju et al. 1996b).
The present results also demonstrate substantial improvements in data analysis. Previous studies utilized multiple data analysis programs and local linear regression for sizing (Shi et al. 1999; Medintz et al. 2000a;). By adapting Genetic Profiler for data analysis, four-color data are easily analyzed and the results are available as both processed data and four-color electropherograms. As seen in Table1, there is a slight discrepancy between predicted amplicon sizes and those actually determined. This is caused by electrophoretic migration differences resulting from the separation of ET-labeled amplicons against mono-labeled single-stranded sizing fragments. The introduction of mobility correction in Genetic Profiler would easily resolve this systematic variation.
The C282Y allele frequency of 3% measured in the population studied here is consistent with the reported frequency ranges of 2.9%–7.5% determined in other reference Caucasian populations (Beutler et al. 1996; Mura et al. 1997; Simonsen et al. 1999). The H63D frequency of 11% compares to reports of frequencies ranging from 12.4% to 16.5% (Beutler et al. 1996; Mura et al. 1997; Simonsen et al. 1999). The S65C frequency of 2% measured compares to frequencies of 1.5% and 2.4% reported previously (Mura et al. 1999; Simonsen et al. 1999; Medintz et al. 2000a). Taken together, these findings confirm the high frequency of these variant HHC alleles in the Caucasian population. Given the high Caucasian carrier frequency (∼1 in 10) of this disease, the huge number of affected people (∼1.5 million in the U.S. alone), and the fact that the best treatment is early detection leading to treatment before physiological damage is apparent, the necessity and utility of a rapid and comprehensive genetic test becomes self-evident. Indeed, the results demonstrated here show that this assay format coupled to high-performance radial CAE microplate analysis will make the rapid screening of large numbers of individuals at large numbers of SNP's possible. The application of this flexible genotyping assay and powerful CAE microplate technology to other health disorders, given the growing number of SNP-linked predispositions to diseases, is also strongly indicated.
METHODS
Samples
DNA samples were obtained from Coriell Cell Repositories. These included the CEPH (Centre d'Estude du Polymorphisme Humain) panel of 100 reference Caucasian samples (part no. HD100CAU), CEPH family no. 00884 as well as CEPH samples NA10859 (universal CEPH donor sample), NA14620, NA14641, and NA14690. The latter three samples had previously been characterized for specific HHC-related mutations [C282Y(845g->a), H63D(187c->g), S65C(193a->t)] and served as positive controls.
Primer Synthesis
ET primers were synthesized and labeled as in Ju et al. (1996) andHung et al. (1998). The donor dye utilized was 3–(ɛ-carboxypentyl)-3′-ethyl–5–5′-dimethyloxacarbocyanine, CYA, which was covalently attached to a modified amino linker at the 5′ end of each ET primer (Ju et al. 1996a; Hung et al. 1998). Acceptor dyes included 6-carboxyrhodamine-110, R110, carboxytetramethylrhodamine, TAM, and 6-carboxy-X-rhodamine, ROX (Molecular Probes). Acceptor dyes were attached to a modified T-nucleotide located within the sequence of each ET primer (Hung et al. 1998). For specific acceptor dye positions within each ET primer sequence, see Table2.
Primer sequences
Allele-Specific PCR
Each sample underwent six separate AS amplification reactions (Hecker et al. 1996; Medintz et al. 2000a), one for each of the six alleles being tested (see Table 2). Separate reactions were utilized because multiple coamplifications (more than two) can result in false PCR products, bias towards a specific product, and misamplifications. Each of the six PCR reactions contained two sets of primers; one set defining the AS amplicon and a second set acting as a PCR control (Medintz et al. 2000a). The AS amplicon is only generated during PCR when that particular allele is present in the template DNA, whereas the PCR control amplicon is always generated and therefore acts as a positive PCR control. The PCR controls are complementary to a sequence located near the 3′ end of the HFE (HLA6) gene located on chromosome 6. Control amplicons were labeled in either an ET-ROX or ET-TAM scheme. Wild-type AS amplicons are all ET-ROX-labeled, whereas the mutant AS amplicons are ET-R110-labeled.
As can be seen in Tables 2 and 3, AS amplicons are defined by both size and color. The AS amplicons elute in the 174–232 bp size range, whereas PCR control amplicons elute in the 244–528 bp size range.
Reaction Primer Mixtures and Expected PCR Products
Reactions utilized PCR Master Mix (Qiagen) with 50 ng of genomic DNA added to each 25 μL reaction volume. For specific primer concentrations and mixtures see Table 3, and for ASPCR thermal cycling conditions, see Supplementary Table 1, available athttp://www.cchem.berkeley.edu/∼ramgrp/supplemental/. Successful PCR was verified on agarose gels prior to CAE analysis.
The presence of mutant alleles in particular samples was verified by reamplifying with a different set of primers and genotyping by restriction analysis as in Mura et al. (1994) and Jazwinska et al. (1996).
Sample Preparation
For each sample, all six ASPCRs were combined and processed using the Qiaquick PCR Purification kit (Qiagen). DNA was eluted with 50 μL of 0.5 x TE buffer. The BioVentures TET MapMarker standard was used for sizing and contains 20 single-stranded DNA fragments labeled with tetrachlorofluorescein (TET). Fragment sizes included 70, 80, 90, 100, 120, 140, 160, 180, 190, 200, 220, 240, 260, 280, 300, 320, 340, 360, 380, and 400 bp. Processed DNA samples were mixed with the TET standard, DI formamide, and 0.1 x TE (ratio of 2.5 sample : 10 formamide : 7.5 TE : 2.5 TET) to yield a final volume of 1.5 μL. Just prior to loading, samples were denatured at 95°C for 4 min and immediately placed on ice.
Microfabrication and Microplate Design
CAE microplates were fabricated at the University of California/Berkeley Microfabrication Laboratory as described previously (Kheterpal et al. 1996; Simpson et al. 1998b; Shi et al. 1999). The design of the chip (Fig. 3) is a modification of one presented previously in that the substrate is now 150 mm in diameter (Shi et al. 1999). Isotropic etching with HF formed channels ∼110 μm wide by 50 μm deep. The distance along the separation capillary from the 250μm twin-T injector to the detection point is 55 mm. The microchannels were coated with polyacrylamide as described by Hjerten (1985) to prevent electro-endosmotic flow. For electrophoresis, the microplates were filled with 3% Long Read Linear Polyacrylamide (LPA) matrix containing 7M urea (Amersham Pharmacia), using a microplate gel loader/pressure washer device (to be presented elsewhere).
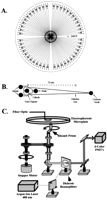
Design of the 96-channel CAE microplate and radial scanner. (A) Mask pattern used to form the 96-channel radial CAE microplate on a 150mm diameter wafer. The black circle in the center of the plate indicates where the rotary scanner interrogates the separations. (B) Enlarged view of a set of individual capillaries showing the common cathode, waste, and anode reservoirs shared by the two capillaries. (C) Schematic of the radial confocal microplate scanner. Adapted from Shi et al. (1999) and Medintz et al. (2000a).
Electrophoresis and Microplate Scanning
Samples were loaded, injected, and electrophoresed as described inShi et al. (1999) and Medintz et al. (2000a). The loaded microplates were placed onto the microplate holder of the rotary confocal scanner and heated at 40°C; a circular electrode array was placed on top of the microplate making electrical contact with all the reservoirs. Samples underwent electrokinetic injection for 100 sec by applying +5 V to the sample reservoir and +425 V to the waste reservoir while ‘floating’ the anode and cathode reservoirs. Separation was immediately carried out following injection by applying +1350 V to the anode reservoir, +200 V at the cathode and +325 V to the sample and waste reservoirs.
During separation, samples were detected within the microplate by the laser-excited, rotary confocal scanner (Fig. 3C). The design and function of the scanner is discussed extensively in Shi et al. (1999). Briefly, the rotary confocal detection system consists of a rotating objective head coupled to a four-color confocal detection unit allowing for four-color analysis (Kheterpal et al. 1996; Shi et al. 1999). A 488-nm beam from an Ar+ laser is used for excitation and detection. Each of the four spectral detection channels have bandwidths of ∼30 nM. The ‘blue’ channel detects from 505 to 530 nM (R110 maximum emission ∼525 nM). The ‘green’ channel detects from 530 to 560 nM (TET max. ∼538 nM). The ‘black’ channel detects from 560 to 590 nM (TAM max. ∼572 nM), and the ‘red’ channel detects > 590 nM (ROX max. ∼620 nM).
Data Analysis
Data for each 96-radial array run were collected and stored as a data appended text (DAT) file written to a specific run folder. Raw data files have to be converted to electrophoretic signal data (ESD) file formats to be processed by the Genetic Profiler (GP) genetic analysis software (Molecular Dynamics).
We utilize a TEXT-to-ESD conversion program in order to accomplish this file format change (Wedemayer et al. 2001). The resulting converted ESD files are imported into GP and processed/sized using a third-order local algorithm. Processed data are accessed as sized data and four-color sized electropherograms. Among the utilities of this analysis software are background subtraction, three-color sizing against a fourth color standard, peak editing, and the addition of multiple run folders to a particular project (Wedemayer et al. 2000).
Acknowledgments
We thank C. Emrich for assistance with microplate fabrication and G. Wedemayer for help with figure preparation and software. We also thank Molecular Dynamics for the Genetic Profilersoftware. This work was supported by NIH Program Project grant P01 CA77664 from Johns Hopkins University, a grant from Amersham-Pharmacia Biotech, NIH grant HG01399, and by the Director, Office of Science, Office of Health and Environmental Research of the U.S. Department of Energy under contract DE FG91ER61125. G.S.'s contribution to this project was supported by a grant from the Kaiser Foundation Research Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL rich{at}zinc.cchem.berkeley.edu; FAX 510 642 3599.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.164701.
-
- Received September 13, 2000.
- Accepted January 2, 2001.
- Cold Spring Harbor Laboratory Press








