
Low-Complexity Regions in Plasmodium Proteins: In Search of a Function
Members of the genus Plasmodium are responsible for malaria, a disease endemic to vast tropical and subtropical areas, causing millions of deaths each year. Of the Plasmodiumspecies that infect humans (P. falciparum, P. vivax, P. ovaleand P. malariae), P. falciparum is the most virulent. Infection can lead to cerebral malaria and to death. Understanding the complex biology and pathogenicity of Plasmodium has been a major effort of the biological and medical community, recently leading to the international project of sequencing the entire P. falciparum genome (for review, see Wellems et al. 1999). Of the 14 chromosomes of P. falciparum, the sequencing of chromosomes 2 and 3 is now complete (Gardner et al. 1998; Bowman et al. 1999). It is expected that the complete genome will allow easier identification of genes responsible for its pathogenicity and will be helpful in the development of effective vaccines. Also, the complete genome will offer an unbiased perspective on the proteome that it encodes, and a unique opportunity to discern the general properties of its coding and noncoding regions. The genome of P. falciparum is unique in many ways. Its DNA is extremely high in A + T content (∼84% for both available chromosomes). The genome is also anomalous in its “genomic signature”, which characterizes the genome composition based on dinucleotide relative abundances (Karlin et al. 1997). In contrast to most other eukaryotic genomes in which the dinucleotide TA is underrepresented, in the Plasmodium genome its relative frequency is in the normal range. However, representation of the pair CC/GG is distinctly high. These and other characteristics render thePlasmodium genome among the most different in signature of all investigated eukaryotic organisms (Karlin and Mrázek 1997, Karlin et al. 1998). The proteome of Plasmodium is as equally anomalous as its genome.
In this issue, Pizzi and Frontali (2001) study the low-complexity elements that dominate many of the proteins of P. falciparum. These authors determine that >90% of all proteins in chromosomes 2 and 3 feature low-complexity regions that can extend to 1.8kb, and that half of all proteins are more than 60% composed of low-complexity regions. These values are much higher than for other eukaryotes. A few of these low-complexity regions are hydrophobic segments conserved between species, but most (∼90%) are predominantly composed of hydrophilic residues. Of these, 20% consist of iterated short oligonucleotides (tandem repeats) and 80% are made of nonrepetitive segments with homopeptide runs of variable lengths (Figure 1) . The bulk of the segments 50–300 amino acids long are nonrepetitive elements pervasively found among informational, metabolic, and housekeeping proteins. An example of such regions is found in one of the two nuclear encoded HSP60 proteins ofPlasmodium (presumably transferred to the mitochondrion) (Brocchieri and Karlin 2000). This sequence is distinguished from hundreds of other sequenced HSP60 genes by having a carboxy-terminal insertion with runs of acidic residues extending ∼90 amino acids. This insertion substitutes a shorter tail of repetitive Gly-Gly-Met elements of unknown function common to most HSP60 proteins. A prototype informational protein with many tandem repeats is 5′–3′ exonuclease (Gardner et al. 1998), in which the inserted element appears to be an exposed loop of 176 amino acids by alignment to a homologous structure. Pizzi and Frontali (2001) align several P. falciparum proteins with available homologs from other organisms, showing that hydrophilic low-complexity regions correspond to unaligned insertions unique to Plasmodium proteins (see also Pizzi and Frontali 2000). Supporting evidence suggests that low-complexity regions often represent rapidly diverging, exposed, non-globular domains. Low-complexity elements have clinical relevance as variable immunodominant epitopes of transmembrane proteins (Reeder and Brown 1996; Newbold 1999). These are part of a strategy for rapid diversification that enables the parasite to evade the immune response of the host by switching among different antigenic phenotypes. Diversification mechanisms include (see Reeder and Brown 1996): (1) possible chromosomal deletion or single mutation events; (2) antigenic population diversity (different alleles), which relates to variability in expression of low-complexity elements containing tandem repeats present in many immunodominant epitopes (e.g., S-antigen, MSA-1, and MSA-2); (3) intergenic recombination, which generates variability at the sexual stage (Kemp 1992; Hill et al. 1995); (4) antigenic switching during maturation is also part of Plasmodium life strategy, as exemplified by the var family of ∼50 genes that encode for the adhesion protein PfEMP1. These genes are variably expressed in different clones and during different stages of the parasite lifecycle, producing distinct host cell-surface phenotypes and adherence properties (Reeder and Brown 1996; Chen et al. 1998; Newbold 1999).

Partial alignment of the ookinete adhesive protein between the Plasmodium falciparum (PLAFA) and the rodent malaria agent Plasmodium berghei (PLABE) sequences, showing two low-complexity elements (lower case, unaligned) between regions of alignment (upper case). The first region includes tandem repeats and the second has homopeptide runs of Asn. Alignment obtained with ITERALIGN (Brocchieri and Karlin 1998). Although multiple alignments are most often used to identify conserved regions of a protein family,Pizzi and Frontali (2001) use it to identify variable regions among homologs. In Plasmodium, variable tandem-repeat regions play a central role in evading the immune system of the host. Do other low-complexity regions have a related function?
The nucleotide composition of Plasmodium coding sequences is certainly influenced by constraints imposed by the protein sequence. The influence of amino acid content in the nucleotide selection of coding sequences refers particularly to the composition of codon position II, which primarily determines the chemical/physical nature of the encoded amino acid. In fact, in the second codon position T corresponds exclusively to hydrophobic residues, whereas A mostly translates to hydrophilic residues. However, many other factors influence codon usage. For example, it has been shown that codon usage also reflects selection for efficiency of translation in connection with tRNA abundances (Sharp and Li 1987; Shields et al. 1988; Sharp 1991; Moriyama and Powell 1997). DNA base-step conformational tendencies may also contribute to codon preferences (Karlin and Mrázek 1996). Furthermore, global genome biases influence the composition of coding sequences. In Plasmodium the strong preference for A + T in noncoding regions is clearly reflected by the A + T composition of coding sequences (Figure2).
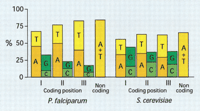
Nucleotide composition of coding and noncoding sequences is compared between chromosomes 2 of Plasmodium falciparum and Saccharomyces cerivisiae. It is evident that in both genomes the A +T content for each category reflects the general genomic bias. The detailed usage is, however, constrained by other selective pressures, particularly in codon positions I and II. In both organisms the A and T content of the third codon position is similar to noncoding regions. The frequency of T in the second codon position, which corresponds to hydrophobic amino acids, is ∼27% in both organisms but the frequency of A is much greater inPlasmodium (50%) than in yeast (35%). However, excluding low-complexity coding regions of Plasmodium gives a frequency of A (36%) similar to yeast, whereas low-complexity regions alone result in a frequency of 55%. Low-complexity regions inPlasmodium have a very different amino acid composition than yeast (Pizzi and Frontali 2001).
The compositional analysis of hydrophilic nonrepetitive low-complexity segments of Plasmodium reveals that they discriminate in favor of residues of greater A + T content. They are enriched in acidic residues (Glu and Asp) but prefer Lys (largely coded by AAA) and significantly more Asn (largely coded by AAT). The greater frequency of Asn compared to Lys cannot be explained by compositional biases of the genome or by any obvious chemical/physical character. Pizzi and Frontali interpret this asymmetry as evidence for the existence of some unidentified factor specifically selecting for Asn. The authors suggest that it might be related to an active role of these elements in the production of immunodominant epitopes. Perhaps the multitude of Asn residues affords multiple alternative sites of glycosylation producing a variable antigenic landscape. Or perhaps the abundance of low-complexity insertions provides a smokescreen against the host immunogenic response. Alternatively, these insertions may only be the by-product of the production of antigenic variability concomitant to repetitive elements, perhaps a consequence of the oxidative stress generated by the Plasmodium metabolism (Francis et al. 1997). However, the ubiquity of the rapidly evolving, non-repetitive, low-complexity regions in Plasmodium genes is astonishing, and it is indeed difficult to believe that they can be simply tolerated as neutral side-products of some other advantageous activity of the parasite.
The composition of the Plasmodium DNA is certainly unique; equally, if not more, special is the composition of its proteins. The biology of Plasmodium is in many respects mysterious and challenging, but complete genomic sequences will be of great value in the effort to understand the properties and relations of its fascinating genome and proteome.
Acknowledgments
I thank S. Karlin for comments on the manuscript. This work was supported by NIH Grants 5R01GM10452–36 and 5R01HG00335–12.
Footnotes
-
E-MAIL luciano{at}gea.stanford.edu; FAX (650) 725-2040.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.176401.
- Cold Spring Harbor Laboratory Press








