
Congenic Mapping of the Type 1 Diabetes Locus, Idd3, to a 780-kb Region of Mouse Chromosome 3: Identification of a Candidate Segment of Ancestral DNA by Haplotype Mapping
Abstract
Type 1 diabetes in the nonobese diabetic (NOD) mouse arises as a consequence of T cell-mediated destruction of the insulin-producing β cells of the pancreas. Although little is known of the events that initiate and subsequently drive β-cell destruction it is clear that the entire process is under complex genetic control. At present 19 loci have been mapped that influence the development of diabetes either at the level of initiation of insulitis or at the level of progression from insulitis to overt diabetes, or both. Previously, we have mapped one of these loci, Idd3, to a 0.35-cM interval on proximal mouse chromosome 3. In the present study we have narrowed the map position of this locus to an interval of 0.15 cM by a combination of novel congenic strains and an ancestral haplotype analysis approach. We have constructed a physical contig in bacterial artificial chromosome (BAC) clones across the minimal interval. Restriction mapping of the BAC contig placed the maximum size of the Idd3 interval at 780 kb between the markers D3Nds36 and D3Nds76. To refine further the Idd3 interval we developed a series of novel single nucleotide polymorphisms (SNPs) and carried out haplotype analysis on DNA from mouse strains known to carry either Idd3susceptibility or protective alleles. This haplotype analysis identified a 145-kb segment of ancestral DNA between the microsatellite marker D3Nds6 and the SNP 81.3. One haplotype of this ancestral segment of DNA is found in mouse strains carrying anIdd3 susceptibility allele and another is found in mouse strains carrying an Idd3 protective allelle. Within the 780-kb congenically defined interval this 145-kb segment represents the most likely location for Idd3. The Il2 gene, which encodes the cytokine interleukin 2 (IL2), maps to this interval and is a strong candidate for Idd3. To investigate whether sequence variation exists in the promoter region of the Il2 gene, which might alter its expression, we sequenced the promoter region of theIl2 gene from mouse strains carrying either an Idd3susceptibility or resistance allele. Two sequence variants were identified, neither of which fell in known regulatory elements within the Il2 promoter. In agreement with this observation steady-state Il2 mRNA levels showed no variation between susceptible and resistant mouse strains. These data suggest that the profound protection from diabetes seen in congenic mice carrying anIdd3 protective allele is unlikely to be due to differences in the level of expression of the Il2 gene. Instead, all of the current data support our hypothesis that Idd3 corresponds to amino acid variation at the amino terminus of Il2.
[Sequence data reported in this paper have been deposited in GenBank and assigned the following accession numbers: AF19594, AF19595, and AF19596.]
Type 1 diabetes results from the autoimmune destruction of the insulin-producing β cells of the pancreas. A breakdown in immune homeostasis leads to a lymphocytic infiltration of the pancreatic islets of Langerhans, a process known as insulitis, which in turn progresses to β-cell destruction and overt disease. Although the molecular events that initiate and subsequently drive the process are uncertain it is clear that disease susceptibility is under complex genetic control.
The nonobese diabetic (NOD) mouse model of type 1 diabetes (Leiter 1998) has greatly facilitated the genetic dissection of type 1 diabetes. At present, 19 loci have been mapped that contribute to the development of type 1 diabetes in the NOD mouse (Leiter 1998; Melanitou et al. 1998; Lyons and Wicker 1999). The approach we have adopted for the fine mapping of these insulin-dependent diabetes (Idd) susceptibility loci in the NOD mouse is one of congenic mapping (Wicker et al. 1994, 1995; Lord et al. 1995; Denny et al. 1997; Podolin et al. 1997).
Using this approach we have previously mapped the Idd3 locus to a 0.35-cM interval on proximal mouse chromosome 3 between the microsatellite markers D3Nds55 and D3Nds40 (Denny et al. 1997). One gene known to map within this small interval is that encoding the cytokine interleukin-2 (IL2) (Denny et al. 1997). A growing body of evidence makes Il2 a strong candidate forIdd3. A series of reports have shown that IL2 plays a central role in the development of self tolerance, with a lack of IL2 being associated with the development of autoimmune disease (Hunig and Schimpl 1998). It has been shown that IL2 is essential for activation-induced cell death of T cells mediated via the Fas pathway, a key mechanism of self-tolerance (Refaeli et al. 1998). We have shown previously that sequence polymorphisms exist between IL2 allotypes from different strains of mice (Ghosh et al. 1993; Denny et al. 1997) and that these polymorphisms, in exon 1 of Il2, segregate with susceptibility to diabetes (Denny et al. 1997). Moreover, one of the polymorphisms, the presence of proline rather than serine at position 6 of the mature IL2 protein, is associated with both the increased glycosylation of IL2 and diabetes susceptibility (Podolin et al. 2000). Despite the observed sequence differences between IL2 allotypes from diabetes-susceptible and -resistant strains no functional difference has been reported. Although the Il2 promoter has been well characterized it is unknown whether variation exists in this region between mouse strains. Any variant that leads to an alteration in expression of Il2 would be a strong candidate for theIdd3 etiological mutation. Consistent with this possibility, reduced secretion of IL2 by mitogen-activated splenocytes has been reported previously in the NOD mouse (Serreze et al. 1989).
In this study we have refined the genetic mapping of Idd3 to a 0.15-cM interval that still encompasses the Il2 gene. We have constructed a physical contig of mouse BAC clones across the new minimal interval and by restriction mapping have determined that the maximum size of the Idd3 interval is 780 kb. Haplotype analysis in mouse strains known to carry either an Idd3susceptibility or protective allele identifies the most likely location of Idd3 as the 145-kb interval between the microsatellite marker D3Nds6 and the single nucleotide polymorphism (SNP)81.3. To identify potential regulatory polymorphisms we sequenced the promoter region of the Il2 gene from mice carrying either susceptibility (NOD and 129) or protective (B6) alleles at Idd3. None of the identified variants within theIl2 promoter region fell in known regulatory elements. In agreement with this, no difference was observed in steady-stateIl2 mRNA levels, as assessed by semiquantitative RT–PCR, between mice carrying Idd3 susceptibility or protective alleles.
RESULTS
Generation of New Variant Microsatellite Markers within theIdd3 Interval
We have described previously the establishment of a YAC framework map across the Idd3 region (Denny et al. 1997). To facilitate the isolation of additional polymorphic markers from within theIdd3 region we identified mouse P1 and BAC clones positive for STSs developed from YACs spanning the Idd3 interval. Mouse P1 and BAC libraries were screened by PCR with the following STSs:B1R18STS, D3Nds36, D3Nds47, D3Nds6, D3Nds56, D3Nds51, D3Nds34, D3Nds46, D3Nds45, and D3Nds40. Four new microsatellite markers, D3Nds76, D3Nds77, D3Nds78, andD3Nds84, were isolated from the clones mP284k17, mP315n15, mP305l10, and mP88b24, respectively. These new microsatellite markers were ordered with respect to D3Nds55, D3Nds6,D3Nds34, D3Nds36, and D3Nds40 by genotyping the 944 progeny of an F2 cross between NOD and the strain NOD.B62 (Lord et al. 1995). The following map order was obtained D3Nds55–(0.2 cM)–D3Nds36–(0.07 cM)–D3Nds84–(0 cM)–D3Nds6–(0 cM)–D3Nds34–(0.08 cM)– D3Nds76–(0 cM)–D3Nds77–(0 cM)–D3Nds78–(0 cM)–D3Nds40 (Fig. 1). Where the marker order could not be resolved genetically the order was determined from the Idd3 region physical map.
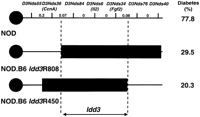
High-resolution genetic map of proximal chromosome 3 aroundIdd3. The lower bars show the genotypes for markers within theIdd3 interval for the congenic strains NOD. B6Idd3R450 and NOD. B6 Idd3R808. The solid bars indicate B6-derived genome and the numbers beside the bars indicate female diabetes frequencies at 7 months of age. Distances between markers are given in cM.
Narrowing of the Idd3 Interval to 0.15 cM
Previously, we have mapped Idd3 to a 0.35-cM interval between, but not including, the microsatellite markers D3Nds55and D3Nds40 (Denny et al. 1997). To more precisely map theIdd3 interval, the NOD.B6 Idd3R808 strain was developed from the previously described NOD.B62 congenic strain (Lord et al. 1995), a strain that carries the Idd3resistance allele. Genotyping the NOD. B6 Idd3R808 strain with polymorphic microsatellite markers that map to the Idd3interval showed that it was NOD derived at D3Nds55 andD3Nds36 and B6 derived at D3Nds84, D3Nds6,D3Nds34, D3Nds76, D3Nds77, D3Nds78, and D3Nds40 (Fig. 1). This mapped the proximal boundary of its congenic segment to the 0.07-cM interval between D3Nds36 andD3Nds84. The frequency of diabetes in females of this strain at 7 months is 29.5% (26/88) compared with 25.4% (17/67) in the NOD.B62 strain (P >0.05) and 77.8% (63/81) in NOD (P < 0.0001). Thus, like NOD.B62, NOD. B6Idd3R808 carries the Idd3 resistance allele. Because NOD. B6 Idd3R808 carries the Idd3 resistance allele the proximal boundary of Idd3 must lie in the 0.07-cM interval between the markers D3Nds36 and D3Nds84 (Fig. 1).
The distal boundary of Idd3 is defined by the congenic strains NOD.B63 and NOD. B6 Idd3R450 (Lord et al. 1995). Typing these strains with the newly developed markers showed that both strains recombine in the 0.08-cM interval between D3Nds34 andD3Nds76 (Fig. 1). Thus, on the basis of this genotyping data the size of the Idd3 locus has been reduced to the 0.15-cM interval between, but not including, the markers D3Nds36 andD3Nds76 (Fig. 1).
Construction of a BAC Contig Across theD3Nds36-to-D3Nds76 Interval
A BAC contig covering the minimal Idd3 interval was constructed as follows. Insert ends of clones, identified by screening the mouse BAC library with the STSs described earlier, were isolated and used to develop new STSs. These new STSs were then used to rescreen the library, identifying additional clones that spanned the gaps between adjacent clusters of clones. A total of 24 clones were isolated that together span the interval between D3Nds36 andD3Nds76 (Fig. 2).
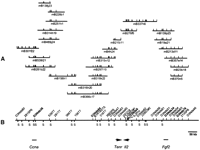
Physical map across the Idd3 minimal interval. (A)SalI fingerprints of BAC clones spanning the interval. (B) SalI restriction map of the Idd3interval; markers were assigned to specific SalI bands by Southern hybridization. Markers shown in bold are polymorphic between NOD and B6. The gene content of the Idd3 interval is shown below, where known the orientation of transcription is indicated by arrowheads (the transcription units are not drawn to scale).
The size of each clone was determined by restriction enzyme digestion (Fig. 2). Each clone was digested independently with bothNotIA and SalI to eliminate errors caused by the comigration of bands of similar size. STSs were assigned to individualSal1 fragments by Southern hybridization and used to identify common bands in overlapping clones. Based on the degree of overlap between individual clones the physical distance betweenD3Nds36 and D3Nds76 is 780 kb (Fig. 2). The proximal boundary of Idd3 lies in the 315-kb interval betweenD3Nds36 and D3Nds84, whereas its distal boundary lies in the 65-kb segment between D3Nds34 and D3Nds76. Thus, the size of the Idd3 locus is between 400 and 780 kb (Fig. 2).
Refinement of the Idd3 Interval by Haplotype Mapping
To refine the Idd3 locus using congenic strains would be impractical given the large number of mice that would have to be bred to identify new recombinants within the interval. Therefore, to further narrow the interval we carried out haplotype analysis on six mouse strains known to carry either the Idd3 susceptibility or protective allele. On the basis of either linkage analysis or congenic mapping the strains 129, SWR, and ABH have been determined to be susceptible at the Idd3 locus, whereas the B6 and NON strains are diabetes resistant at the locus (Denny et al. 1997; Podolin et al. 2000).
These strains were genotyped for all the microsatellite markers and SNPs known to be variant between NOD and B6 that map to the 780-kbIdd3 region (Table 1). In addition, nine more novel SNPs were identified in the six strains by sequencing STSs derived from BAC clone ends (Table 1). A comparison of the NOD and 129 mouse strains, both of which carry an Idd3 susceptibility allele, shows that they have identical genotypes at all of the markers typed with the exception of D3Nds36 (Table 1). This data confirms the congenic mapping data and identifies anIdd3-susceptible haplotype. The sequence identity between the two strains at multiple variable sites indicated the common ancestry of this segment of DNA between the NOD and 129 strains. Analysis of the two strains that carry an Idd3-protective allele, namely B6 and NON, reveals that they also have identical genotypes, but their shared ancestral haplotype is distinct from that found in susceptible strains at all markers (Table 1). This identifies anIdd3-protective haplotype and narrows the Idd3interval to the 365-kb region between D3Nds84 andD3Nds76. Analysis of the ABH strain, which has anIdd3-susceptible allele, shows that it carries theIdd3-susceptible haplotype between the markers 229.1and D3Nds76 (Table 1). These data place the proximal boundary of Idd3 in the 4-kb interval between the microsatellite markerD3Nds6 and the SNP 229.1. The SWR mouse strain has a recombinant haplotype; it has the susceptible haplotype at the markers229.1 through 81.2 and the protective haplotype at the markers 81.3 through 51.2 (Table 1). Because SWR has an Idd3-susceptible allele this gives the most likely location of Idd3 as the 145-kb region between, but not including, the microsatellite marker D3Nds6 and the SNP81.3 (Table 1). As the G allele of the SNP −694 is carried on both the protective and susceptible haplotypes it can be excluded as a candidate for the Idd3 etiological mutation. Of the four genes known to map to the 780-kb interval, namely Ccna, Tenr, Il2, and Fgf2, only Il2 maps to the 145-kb candidate region.
Genotypes at Microsatellite and SNP Markers Across theIdd3 Interval in Mouse Strains Possessing EitherIdd3 Susceptibility or Protective Alleles
Analysis of Il2 Gene Expression in Mouse Strains CarryingIdd3 Susceptibility or Protective Alleles
The promoter regions of the Il2 genes from NOD, B6, and 129 were sequenced to identify any polymorphisms that might lead to differences in transcription between mice carrying either Idd3susceptibility or resistance alleles. The B6 promoter sequence was determined by sequencing a cosmid clone, mC1h5, that contains the entire IL2 structural gene. Based on this sequence data PCR primers were designed to amplify the entire Il2 promoter region from nucleotide −740 to nucleotide +100. This region has been shown previously to encompass all the regulatory elements required for transcription of Il2 (Serfling et al. 1995). Using this PCR-based approach, the sequences of the NOD and 129 Il2promoters were determined and aligned to that of B6. Two polymorphisms were detected, one at position −694 the other at position −674. In both cases the polymorphisms were A-for-G substitutions, with NOD and 129 having the A allele and B6 the G allele. In neither case did the polymorphism occur in a known transcription factor binding site.
To extend the evidence against the presence of functional regulatory allelic variants, the relative levels of transcription of the NOD, B6, and 129 IL2 genes were measured. Total RNA from mixtures of either NOD and NOD.B6 Idd3R450, or NOD.B6 Idd3R450 and NOD.129Idd3 splenocytes stimulated with ionomycin and PMA was used as the template for RT–PCR amplification with the microsatellite markerD3Mit21. D3Mit21 amplifies a variant CAG repeat in exon 1 of Il2. The relative amount of RNA from each allele was quantified by analyzing the fluorescently labeled PCR products on an ABI 373 automated sequencer and calculating the ratio of the two products. As a control for preferential amplification genomic DNA was amplified with D3Mit21 at the same time. No difference was observed in the steady state level of Il2 RNA from either the NOD or the 129 allele compared with that from the B6 allele (ratio of NOD Il2 RNA to B6 Il2 RNA equals 0.89 ± 0.12 and ratio of 129 Il2 RNA to B6 Il2 RNA equals 1.06 ± 0.07, Fig. 3). Taken together with the sequencing data this suggests that the reduction in diabetes conferred by Idd3-protective alleles is not due to differences in the level of transcription of the Il2 gene.
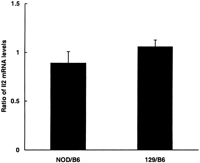
Transcriptional analysis of the Il2 gene from mouse strains carrying either Idd3 susceptibility or protective alleles. RelativeIl2 mRNA levels were determined by semiquantitative RT–PCR.
DISCUSSION
The publication of the results of genome-wide scans for type 1 diabetes susceptibility genes in both the NOD mouse and humans (Todd et al. 1991; Ghosh et al. 1993; Davies et al. 1994; Hashimoto et al. 1994;Concannon et al. 1998; Mein et al. 1998) has raised expectations that within a few years we will know the identity of the genes that predispose to the disease, have a better understanding of the pathological mechanisms underlying it, and, as a result, have identified targets for therapeutic intervention. Although the fine mapping of diabetes susceptibility genes in humans awaits further advances in technology, the genome sequence, a catalog of SNPs, and additional DNA samples from larger patient collections, fine mapping in the NOD mouse has proved more tractable (Lyons and Wicker 1999).
A number of experimental strategies for fine mapping quantitative trait loci (QTL) in mice have been proposed (reviewed in Darvasi 1998). The approach we have adopted to fine map Idd loci in the NOD mouse is one based on the congenic strategy pioneered by Snell in the 1940s during his work on the H2 locus (Snell 1948). Over the past 5 years we have described the production of a series of congenic strains that, initially, confirmed the linkage mapping of Idd3 and, subsequently, systematically fine mapped it to a 0.35-cM interval (Wicker et al. 1994; Lord et al. 1995; Denny et al. 1997). In this study we describe the mapping of the Idd3 locus to a 780-kb interval. This is the first time congenic mapping has been used to map a QTL to an interval less than one megabase in size, confirming the power of the congenic strategy to map loci to intervals amenable to systematic gene identification. Ultimately, two factors limit the resolving power of congenic mapping: the density of the genetic map in the region of interest and the ability to generate and screen large populations of mice.
The current microsatellite map of the mouse genome is dense enough that variant markers between any two inbred mouse strains can be found approximately every centimorgan. To achieve a greater resolution will in most cases require, as was the case for Idd3, the generation of novel, interval-specific markers, which is a time-consuming process. The production of a dense, evenly-spaced SNP map, similar to that currently being generated in man, would greatly alleviate this problem and speed the mapping process.
The numbers of mice required to reduce a congenic interval to any particular size can be theoretically determined (Darvasi 1997). In theory to reduce an interval from 4 cM to 0.15 cM would require the screening of ∼1300 mice. The screening of this number of mice is a realistic proposition; in fact in the present study we screened 944 F2 mice to identify the two recombinant events that reduce the Idd3 interval to 0.15 cM. To achieve a similar level of resolution (with a 95% confidence interval) using a conventional intercross strategy would require 40,000 F2 progeny to be screened.
An alternative strategy for the fine mapping of QTL to interval sizes amenable to positional cloning has been described recently (Talbot et al. 1999). This approach uses an outbred stock of mice derived >30 years ago from an eight-way cross of inbred mouse strains and in theory has a 30-fold increase in resolving power when compared to a conventional F2 cross. However, the general applicability of this approach to fine mapping QTL is debatable as the ability to detect QTL is extremely sensitive to the allele distribution among the eight parental strains at each marker. Moreover, the approach can only map the QTL into a statistically defined confidence interval. The congenic approach on the other hand produces a defined interval that must contain the QTL. Furthermore, the strategy of Talbot et al. (1999)would be compromised if there were two closely linked but separate loci in the region under the linkage curve.
Once an interval has been mapped to a size such that markers can be positioned relative to each other with some precision haplotype, mapping becomes a powerful tool to refine the interval size. By identifying chromosome regions shared identical by descent (IBD) it is possible to use ancestral recombination events to provide additional mapping information. One important assumption is that all strains carrying a susceptibility or protective allele at a particular locus carry the same allele. Given the observed degree of allele sharing IBD among the susceptible and resistant strains employed in the present study this assumption is likely to be valid for the Idd3locus. Given the size of the interval and marker density it is likely that the observed allele sharing is IBD and not merely identical by state (IBS). If the observed sharing were to be IBS it would invalidate this approach. Another assumption is that within the 0.15-cM interval there is only one locus responsible for the Idd3 effect.
The Il2 gene, the only gene currently known to lie within the 145-kb minimal interval, is a very strong candidate for Idd3. Mice that are deficient in the action of IL2, either through the targeted disruption of the Il2 structural gene itself or the genes encoding the α or β chains of its receptor, develop a variety of autoimmune diseases (Hunig and Schimpl 1998). Studies in IL2 and IL2 receptor knockout mice have shown that the cytokine plays a nonredundant role during Fas-mediated apoptosis of activated T cells, one of the central processes in immune homeostasis (Van Parijs et al. 1997; Refaeli et al. 1998). Variants within the coding or regulatory elements of the Il2 gene that alter the expression or function of IL2 would be strong candidates for the Idd3 etiological mutation.
As described in this study we found no evidence of regulatory variants that alter the expression of the Il2 gene, however, sequence variation has been described previously in the coding sequence of IL2 allotypes from different mouse strains (Chesnut et al. 1993; Ghosh et al. 1993; Matesanz and Alcina 1996, 1998; Denny et al. 1997). One sequence variant, a proline/serine substitution at position 6 of the mature IL2 protein, has been shown to correlate with diabetes susceptibility or resistance at Idd3. Mouse strains carrying an Idd3 susceptibility allele, such as NOD or 129, have a proline, whereas strains carrying an Idd3-protective allele, such as B6, have a serine (Denny et al. 1997; Podolin et al. 2000). We have shown recently that different IL2 allotypes have different glycosylation profiles and that this also correlates with the presence or absence of proline at position 6 (Podolin et al. 2000). Although these glycosylation differences have no effect on the ability of IL2 to stimulate proliferation of IL2-dependent cell lines (Podolin et al. 2000) it is conceivable that they may affect the half-life of the molecule leading to a functional deficiency in circulating IL2. It has been shown recently that IL2 can bind to heparan sulfate in vivo and that this bound IL2 is functional in both promoting T-cell activation and stimulating activation-induced cell death (Wrenshall and Platt 1999). This binding to heparan sulfate may well be altered by differences in glycosylation, thereby influencing in vivo availability of IL2.
Using a combination of congenic mapping and haplotype analysis we have narrowed the most likely location of Idd3 to a 145-kb segment of DNA that contains the variant Il2. Although our genetic mapping data does not conclusively prove that the Il2 gene isIdd3, the current lack of evidence for a functional difference between IL2 allotypes from mouse strains with Idd3susceptibility or protective alleles does not exclude it. Ultimately, the only definitive way to establish whether Il2 isIdd3 will be to construct a “knock-in” mouse in which Il2 from NOD is replaced with the variant gene from a diabetes-resistant mouse such as B6.
METHODS
Animals
NOD/MrkTacfBR (NOD) mice were purchased from Taconic Farms, Inc. (Germantown, NY). The following congenic strains were used in this study: NOD.B62 (N11F2-3), NOD.B63 (N11F2-3), NOD. B6 Idd3R450 (N13F2-3), NOD. B6 Idd3R808 (N13F2-3), and NOD.129 Idd3 (N6F2-4). The derivation of congenic strains NOD.B62, NOD.B63, NOD. B6 Idd3R450, and NOD.129 Idd3 has been described previously (Lord et al. 1995;Denny et al. 1997; Podolin et al. 2000). The NOD. B6 Idd3R808 congenic strain was developed by backcrossing NOD.B62 to NOD and intercrossing the resulting F1 progeny. The F2progeny were genotyped with markers within the Idd3 interval and appropriate recombinants backcrossed to NOD. Suitable F1progeny were intercrossed to produce homozygous animals. The congenic strain was maintained by brother–sister mating. All mice were housed under sterile, specific pathogen-free conditions.
Assessment of Diabetes
Mice were monitored for the development of diabetes as described previously (Wicker et al. 1994).
STS Mapping and Isolation of Novel Microsatellites
Mouse BAC (Research Genetics) and P1 Imperial Cancer Research Foundation libraries were screened by PCR according to the suppliers' instructions. Clone insert ends were rescued by vectorette PCR and STSs developed as described previously (Denny et al. 1997). Novel microsatellite markers were isolated from P1 clones using a PCR-based vectorette approach as described previously (Merriman et al. 1997). Primer sequences for the microsatellite and STS markers described in this paper are available on the web at http://diesel.cimr.cam.ac.uk/todd.
PCR Analysis
STS, fluorescent, and nonfluorescent PCR reactions were performed and analysed as described previously (Denny et al. 1997).
Restriction Enzyme Mapping of BAC Clones
BAC DNA was prepared using a standard alkaline-lysis protocol. Aliquots of 1 μg of BAC DNA were digested for 1 hr at 37°C with 10 units of either NotIA or SalI. Following digestion, DNA fragments were separated by pulsed-field gel electrophoresis. Gels were run at 200 V for 15.2 hr at 14°C with pulse times ramped from 0.2 sec to 21.8 sec. Following electrophoresis and visualization by ethidium bromide staining, DNA fragments were transferred to nylon filters by capillary action and fixed by UV cross-linking. Filters were hybridized with [γ-32P]-labeled oligonucleotides.
Il2 Promoter Sequencing
The cosmid mC1h5 was sequenced using a random shotgun approach essentially as described in Bankier et al (1987). Sequence data was assembled using the program GAP4 (Bonfield et al. 1995). This sequence data has been submitted to GenBank and assigned the accession number AF19596. Two sets of PCR primers were designed to amplify theIl2 promoter. IL2.Pro1 (5′-ATGAAAGTGCAACTAGAGCAC-3′) and IL2.Pro3 (5′-GAGACACAAAAGTAACTCATG-3′) give a 444-bp product spanning nucleotides 3773–4217 of the mC1h5 sequence and IL2.Pro2 (5′-CTTTTCATCTATCTCCTCTTGC-3′) and IL2.Pro4 (5′-GACAAGGAGCACAAGTGTCAAT-3′) amplify a 500-bp product spanning nucleotides 4115–4615. Following amplification, PCR products were gel purified and then sequenced, with the amplification primers, using an ABI Prism dye terminator cycle sequencing kit (PE Biosystems, Warrington, UK) according to the manufacturer's instructions. Sequence data for the NOD and 129 Il2 promoter regions have been submitted to GenBank and assigned the accession numbers AF19594 and AF19595, respectively.
Generation of IL2-Containing Splenocyte Cultures
One million mouse spleen cells were stimulated with 10 ng/ml of PMA and 400 ng/ml of ionomycin (Calbiochem, San Diego, CA) for 4 hr as described previously (Chen et al. 1994).
Quantification of Il2 mRNA Levels
Total RNA was extracted from 4 × 107 splenocytes stimulated with ionomycin and PMA as described above using an RNeasy RNA extraction kit (Qiagen, Crawley, UK) following the manufacturer's instructions. First-strand cDNA was synthesized from 1 μg of total RNA using a M-MLV H− RT cDNA synthesis kit (Life Technologies, Glasgow, UK) according to the manufacturers instructions. The cDNA was diluted 1/100 and 1 μl was used as template for amplification withD3Mit21 (primer sequences for this microsatellite marker are available on the web athttp://www-genome.wi.mit.edu/cgi-bin/mouse/index). Following amplification the fluorescently labeled PCR products were analyzed on an ABI373 automated sequencer as described previously (Denny et al. 1997). The amount of Il2 mRNA was quantified using Genotyper software (PE Biosytems).
Acknowledgments
This work was funded by grants from the UK Medical Research Council, the British Diabetic Association, and the Wellcome Trust.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.








