
Sequence and Analysis of Chromosome I of the Amitochondriate Intracellular Parasite Encephalitozoon cuniculi (Microspora)
Abstract
A DNA sequencing program was applied to the small (<3 Mb) genome of the microsporidian Encephalitozoon cuniculi, an amitochondriate eukaryotic parasite of mammals, and the sequence of the smallest chromosome was determined. The ∼224-kb E. cuniculichromosome I exhibits a dyad symmetry characterized by two identical 37-kb subtelomeric regions which are divergently oriented and extend just downstream of the inverted copies of an 8-kb duplicated cluster of six genes. Each subtelomeric region comprises a single 16S–23S rDNA transcription unit, flanked by various tandemly repeated sequences, and ends with ∼1 kb of heterogeneous telomeric repeats. The central (or core) region of the chromosome harbors a highly compact arrangement of 132 potential protein-coding genes plus two tRNA genes (one gene per 1.14 kb). Most genes occur as single copies with no identified introns. Of these putative genes, only 53 could be assigned to known functions. A number of genes from the transcription and translation machineries as well as from other cellular processes display characteristic eukaryotic signatures or are clearly eukaryote-specific.
[The sequence data described in this paper have been submitted to the EMBL data library under accession no. AL 391737.]
The eukaryotic phylum termed Microspora is composed of more than 1000 amitochondriate unicellular species which are all obligate intracellular parasites found throughout the entire animal world, including humans. Many of the Microspora species exhibit a wide host range, parasitizing a large number of mammalian species. The clinical importance of these opportunistic pathogens in man was discovered in immunodepressed patients with the emergence of AIDS and through the evolution of surgical and medical technologies, mainly in transplantation and cancer chemotherapy involving immunosuppression. Recently, microsporidiosis also has been detected in immunocompetent patients (Sandfort et al. 1994; Raynaud et al. 1998). Evidence for waterborne transmission of several species of microsporidia has been reported (Dowd et al. 1998) and the zoonotic potential of Encephalitozoon cuniculi genotypes in mammals has been demonstrated (Mathis et al. 1997).
Species of the Encephalitozoon genus were shown to harbor the smallest known eukaryote nuclear genomes with a size of <3 Mb (Biderre et al. 1995, 1999b). However, little is known about the organization of microsporidian DNA sequences, except for the rDNA transcription unit, which was first claimed to have a prokaryote-like organization because of the small size of rRNAs (16S and 23S) and fusion of the 5.8S sequence to the 5′ end of 23S coding region (Curgy et al. 1980; Vossbrinck and Woese 1986). Models of secondary structure clearly showed a strong reduction of several domains within the microsporidian 23S rRNA but typical eukaryotic features of this molecule were retained (De Rijk et al. 1998; Peyretaillade et al. 1998a; Van de Peer et al. 2000). A few protein-coding genes also have been sequenced. To the list published by Weiss and Vossbrinck (1999), can be added genes encoding the largest subunit of the RNA Pol II (Hirt et al. 1999), a TATA box-binding protein (Fast et al. 1999), and a spore wall protein (Bohne et al. 2000). Most sequencing data were used for phylogenetic purposes and led to controversial hypotheses about the evolutionary origin of microsporidia. Using phylogenetic analyses based on the sequences of small-subunit rRNA and two translation elongation factors (Vossbrinck et al. 1987; Kamaishi et al. 1996a,b), these amitochondriate organisms are viewed as early-diverging eukaryotes. However, the concept that microsporidia could represent primitively amitochondriate eukaryotes was not supported by the finding of microsporidian genes encoding a mitochondrial HSP70 homolog (Germot et al. 1996; Hirt et al. 1997; Peyretaillade et al. 1998b). In addition, phylogenetic trees constructed with four different protein-coding genes were all suggestive of a late origin and a relationship with fungi (Edlind et al. 1996; Keeling and Doolitle 1996; Fast et al. 1999; Hirt et al. 1999). Only three genes encoding microsporidia-specific structural proteins have been reported (Delbac et al. 1998; Keohane et al. 1998; Bohne et al. 2000).
The analysis of a 4.3-kb sequence contig from chromosome I provided preliminary information on the chromosomal arrangement of protein-coding genes, revealing a strong reduction of intergenic spacers (Duffieux et al. 1998). The occurrence of gene length variability has also been documented through the interspecific comparison of microsporidian rRNA sequences (Weiss and Vossbrinck 1999) and suggests that strong selection pressures may have driven theE. cuniculi into nuclear genome close to the minimal gene complement for a “selfish” eukaryote dependent on a host organism. To test this concept and to obtain new insights into microsporidian biology and evolution, a whole genome sequencing project was initiated in 1998. Here, we report the complete sequence of E. cuniculichromosome I and highlight some features of its organization and gene content.
RESULTS
General Features
The complete sequence of E. cuniculi chromosome I was determined using a whole shotgun strategy followed by targeted gap closure and polishing. The proposed numbering in nucleotides of the chromosome for a precise analysis, 199,956 base-pairs (bp), extends from the 3′ end of the large subunit (LSU) rDNA gene at one extremity to the 3′ end of the second LSU rDNA associated with the other extremity. The analyzed part of the chromosome does not take into account either the telomeric repeats nor the multiple subtelomeric repeats in proximal ends.
Because the highly repeated sequences are very short, their assembly cannot be considered to reflect the exact full sequence organization. The telomeric repeats characterized in this study were estimated to be situated between 8.5 kb and 9.5 kb from the end of the 23S rDNA gene (Fig. 1A) as deduced by physical mapping (Brugère et al. 2000a). As shown in Figure 1A, the chromosome is composed of two distinct regions: an ∼150-kb central region composed essentially of unique sequences and comprising most, if not all, protein encoding sequences (CDS1–CDS132), and two ∼37-kb subtelomeric divergent regions, each including one rDNA transcription unit composed of a small and a large rDNA gene, the 5′ of the latter being fused to 3′ of the 5.8S gene. The G + C content is 47.30% for the coding region, slightly lower (43%) in the intergenic regions and higher (52.85%) in the overall telomeric and subtelomeric regions (Fig. 1B). Analysis using the motif discovery programMEME revealed A + T rich consensus transcription promoting sequences (AAATGACA; ATAAAAAA) located in the 50 bp region upstream of the putative ATG in the 53 genes encoding proteins with known functions (see below). The presence at the junction of the subtelomeric region and the core region of a strict duplication of an 8-kb cluster comprising six potential genes including aminopeptidase, dihydrofolate reductase, thymidylate synthase, serine hydroxymethyl transferase, an ABC transporter, and a yet unknown protein is also observed. This gene duplication extends the dyad symmetry of the chromosome extremities to over >45 kb.
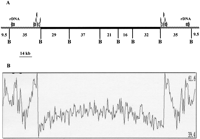
Scheme of Encephalitozoon cuniculi chromosome I organization. (A) The symmetrical organization of chromosome extremities, the location of the two rDNA transcription units in the subtelomeric region (gray), and the presence of a six putative CDSs duplication at the junction of the core and subtelomeric region (see Results) are illustrated. The central black trait depicts the core region composed mostly of unique genes. B represents BssHII restriction sites used for physical mapping and sequence confirmation in this study. (B) G + C percentage along the chromosome calculated in a 500-nt window with 100-nt progression increments. The highest and lowest G + C percentage are shown on the right of the panel.
Telomere Repeats, Telomere Associated Sequences, and Tandem Repeats
Telomeric sequences were identified in a telomere-enriched library constructed as described in Methods. The insert extremities were sequenced and in ∼12% of the clones, repetitive sequences were detected next to the NotI adapter used for cloning. The repetitive elements are composed of alternate G(A or G)GCCT(C or T)CT, GAGCCTTGTTT, and GAGACGCAGTGTTGCCAGGATG. In the pBAM recombinant library (genomic DNA digested by CviJI, see Methods), ∼0.1% of the clones exhibited the same repeats but none were found in the pBAC library (genomic DNA digested by Sau3A, see Methods). This could be explained by the fact that the CviJI cuts in PuGCPy, i.e., in sequences within telomeric repeats. To verify that these repeats were indeed telomeric, they were used as probes in Southern blot hybridization experiments, following Bal31 digestion kinetics experiments (see Methods). As shown in Figure2, the genomic DNA that hybridizes with the repeats probe was completely degraded by the Bal31 exonuclease in 7.5–8 min at digestion rates of ∼155 bp/min. This demonstrates both that the repeats are telomeric and that the size of the region is between 1100 bp and 1200 bp.
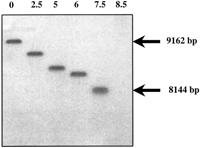
Bal-31 digestion blot analysis of telomere repeats.Encephalitozoon cuniculi total genomic DNA was digested byBal-31 nuclease for 0, 2.5, 5, 6, 7.5, 8.5 min (lanes1–6, respectively) and subsequently digested withBssHII. This was followed by size-fractionation on agarose gel electrophoresis and Southern blotting onto a nylon membrane. The blot was then hybridized with 32P-labeled telomere repeat oligonucleotides as probes and autoradiographed for 5 h.
It should be emphasized that our study of the telomeric repeats was not restricted to chromosome I alone. The organization of chromosome ends was analyzed through both physical mapping (Brugère et al. 2000a) and sequencing of other chromosome ends. This demonstrated very limited if any variation in the subtelomeric sequences with an identical distance between the BssHII site and the chromosome end. Thus, the telomere repeat probes hybridized to all 11 chromosomes in Southern blot experiments (data not shown) and the compact size of the bands revealed in the blots after Bal31 digestion of all 22 chromosome extremities indicates a low variability of repeat length (≤50 bp).
The search for tandem repeats in the subtelomeric region situated between the rDNA cluster and the core [to the first coding DNA sequence (CDS) encoding aminopeptidase] was performed using the Tandem Repeats Finder Program and reveals a highly structured region with 11 repeat types shown in Table 1. The largest repeat is 30-bp long and is present in 2.4 copies; the smaller one is the ACACACC present in 3.9 copies. Two regions devoid of tandem repeats (8291–16002; 16050–18599) could be potentially transcribed and eight putative open reading frames (ORFs) without significant identities to known proteins were detected by Glimmer. A strict conservation of this organization is shown in the other extremity. Only five single nucleotide polymorphisms (position 10193, 10233, 10372, 10620, 10630) were found. The size of the subtelomeric region between the telomere repeats and the rDNA cluster obtained after sequence assembly was of only 3.5 kb instead of ∼9.5 kb deduced by physical mapping (Brugère et al. 2000a). This underestimation is attributable to the presence of short highly repetitive sequences, preventing proper assembly and limiting the interest of a precise description.
Tandem Repeat Organization of the Internal Subtelomeric Region of Chromosome 1 ofE. cuniculi
Gene Content
Using Glimmer as a tool for gene detection, 131 putative ORFs were identified as schematized in Figure3. An additional ORF (CDS 95) was identified by the Prodom BLAST and Wise2programs and encodes for a putative ring-box-like protein. The size of this CDS (99 amino acids) was below the detection threshold ofGlimmer fixed at 100 amino acids. Other small ORFs with no homologies or characteristic signatures will remain undetected under these conditions. Genome analysis depicts a highly compact organization with a mean gene density of one gene per 1.14 kb. Intergenic regions are very small with a maximum of 639 bp between CDSs 40 and 41 and a mean distance of 116 bp. Some ORFs seem to overlap each other but in all such cases at least one of the ORFs remains ill-defined and potentially encodes a hypothetical protein with an undetermined function. Examples of possible overlaps were observed between a serine hydroxy methyl transferase gene (CDSs 4 and 129) and thymidylate synthase (CDSs 3 and 130). However, even this possibility seems remote because multiple alignments using CLUSTAL W applied to the putative serine hydroxy methyl transferase gene indicate that the most probable initiation codon is located 196 amino acids downstream of the putative start codon determined by Glimmer. This unlikely overlap was therefore not presented in Figure 3. The number of ORFs is in the same range on both DNA strands with 44% (58 CDSs representing 66,876 bp) on the Watson strand (upper strand) and 56% (74 CDSs representing 73,541 bp) on the Crick strand (lower strand). The largest CDS (CDS 20, 3752 bp) encodes a putative helicase with a molecular weight of 144,575 Da. CDS 84 likewise encodes a large protein (141,454 Da) with an unknown function but containing five putative transmembrane domains. A total of 11 putative proteins harbor transmembrane domains. Among them, is chitin synthase (CDS 126), which must play a major role in spore wall formation in microsporidia. Analysis of the third largest protein (115,974 Da) encoded by CDS 112 reveals the presence of a calcium-binding site suggesting a role in an as yet undetermined regulatory process. The CDSs coding for proteins with either putative or unknown functions represent ∼60% of the total ORFs identified. Table 2 presents a functional classification of the proteins encoded on chromosome I. We note a high proportion (∼20%) of genes implicated in replication, transcription, and translation processes. Ten putative proteins with zinc-finger (CDSs 22, 28, 42, 59, 60, 62, 67, 95, 101, 106) or s-antigen (SWI3, ADA2, N-COR, and TFIIIB) domains are observed and probably function in regulatory mechanisms of nucleic acids synthesis. Contractile domains were identified in three putative proteins (CDSs 26, 41, 107) and could be related to the myosin family. Of the three sugar metabolism genes identified, two belong to the trehalose pathway (CDS 64 alpha, alpha-trehalose-phosphate synthase, and CDS 71 trehalose-6P phosphatase). Codon usage was studied in individual genes (putatively high and low expression rates) as well as for the whole ORF population. On the whole, the codon usage resembles that of a standard unicellular eukaryote (results not shown). Fifteen additional ORFs with no known homology were detected in the subtelomeric region byGlimmer and of these only eight CDSs that are not part of the tandem repeat sequences are shown in Figure 3. Two probable tRNA genes (Gly, Ser) were detected by the tRNAscan-SE program and are located at nucleotide positions 50,585 and 88,026, respectively. The tRNASer gene contains an extra U residue between the acceptor and the D-loop stems, in position 10 of the classical cloverleaf model. No intron was detected in these two tRNA genes.

Putative gene map of Encephalitozoon cuniculi chromosome I. Predicted coding regions are shown with the arrows indicating the transcription orientation; the black box depicts the rDNA transcription unit; the chevrons depict the telomeres. Genes are color coded according to broad role categories as shown in the key.
Classification of E. cuniculi Genes According to Clusters of Orthologous Groups of Proteins (COGs)
DISCUSSION
Large-scale sequencing in genomes from eukaryotic parasites to date is limited to two complete chromosomes from Plasmodium falciparum(Bowman et al. 1998; Gardner et al. 1998) and one fromLeishmania major (Myler et al. 1999), and to a large contig from Trypanosoma cruzi (Andersson et al. 1998) and a high throughput EST project in Toxoplasma gondii (Ajioka et al. 1998). The characteristic feature of the chromosome structure ofE. cuniculi is the highly symmetrical organization of the two subtelomeric regions that occupy almost one third of the chromosome length and consist of one rDNA unit with a closely associated cluster of six genes each. A similar organization has been described in the vestigial genome of a cryptomonad nucleomorph (Zauner et al. 2000) and of a chlorarachniophyte nucleomorph, with a centripetal orientation of the rDNA unit in the latter organism (Gilson and McFadden 1997). The telomeric repeats of the nucleomorph chromosomes of Guillardia theta [(AG)7AAG6A, Zauner et al. 2000] like those of a number of fungi (C2–3ACA1–6 inSaccharomyces; ACAC2ACATAC2 TA2TCA3TC2GA in Kluyveromyces; and C1–6G0–1 T0–1GTA1–2 inSchizosaccharomyces) differ from the standard 5–8 nucleotide repeats of other eukaryotes. In E. cuniculi, the telomeric repeats are also relatively complex but it would be premature to conclude some close phylogenetic relationship with the previous organisms.
The understanding of the sequence organization in the subtelomeric region around the rDNA unit will be of interest for precise localization of the sites of chromosomal rearrangement that have been predicted from studies on intraspecies karyotype variability (Biderre et al. 1999a; Brugère et al. 2000b). Moreover, the phenotype of some parasitic protozoans can be affected by subtelomeric rearrangements as demonstrated for knob formation and gametocyte production in P. falciparum (Pologe and Ravetech 1986; Day et al. 1993). As in P. falciparum and P. berghei, theE. cuniculi chromosome structure is characterized by the conservation of the two subtelomeric regions. Antigenic variation inTrypanosoma brucei is also linked to chromosome end reorganization by gene conversion (for review, see Pays and Nolan 1998). In Microsporidia, however, programmed rearrangements inducing phenotype variability have not yet been reported and extensive studies will be needed to understand their physiological functions (centromeres? replication origins? recombination hot-spots, etc.).
The genes in chromosome I of E. cuniculi are highly packed with a density of one gene per 1.14 kb which is higher thanSaccharomyces cerevisiae (one per 2 kb; Goffeau et al. 1996),L. major (one per 3.26 kb; Myler et al. 1999), P. falciparum (one per 4 kb; Gardner et al. 1998; Bowman et al. 1998),T. cruzi (one per 4.5 kb; Andersson et al. 1998) and C. elegans (one per 5 kb; The C. elegans Sequencing Consortium 1998). This denotes, at least in part, the scarcity ofE. cuniculi introns. The CDS mean size, estimated at 1070 bp, partly reflects a reduction in the mean protein size. Proteins such as actin and ribosomal proteins remain highly conserved in length but some enzymes of intermediary metabolism appear to be greatly shortened, such as those involved in trehalose biosynthesis. The molecular weight for the trehalose-6-phosphate phosphatase of S. cerevisiae is 103 kDa compared to 82 Da for E. cuniculi. This is reminiscent of the previously described reduction in the 16S and 23S rRNA sequences (Curgy et al. 1980).
The gene content of chromosome I suggests some interesting physiological features. Two genes coding for enzymes implicated in trehalose biosynthesis (alpha, alpha-trehalose phosphate synthase, and trehalose-6P phosphatase) were found. Trehalose is known to represent a large fraction of stored carbohydrates in fungi. Increased osmolyte concentration through the cleavage of trehalose into two glucose molecules was proposed as responsible for the pressure increase required for initiating microsporidian spore germination, a crucial step for the entry of the parasite into a new host cell (Undeen 1990). It seems likely that this disaccharide also plays a role as a glucose energy reserve for possible degradation through a glycolysis pathway in this amitochondriate organism (Weidner et al. 1999). High concentrations of trehalose in spores of both aquatic and terrestrial microsporidian species (Undeen and Vander Meer 1999) may indicate a long-term energy storage facility due to its protective properties, which have been described by Singer and Lindquist (1998). Recently, Yfh1, a yeast homolog of mammalian frataxin, was found to be important in the efflux of iron from mitochondria (Radisky et al. 1999) and two other yeast proteins (NFU1 and ISU1) related to NifU protein play a role in iron homeostasis in assembly, insertion, and/or repair of mitochondrial Fe-S clusters (Schilke et al. 1999). Putative frataxin and NIFU protein identified on chromosome I would be valuable for studies on the evolution of iron homeostasis in amitochondriate organisms. Because of their involvement in the various microsporidian life stages, the presence of myosin, dynamin and actin, which are typical eukaryote genes on chromosome I is also of significant interest. In a recent electron microscope immunocytochemical study, actin was demonstrated to be located mainly at the periphery of the developmental cell stages including mature spores (Bigliardi et al. 1999). A possible involvement of actin in the biogenesis of the complex invasive apparatus, especially the long extrusome-like structure known as the polar tube, has been suggested.
Very little is known about microsporidian transcriptional processes. Only a single mRNA encoding a spore wall protein with 5′ and 3′ UTRs 9 and 165 nucleotides long, respectively, has been described in microsporidia (Bohne et al. 2000). Identification of a gene encoding a TATA binding protein and an A + T rich consensus sequence in intergenic regions emphasizes the probable transcriptional regulatory implications of TATA box recognition. No polycistronic gene organization like that of trypanosomatid and kinetoplastid protein-coding genes (Andersson et al. 1998; Myler et al. 1999) has been found and E. cuniculi chromosome I encodes mostly monocistronic transcription units. Only one gene cluster is observed, the two adjacent putative thymidylate synthase and dihydrofolate reductase genes. These two genes, which encode a monofunctional peptide, are independent in both fungi and animals but are fused in plants and protozoa (Beverly et al. 1986; Cella et al. 1988). One spliceosomal-type intron has been described in a ribosomal protein gene located on E. cuniculi chromosome X (Biderre et al. 1998) but no possible intron was detected in the chromosome I sequence. Indirect evidence that microsporidia harbor genes with introns was previously suggested by the discovery of divergent U2 snRNA in Vairimorpha necatrix (DiMaria et al. 1996) and more recently with U2 and U6 snRNA gene descriptions in Nosema locustae (Fast et al. 1998). However, cis-splicing does not seem a common phenomenon in microsporidia unlike P. falciparum chromosomes 2 and 3 in which half of the genes have been predicted to contain at least one intron. Other protozoans such as Paramecium aurelia orTetrahymena thermophila contain a small number of genes with introns (<1 %). We cannot exclude however, the presence of short introns in-frame in 5′ and 3′ gene regions and characterization of cDNA will be of great help. A supernumerary U base is observed in the D stem of the tRNASer gene. Surprisingly, a tRNASer of the cryptomonad nucleomorph bears two introns, the first of which (intron 1, 10 nucleotides) occupies a unique position in the D loop whereas intron 2 typically resides in the anticodon loop as observed in other eukaryotic tRNAs (Zauner et al. 2000). The first base of intron 1 of the nucleomorph tRNASer is a U residue and could correspond to the supernumerary U residue of E. cuniculitRNASer which would hence be a remnant of an intron eliminated incompletely during evolution.
A better understanding of the microsporidian transcriptional and translational machineries as well as that of its metabolic pathways should greatly progress with the determination of the complete genome sequence and should accelerate drug discovery based on new therapeutic targets. Microsporidia infection is treated today by two drugs, albendazole and fumagillin, with limited success (Didier 1997). TNP-470, a semisynthetic analog of fumagillin of lesser toxicity, seems a promising treatment of some types of microsporidiosis (Coyle et al. 1998). Recently, methionine aminopeptidase-2 was identified in yeast as the cellular target of fumagillin (Sin et al. 1997) and the three-dimensional structure of the protein complexed to its fumagillin ligand determined (Liu et al. 1998). A methionine aminopeptidase is found on chromosome X of E. cuniculi and may contribute to structure-based microsporidiosis drug design. Genomics may lead to the revelation of new genes implicated in pathogenesis and relationships with host cells as well as to the development of better diagnostic tools and new treatments.
E. cuniculi has been presumed to be a diploid organism and some evidence for allelic chromosomes of different size was derived from hybridization data with different isolates (Biderre et al. 1999a;Brugère et al. 2000b). In the strain I mouse isolate used for the systematic sequencing project, only chromosome III exhibits potential allelism deduced from differential migration in agarose gels. The absence of flagrant allelic variation and the minimal amount of sequence polymorphism within the chromosome I could be explained by frequent recombination between homologous chromosomes in the context of asexual reproduction. An identical situation has been described for chromosome I of L. major (Myler et al. 1999). No characteristic sequence or (A + T) rich region corresponding to a putative centromere has been identified on chromosome I. The sequencing of the whole E. cuniculi genome should in the near future clarify whether the low degree of gene redundancy on chromosome I also prevails in the 10 larger chromosomes. The identification of major potential metabolic pathways is expected and the extent of a possible loss of genetic information related to the parasitic life style will be evaluated.
METHODS
Genome DNA Cloning, Nucleotide Sequencing and Sequence Validation
The reference mouse isolate of E. cuniculi (GB-M1; Biderre et al. 1994, 1999a) used throughout this work was provided by Professor E.U. Canning (Imperial College of Science, Technology and Medicine, London, UK). Parasite cells were grown on Madin Darby Canine Kidney (MDCK) cells as described (Beauvais et al. 1994). E. cuniculispores were isolated from infected cultures and DNA purified as described previously (Duffieux et al. 1998). Two libraries were constructed: (1) a 2.3 × 104 clone library in the plasmid vector BAM3 (Dr. Roland Heilig, unpubl. construction derived from pBluescript II KS+, Stratagene) harboring total genome DNA which was restricted randomly by CviJI into 3-kb fragments that were inserted into the unique SmaI site, and (2) a 2 × 105 clone library using a slightly modified Bacterial Artificial Chromosome (BAC) vector, pBeloBAC11, in which randomly restricted Sau3A 20–30 kb fragments were inserted into the unique BamHI site. For the latter library, spores were embedded in agarose and following proteinase K lysis, DNA was purified by AFIGE (Asymmetric Field Inversion Gel Electrophoresis) pulsed field electrophoresis using a Fige Mapper (BioRad) apparatus. For both libraries recombinant material was electroporated intoEscherichia coli K-12 DH10B electro-competent bacteria (GIBCO-BRL, Life Technologies) in 0.1 cm cuvettes using a Micropulser apparatus (BioRad). Electroporation conditions were 17.5 kV/cm, 25 μF, and 200 Ω. Extraction of plasmid or BAC DNA and sequencing of the recombinant material ends were performed according to standard procedures. Sequences were established using both ABI 377 (PE-Applied Biosystems; dye terminators) and LICOR 4200 (LICOR; dye primers) sequencers. Sequences were assembled using Phred, Phrap (Ewing and Green 1998; Ewing et al. 1998), andConsed (Gordon et al. 1998) software. Finishing, mainly gap filling, and resequencing regions of poor quality (polishing) were performed according to standard methods. Each nucleotide was sequenced at least three times either on both strands or using both dye primer and terminator chemistries.
The assembly of the contigs and their integrity were verified after in silico construction of the “minimum tiling path.” Relevant recombinant BACs were analyzed by four restriction endonucleases (BamHI, BglII, HindIII, and XhoI) to confirm their integrity. Also, the restriction map forBssHII and MluI sites was compared to that published by Brugère et al. (2000a). Assignment of contigs to chromosome I was based on known genetic markers (Biderre et al. 1997) and/or by hybridization of Southern blots of E. cuniculi chromosomes separated by pulsed field gel electrophoresis. Blots were hybridized using standard procedures with specific 35-residue-long oligonucleotides labeled with [α-32P]dCTP using terminal transferase (Amersham Pharmacia Biotech).
Sequence Analysis
The chromosome I consensus sequence was examined for putative ORF using the Glimmer program (Salzberg et al. 1998). The predicted amino acid sequence (CDS) from each putative ORF was used forBLASTP and PSI-BLAST searches in nonredundant protein databases (www.ncbi.nlm.nih.gov/cgi-bin/BLAST). Protein domains were determined using Prodom BLAST with graphical output (http://protein.toulouse.inra.fr/prodom/blast_form.html) andWise2 (DNA versus Pfam) (http://www.sanger.ac.uk/Software/Wise2/pfamsearch.shtml). Protein structural features were delineated with the SMART program (http://smart.embl-heidelberg.de/) and the PredictProtein server (http://www.embl-heidelberg.de/predictprotein/). Tandem repeats were identified by Tandem Repeats Finder Program (Benson 1999;http://c3.biomath.mssm.edu/trf.html) and repeats were screened against a library of repetitive elements using RepeatMasker(http://ftp.genome.washington.edu:80/cgi-bin/RepeatMasker). Multiple sequence alignments were constructed withCLUSTAL-W (Thompson et al. 1994). Transfer RNAs were identified with tRNAscan-SE (Lowe and Eddy 1997). Motifs discovery was done by the MEME program (http://meme.sdsc.edu/meme/website/). The 199956-bp sequence of chromosome I was submitted to EMBL under the accession no. AL391737.
Telomere-Repeat Determination and Telomere-Associated Sequence Analysis
Total genomic DNA was treated by Mung Bean nuclease, ligated to aNotI adapter, digested by EcoRI, dephosphorylated and inserted into pGEM-11Zf(+) vector (Promega), restricted byNotI and EcoRI, and gel purified. The recombinant plasmids were sequenced as described. Total E. cuniculigenomic DNA was digested by Bal31 nuclease (New England BioLabs) in a controlled kinetics experiment, restricted byBssHII, electrophoresed on a 0.4 % agarose gel and blotted following acid depurination onto Hybond-N+ nylon membranes (Amersham Pharmacia Biotech) in 0.5 M NaOH. The blots were then hybridized with oligonucleotides labeled with [γ-32ATP] by T4 polynucleotide kinase, washed, and submitted to autoradiography against Kodak X-Omat AR films at −80°C with intensifying screens. Oligonucleotides (5′GGGTCTTCTGGG TCTTCTGGGCCTCCT3′, GGGCCTCCGAGCCTTCTGAG CCTTCT, GGGCTTCCTGAGCCTTCTGGGCCTTCT, GAGCC TTGTTTGAGACAGTGTTGCCAGGATGT, and GAGACGCAG TGTTGCCAGGATGTGGG) were designed according to the sequence of the repeats determined in pGEM-11Zf(+) recombinant plasmids.
Acknowledgments
We thank G. Méténier for critical reading of the manuscript, P. Brottier and P. Wincker for coordinating the sequence work, B. Chebance and R. Guerry for technical assistance, and Susan Cure for expert reading of the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Present address: Laboratoire de Résonance Magnétique Nucléaire, Département de Chimie Structurale et Organique, Ecole Polytechnique, 91128 Palaiseau Cedex, France.
-
↵5 Corresponding author.
-
E-MAIL pierre.peyret{at}lbp.univ-bpclermont.fr; FAX 33-47340-7670.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.164301.
-
- Received September 8, 2000.
- Accepted November 8, 2000.
- Cold Spring Harbor Laboratory Press








