
Is There a Bias in Proteome Research?
Abstract
Advances in technology have enabled us to take a fresh look at data acquired by traditional single experiments and to compare them with genomewide data. The differences can be tremendous, as we show here, in the field of proteomics. We have compared data sets of protein-protein interactions in Saccharomyces cerevisiae that were detected by an identical underlying technical method, the yeast two-hybrid system. We found that the individually identified protein-protein interactions are considerably different from those identified by two genomewide scans. Interacting proteins in the pooled database from single publications are much more closely related to each other with respect to transcription profiles when compared to genomewide data. This difference may have been introduced by two factors: by a selection process in individual publications and by false positives in the whole-genome scans. If we assume that the differences are a result of false positives in the whole-genome data, the scans would contain 47%, 44%, and 91% of false positives for the UETZ, ITO-core, and ITO-full data, respectively. If, however, the true fraction of false positives is considerably lower than estimated here, the data from hypothesis-driven experiments must have been subjected to a serious selection process.
With the development of technology-driven high-throughput methods in functional genomics and proteomics, it has become possible to analyze transcriptional profiles and protein interactions on a genomic scale (Lockhart and Winzeler 2000).
We addressed the question of whether systematic differences occur in the outcome between many single experiments and genomewide approaches in the field of proteomics. Knowledge about protein-protein interaction is important for the identification of new regulatory networks and, for example, is used to predict functions of proteins (Marcotte et al. 1999).
Protein-protein interaction may be detected by means of the yeast two-hybrid system (Fields and Song 1989). We have compared three large datasets of protein-protein interactions in Saccharomyces cerevisiae detected by yeast two-hybrid experiments. We looked at transcription correlations of interacting pairs in the three protein-protein interaction databases.
The first dataset “UETZ” consisting of 1519 interactions was obtained from a genomewide screen in which nearly all of the ∼6000 open reading frames (ORFs) in S. cerevisiae were examined (Uetz et al. 2000). A second genomewide scan was analyzed with its 841 “ITO-core” pairs (core dataset with more than three hits in the scan) and its 4549 “ITO-full” pairs (full dataset) of interacting proteins (Ito et al. 2001). The third dataset: “MIPS” consisting of 1082 yeast two-hybrid protein-protein interactions was obtained from the MIPS database (Mewes et al. 2000). The MIPS data represent yeast two-hybrid reports pooled from 174 single publications.
RESULTS AND DISCUSSION
If two proteins show a physical interaction, their transcription profiles may correlate. We have estimated the degree of this relation. For all datasets, we calculated the distribution of the correlation coefficients of the transcription profiles of interacting protein-protein pairs and randomly selected pairs. Transcription data were obtained from three independent genomewide transcription microarray experiments in S. cerevisiae: the cell cycle analysis (Cho et al. 1998), sporulation (Chu et al. 1998), and response to pheromone (Roberts et al. 2000). As expected, there is an excess of positive correlation coefficients in all cases (Fig. 1). Analysis of each of these three independent whole-genome transcription profiles revealed that there is a significant higher fraction of positively correlated transcription of protein-protein interactions in the MIPS database compared to the protein-protein-interactions of the whole-genome screens (Fig. 1, Table1).
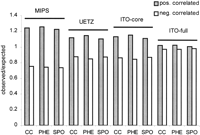
Comparison of distributions of positive and negative correlation coefficients of transcription profiles of interacting protein-protein pairs depicted as ratios of observed number (yeast two-hybrid pairs) and expected number (random pairs) in the genomewide screens (UETZ, ITO-core, ITO-full) and in the pooled single yeast two-hybrid experiments (MIPS). Profiles of interacting proteins from hypothesis-driven experiments show significantly higher positive correlation when compared to the genomewide scans. Transcription data come from three independent genomewide transcription microarray experiments: response to pheromone (PHE), cell cycle (CC), and sporulation (SPO).
χ2-Statistical Analysis of Three Independent Whole-Genome Transcription Profiles
One major source of the differences could be attributed to false positive interactions in the whole-genome scans. Assuming that false positives in the genomewide scans were the only source for the statistical differences obtained, the false positive quantity can be estimated by adding a fraction of random pairs y to the MIPS database such that the statistical property of the genomewide scan is matched. We have estimated this fraction y with respect to transcription correlation. The result of the analysis is given in Table2. The values of y differ slightly for the three transcription experiments. One reason for this might be found in the biological nature of the transcription profile experiment; that is, that the pheromone differs from cell cycle and sporulation, thereby changing different classes of genes in their activity. To obtain an estimate for the possible variation caused by the variability in the transcription profiles we calculated the standard deviation fory for each experiment and dataset by means of a bootstrapping algorithm (Table 2).
Estimation of False Positives in Whole-Genome Scans
Interestingly, the ITO-full dataset shows much higher y values compared to ITO-core and the UETZ data. The less stringent filtering in the ITO-full data compared to the subset ITO-core (containing only pairs with more than three hits in the screen) appears to influence the magnitude of y tremendously. To obtain further insight we analyzed the difference set of ITO-full minus ITO-core. With respect to transcription correlation a significant difference of the ITO difference set to random pairs no longer occurred (SPO, χ2 = 0.4, P = 0.52; CC, χ2 = 0.0068, P = 0.93; PHE, χ2 = 0.0067,P = 0.93), indicating that most of the true positive pairs are already contained in the ITO-core set.
In summary, protein-protein interaction pairs from many single experiments in hypothesis-driven research show a much closer relationship to each other with regard to transcription profiles when compared to a genomewide scan. The difference may have been introduced by a selection process in individual publications and by false positives in the whole-genome scan. The differences calculated can be completely explained, if the whole genome scans of UETZ, ITO-core and ITO-full would contain approximately 47%, 44%, and 91% false positives, respectively.
If it can be shown that the fraction of false positive interactions is much smaller than estimated here, our analysis points to a serious selection bias of the hypothesis-driven single experiments in the MIPS database. This selection bias may have been introduced by the failure to report interactions which cannot be understood from previous publications, or by failing to perform the experiment for such pairs in the first place. This, in turn, may have undermined the biological relevance of the reported interactions. The initial hypothesis of the researcher about protein function and other properties may have influenced each single experimental design and interpretation of results, leading to a biased protein-protein interaction pool in the literature.
The identification of sources for the tremendous differences between the datasets is critical, because they are the basis for subsequent research such as the prediction of protein function (Marcotte et al. 1999).
METHODS
Interaction Databases
Data of the genomewide screen performed by Uetz et al. (2000) were obtained from http://depts.washington.edu/sfields, including additional data downloaded July 9, 2000. The ITO-core and the ITO-full data of the screen performed by Ito et al. (2001) was obtained fromhttp://genome.c.kanazawa-u.ac.jp/Y2H. Pooled data of yeast two-hybrid experiments were obtained from the Munich Information Center for Protein Sequences (MIPS) site at http://www.mips.biochem.mpg.de, downloaded October 6, 2000.
Data Processing
Alias names were translated into unique ORF names using translation at http://www.proteome.com/databases/YPD/YPDsearch-quick.html and athttp://www.mips.biochem.mpg.de/proj/yeast/search/index.html. Self-interactions have been excluded in all datasets and all calculations. Parsing of data was performed in the open-source Linux environment (http://www.suse.de).
Estimation of False Positives
Assuming that false positives in the genomewide scan were the only source for the obtained statistical differences, the fraction can be estimated by adding a fraction of random pairs y to the MIPS database such that the statistical property of each of the genomewide scans is matched. To estimate the variability of each measure ywe performed a bootstrapping by which 1000 × 50% of the MIPS data was randomly chosen for each calculation of y. The mean and the standard deviation of each distribution of y were used for further analysis and presentation.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL mrowka{at}rz.hu-berlin.de; FAX 49-30-450 528972.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.206701.
- Cold Spring Harbor Laboratory Press








