
A Clone-Array Pooled Shotgun Strategy for Sequencing Large Genomes
- Wei-Wen Cai1,2,
- Rui Chen1,2,
- Richard A. Gibbs1,2,5, and
- Allan Bradley1,3,4
Abstract
A simplified strategy for sequencing large genomes is proposed. Clone-Array Pooled Shotgun Sequencing (CAPSS) is based on pooling rows and columns of arrayed genomic clones, for shotgun library construction. Random sequences are accumulated, and the data are processed by sequential comparison of rows and columns to assemble the sequence of clones at points of intersection. Compared with either a clone-by-clone approach or whole-genome shotgun sequencing, CAPSS requires relatively few library constructions and only minimal computational power for a complete genome assembly. The strategy is suitable for sequencing large genomes for which there are no sequence-ready maps, but for which relatively high resolution STS maps and highly redundant BAC libraries are available. It is immediately applicable to the sequencing of mouse, rat, zebrafish, and other important genomes, and can be managed in a cooperative fashion to take advantage of a distributed international DNA sequencing capacity.
Advances in DNA sequencing technology in recent years have greatly increased the throughput and reduced the cost of genome sequencing. Sequencing of a complex genome the size of the human is no longer a question of feasibility but of the selection of the most efficient, economical, and practical strategy. Two competing strategies have been used to generate a draft sequence of the human genome: clone-by-clone (CBC) sequencing and the whole-genome shotgun (WGS) strategy (International Human Genome Sequencing Consortium 2001; Venter et al. 2001). The CBC strategy is being used by publicly supported sequencing centers around the world and has produced highly accurate sequences of Escherichia coli (Blattner et al. 1997), yeast (Goffeau et al. 1996), Caenorhabditis elegans (The C. elegans Sequencing Consortium 1998), and human chromosomes 22 (Dunham et al. 1999) and 21 (Hattori et al. 2000). These successes benefited from the prior construction of sequence-ready maps. For other projects underway, such as the sequencing of the mouse and rat genomes, for which map resources are relatively scarce, the advantage of this strategy is less obvious.
An alternative clone-by-clone strategy that does not depend on a sequence-ready map has been described (Venter et al. 1996), but the most radical genome sequencing method is the WGS strategy (Weber and Myers 1997). WGS obviates the need for a sequence-ready map, but relies heavily on immense computational power for assembling random shotgun reads into long continuous-sequence contigs, which are finally anchored to chromosomes using other mapped sequence information (Venter et al. 1998). Success in applying this strategy to sequence the 120-Mb euchromatic portion of the Drosophila genome provided a proof of principle for WGS (Adams et al. 2000). This impressive achievement did not, however, guarantee that the strategy would work on the human or mouse genomes. Each is more than 20 times larger than theDrosophila genome, and the computational requirements to perform the necessary pairwise comparisons increase approximately as a square of the size of the genome (see ). Indeed, the reported experience with the Drosophila WGS (Myers et al. 2000) indicated that the achievable computational power would not be sufficient to assemble the human genome sequence purely from shotgun random reads. This question is not resolved because the human genome has not so far been assembled by a WGS method; instead, binned sequence reads from individual BACs in the public database have been used to anchor WGS reads to resolve ambiguities and lower the computational load (Venter et al. 2001).
Clone-Array Pooled Shotgun Sequencing
Here we propose an alternative strategy for large-scale DNA sequencing, Clone-Array Pooled Shotgun Sequencing (CAPSS). BAC clones representing a complete genome are organized in a two-dimensional array format. DNA from each BAC is pooled with clones in associated rows and columns, and shotgun libraries are prepared from each pool. Sufficient random reads are collected from each library to generate four- to fivefold coverage of each of the BACs in a row or column. Cross assembly of random reads between pairs of columns and rows results in sequence contigs of 8- to 10-fold coverage that belong to specific BACs at the points of intersection (e.g., W in Fig.1). Each assembled BAC can then be finished using present methods for directed sequencing of individual subclones.
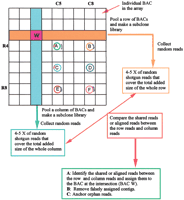
General Clone-Array Pooled Shotgun Sequencing (CAPSS) strategy. Genomic clones (e.g., BACs) are organized in a two-dimensional array, and pools of DNA from each row and column are converted to a subclone library for sequencing. The sequence assembly of each clone is generated by cross assembly of each row and column, shown as clone W in this schema. Clones A–F exemplify possible complications from other overlapping sequences in the array. The colors in clones A–F represent unique sequences. Here clone A and clone C share sequence, as do B and E. Cross assembly of R4 + C5 will yield assembly from BAC A, and will include reads from the overlap in BAC C. Clones B and E will also generate contigs from both assemblies of R4 + C5 and R8 + C8. The generation of contigs at multiple locations in the grid distinguishes overlap that does not originate from the clone at the row/column intersection. Circles in A–F represent a perfect complete sequence contig, with colors coding for different sequences. Note that the shared sequence contig (in blue) between clone B and clone E will lead to assignment of the same contig to clone A and F, shown as an independent contig (in blue).
The scheme in Figure 1 shows that CAPSS retains the advantages of both CBC and WGS strategies, while overcoming their limitations. Pooling BACs dramatically reduces the effort required for constructing and managing subclone libraries. To sequence the human genome using the CBC strategy, for example, at least 22,000 subclone libraries from individual BACs of 150 kb (assuming 10% overlaps) are required. However, if these BACs were organized and managed in a 148 × 148 two-dimensional format (Fig. 1), only 296 subclone libraries would be needed, considerably reducing the labor and management effort.
For a complete sequence, CAPSS ultimately requires the same average 8- to 10-fold DNA sequence coverage across the entire genome as CBC or WGS approaches (∼6.0 × 107 reads/3.0 Gb); however, the reads can be assembled progressively with a modest amount of computational power. In the example of the 148 × 148 array for the human genome, ∼203,000 reads are accumulated from each sublibrary. Assembly of a pool of any row with any column (406,000 reads) requires ∼1.8 × 104-fold more computation than assembly of a single typical BAC, which is still a formidable task. Prior independent assembly of reads from each row and column in an array will, however, dramatically reduce peak computational requirements, as assembly of each intersecting BAC can be accomplished by comparison of these intermediate results, obviating the need to reiterate many computationally expensive pairwise comparisons. A 203,000-read assembly represents ∼4.5 × 103 times the load of a single BAC assembly and can be readily achieved in ∼16 h on an 800-MHz dual PIII processor-board with adequate RAM. A single such device costs < $20,000 (US), and although less expensive machines can be applied with lower performance, slightly more costly computers can dramatically speed the result. This is one of the major advantages of CAPSS. The computational power needed to assemble each sublibrary pair in pooled columns or rows is only 1/90,000 of the power required for the WGS strategy assembly (Table 1; see ). When the time scale for the requirement for assemblies in a large genome project are distributed over 1 yr, further economies of CAPSS relative to WGS are apparent (see ; Fig. 2).
Computational Requirements for Assembly of a 3-Gb Genome
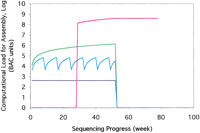
Computational load for different sequencing strategies. (Red) WGS; (green) CAPSS, 22,000 BACs in a single 148 × 148 array; (turquoise) 21,600 minimally overlapping BACs sequenced in six smaller 60 × 60 arrays; (blue) CBC strategy for a total of 22,000 clones. See for details.
Practical Aspects of Sequence Assembly in CAPSS
Computer simulation data (not shown) clearly indicate the feasibility of the CAPSS strategy. To optimize the sequence read assignment in CAPSS, however, there are three additional aspects to the strategy that can be considered. First, paired-end subcloned sequencing improves the efficiency of the assembly. In the paired-end scheme, a clone is considered positioned in an assembly whenever at least one of its end reads has significant match. Because the chance of both ends of a clone being in a repetitive region is relatively small, the likelihood that a clone will be positioned is increased when sequences from both ends of a clone are used for comparison.
Second, in order to achieve a high assembly efficiency and a low error rate, it is best to use reads that have been masked with a program to remove repetitive sequences (e.g., RepeatMasker; A.F.A. Smit and P. Green,http://ftp.genome.washington.edu/RM/RepeatMasker.html) to search against unmasked contigs. Repeat-masking however, imposes an additional computational load. To minimize computation time, a two-step protocol can be used in which, following comparison between unmasked reads and repeat-masked contigs, remaining reads are repeat-masked and compared with the unmasked contigs.
Third, incorrect assignment of reads often results from matching between a short stretch of sequence and a contig. Usually such a match has a relatively low score and tends to be assigned to clones containing a high number of repetitive sequences. These errors can be reduced dynamically by adjusting the score used to filter the match results and developing procedures for manual examination of matches that generate ambiguous matches producing conflicting assemblies.
Possible Problems with CAPSS
Possible unequal representation of the amount of DNA from each clone is the primary pitfall for CAPSS. Our experience in routinely pooled cDNA clones for sequencing by a concatenation procedure (Yu et al. 1997) has shown the issue of equalizing clone representation in mixed libraries to be easily manageable. If needed, a highly accurate clone-DNA concentration measurement step could be added, such as quantitative PCR of multiple dilutions of each stock. It is unlikely, however, that this level of effort will be needed.
In addition, the main source of this operational difficulty in CAPSS would be the variation in yield of BAC DNAs in standard preparation protocols. This variation is dramatically less, however, when smaller BACs are generated. In our work, BACs <120 kb have a higher yield, inversely proportional to the BAC length. Because CAPSS does not require the longer BACs that are usually desired for conventional mapping activities, high yields can be expected. Additional advantages of this may include efficient use of shearing protocols for BAC library construction, thus avoiding the bias of representation caused by nonrandom distribution of restriction sites.
Simple DNA repeats will not confound CAPSS assemblies although long, low frequency repeats can generate the same kind of ambiguities that are found in the CBC approach. The remedies for these complications are also the same as for CBC sequencing. The generation of double-ended sequences from subclones allows the formation of physical scaffolds along the length of each contig. This methodology was pioneered by the use of Sequence Mapped Gaps (SMGs) in the first automated shotgun sequencing of a human cosmid (Edwards et al. 1990), and has since been used in other schemes to resolve ambiguous assemblies (Gibbs 1995). In extreme cases, single BACs in arrays can be addressed individually to resolve the ambiguous assemblies.
CAPSS and 3-Gb Genomes
Application of CAPSS to large genomes for which no complete sequence-ready map exists further illustrates the power of the method. For example, a 140 × 140 array would be suitable for sequencing the mouse genome, where accumulated efforts over the past decade have resulted in a high resolution genetic map and an STS-based physical map with ∼12,000 markers. Of these markers, ∼2800 have been used to identify corresponding BAC clusters across the genome (Cai et al. 2001). In addition, BAC-end sequencing representing 10-fold clone coverage is underway (Battey et al. 1999). This example therefore represents a CAPSS approach to a real problem.
Each of the presently available murine BACs is of the average size 200 kb, and an array containing ∼20,000 clones represents ∼4.0 Gb, or 1.3-fold of genomic coverage. Accumulation of 6.0 × 107sequence reads for the entire collection yields ∼215,000 reads per row or column, and provides an average total of 3000 reads that originate from each BAC at the points of intersection, or ∼7.5-fold coverage per BAC. This is sufficient to enable assembly of large contigs from each clone at points of intersection, but represents less coverage than the 8- to 10-fold that would be ideally achieved in an array formed by only minimally overlapping BACs.
Further coverage of each BAC will be automatically generated within the matrix of the assemblies that are completed for the entire array. Figure 1 shows that fortuitous overlap with other clones in the same row or column directly increases the depth of sequence coverage in the assembly of the BAC at the point of intersection. These overlapping fragments can be distinguished from a second class of contigs within each assembly that contain reads from both rows and columns but are derived from pairs of overlapping clones, neither of which is at the row/column intersection. The reads in these unrelated overlaps are also found in contigs from the cross assemblies for which each unrelated BAC is the primary assembly target. A simple computer routine is sufficient to correlate these events and ultimately assign each initial contig to its correct final assembly based on the contig positions in the different row-versus-column assemblies.
Reduced array structures can also be applied to further simplify the analysis of the mouse genome example. For example, the 2809 BACs from the presently available BAC framework map can first be sequenced in a 53 × 53 format. Because these BACs do not overlap with one another, sequence contig assignment will be unambiguous. When each BAC in this array is assembled, a second set can be identified by physical mapping or BAC end sequence assignment. After 6 iterations of this process, the total sequences would provide 1.2-fold coverage of the genome. Alternatively, mapped BACs can be combined with a selection of random BACs to form a slightly larger array. After these BACs are sequenced, further selection and sequencing of minimally overlapping BACs will complete the whole genome. As a general strategy, the use of these subarrays provides the advantages of CAPSS while obviating any possible operational problems arising from unexpected clone overlap in poorly mapped genomes. The smaller arrays also present a more manageable logistics problem for existing sequencing centers.
A CAPSS Key for WGS Assembly
A CAPSS approach can be also used in combination with whole-genome shotgun sequencing to enable a complete genome assembly. The basic principle is that the CAPSS data can provide an initial assembly of each clone, and these contigs could be used to select sequence reads from a pool of WGS data for subsequent cycles of clone-linked assemblies. This is a particularly attractive strategy as it maximizes the diversity of sequence data that can be combined to produce a final genome assembly.
The combination of CAPSS and WGS data may be the best solution for analyzing large genomes that have very little mapping data available. This strategy would use arrays that contain sufficient clones to ensure complete genomic coverage, and the DNA sequencing effort would be divided between the CAPSS and WGS components. For example, a 3-Gb genome for which an average 2-fold BAC clone coverage array of 200 × 200 clones is constructed could have 40 million reads produced for the entire array. CAPSS assemblies in this case would have ∼6- to 7.0-fold coverage at the points of intersection of rows and columns, which would be predicted to generate contigs of sufficient length to localize the information from a further 2.0 × 107 WGS reads.
Further Advantages of CAPSS
There are several further technical and logistic advantages of using CAPSS to sequence complex genomes. First, unlike WGS sequencing, each project will progressively yield regions with full sequence coverage. As each new row or column is completed, all intersecting BACs are fully covered, and consequently clones of high biological interest can be prioritized for early finishing. In addition, gap closing can proceed in parallel with sequencing. This is an important advantage because subclone archives need not represent the whole genome, as they do in the WGS method.
Second, in CAPSS, because the number of subclone libraries from pooled BACs increases only with the square root of the number of clones in the BAC array, larger numbers of BACs with relatively small insert sizes (∼100 kb) can be used. This is an extremely useful technical advantage as library construction and growth of these smaller clones is considerably easier than for larger inserts. Recent development of an inducible multicopy BAC cloning vector (Wild et al. 1996) raises the possibility of pooling clones before growth, which would even further simplify CAPSS.
Finally, many sequencing centers can participate in different phases of a CAPSS project independently. Large centers can focus on sequencing multiple rows or columns of BACs to completion and assemble the sequence contigs assigned to individual BACs progressively, and smaller groups can close gaps in those BACs of their scientific interest. This is an important advantage because it follows the present international trend of allowing the cultivation of both small and large sequencing centers.
Acknowledgments
W.W.C. is a Jane Coffin Childs Memorial Fellow; R.A.G. is supported by grants from the NHGRI, NHLBI, and the DOE; and A.B. is a Howard Hughes Medical Institute Investigator and also supported by NCI. We thank Guangbin Luo and George Weinstock for helpful discussions.








