
Abstract
The identification of many of the transcribed genes in man and mouse is being achieved by large scale sequencing of expressed sequence tags (ESTs). Attention is now being turned to elucidating gene function and many laboratories are looking to the mouse as a model system for this phase of the genome project. Mouse mutants have long been used as a means of investigating gene function and disease pathogenesis, and recently, several large mutagenesis programs have been initiated to fulfill the burgeoning demand of functional genomics research. Nevertheless, there is a substantial existing mouse mutant resource that can be used immediately. This review summarizes the available information about the loci encoding X-linked phenotypic mutants and variants, including 40 classical mutants and 40 that have arisen from gene targeting.
Mammalian X-linked traits are easily recognized by their inheritance patterns and their mode of expression. Whereas hemizygous males carry one copy of X-linked loci and suffer from the full effect of any mutation, heterozygous females carry two copies and have a phenotype that reflects the relative expression, as determined by X-inactivation status, of the mutated and normal copies of the gene (Lyon 1999). The X chromosome is also unusual in that X linkage of genes is almost totally conserved in eutherian mammals (Ohno 1973). Therefore, disorders that are X linked in man are also X linked in the mouse, which leads to the ready identification of mouse models of human X-linked disease. The existing mouse mutant resource, which comprises well over 1000 different stocks and strains, has been exploited to investigate gene function and disease pathogenesis associated with X-linked and autosomal loci (Paigen 1995; Bedell et al. 1997)5. The past decade has seen a revolution in the ability to deliberately introduce mutations into mouse genes by homologous recombination (Fisher 1997; Müller 1999; Roths et al. 1999) and, as a result, the number of mouse X-linked traits has doubled. Although there are earlier reviews (Davisson 1987; Miller 1990), no comprehensive summary of existing mouse X-linked phenotypes has been published recently. The primary aim of this review is to describe the current status of the phenotype map of the mouse X chromosome for those working in the field of genome research with an interest in X-linked disease.
Analyzing mouse mutants at the molecular and phenotypic level is one of the most powerful ways of understanding gene function in mammals. Apart from a few notable exceptions, in which mutations exist in mouse genes whose homologs are known to be responsible for human disease, there are considerable phenotypic similarities between the mouse and human disorders, and the mutant mouse provides an animal model for understanding disease pathogenesis and for assessing therapeutic regimes. Therefore, it is likely that most of the remaining mouse phenotypes will provide a valuable resource for identifying the molecular basis of a homologous human disease.
Comparative Map of the Human and Mouse X Chromosomes
The initial stage of assessing and identifying mouse models for human disease from the existing mouse mutant resources involves careful characterization of the phenotype associated with the mutant locus and predicting the position of its human homolog on the man–mouse comparative map.
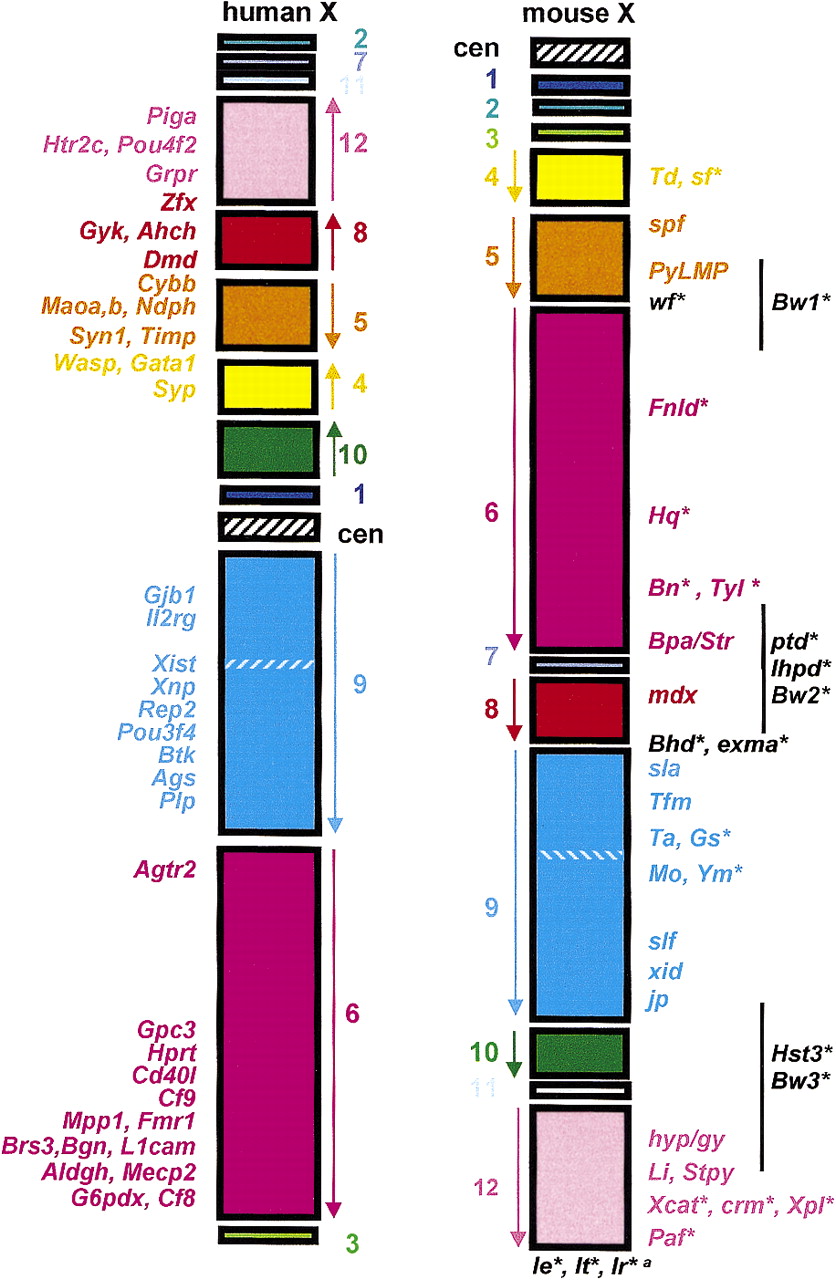
The mapping of >130 conserved loci on the X chromosomes of both mouse and man (Boyd et al. 1998, 1999) has confirmed the prediction that X linkage of genes is preserved in mammals (Ohno 1973). However, when the relative positions of loci on the human and mouse X chromosomes are compared, it can be seen that subchromosomal blocks of homologous loci have been rearranged with respect to each other during the 80 million years of evolutionary time that separate the two species. It is important to understand these rearrangements fully, because an identical comparative map position is an important criterion for identifying and confirming mouse models for human genetic disease. More than a decade ago, five distinct homologous blocks of loci or conserved segments were acknowledged as sharing homology on both the human and mouse X chromosomes (Searle et al. 1987, 1989; Amar et al. 1988; see Fig.1 and Table 1 for the current status of the comparative map). The human X chromosome long arm (Xq) was recognized as being split into two major blocks on the mouse X chromosome. There are only two minor modifications to this Xq comparative map, the identification of a 600-kb inversion around Xist (Rougelle and Avner 1996) and the mapping of the synaptobrevin like locus (Sybl1) to the proximal region of the mouse X chromosome (D'Esposito et al. 1997). In contrast, it soon became apparent that the three conserved segments thought to comprise the human X chromosome short arm (Xp) could be apportioned into five major and four minor conserved segments on the mouse X chromosome (Laval and Boyd 1993; Blair et al. 1994, 1995,1998b; Blaschke and Rappold 1997). Twelve conserved segments have now been identified on the man–mouse X chromosome comparative map (Fig.1), and additional regions of homology may be defined as conserved genes are mapped to a higher resolution (Ehrmann et al. 1998). Nevertheless, for most of the X chromosome, once the position of a locus is known on the human X chromosome, its position on the mouse X chromosome can be predicted with reasonable accuracy and vice versa. The evolutionary breakpoint regions remain the only areas of uncertainty and these, because they have been subjected to multiple rearrangements during evolution, can be expected to have a complex structure and this has been borne out by recent mapping data (Dinulos et al. 1996; Blair et al. 1998b; Disteche et al. 1998). Over 130 genes and conserved loci have been regionally mapped on both the human and mouse X chromosomes and form the framework for constructing the comparative map (Table 1). This map has been invaluable in identifying human diseases and candidate gene loci for many of the classical mutants that have been recovered from mouse colonies over the years.
List of X-Linked Genes and Conserved Sequences

|

|
[i] Included are only those loci that have been regionally mapped on both the human and mouse X chromosomes with sufficient resolution to contribute to the man–mouse comparative map (see Fig. 1). This list is given in order of loci from the mouse centromere to the telomere. The rectangles encase conserved segments, in which locus order is preserved in man and mouse, with the exception of a small inversion around Xist (see text). Note that the relative order of segments 1–3 has not been established, and eIF2γ (Ehrman et al. 1998) has not been sufficiently well positioned in the mouse to include here, although it might represent a novel conserved segment. The location of each locus on the mouse X chromosome is given in column 1 and indicates the distance in cM from the centromere. When a locus also has been mapped by in situ hybridization, the cytogenetic location on the mouse X chromosome is given in brackets before the genetic map position. Mapping data are taken from the literature (Boyd et al. 1998), except for the Sat locus (H.J. Blair, unpubl.). The cytogenetic position of each locus is given for its location on the human X chromosome as, because of the large number of translocation breakpoints and rearrangements affecting the human X chromosome, this provides a more useful reference than a genetic map position. For a visual appreciation of the relative physical size of the conserved segments and their locations on the human and mouse chromosomes, see Fig. 1.
Spontaneous and Induced X-Linked Mouse Mutants and Variant Traits
Thirty-eight X-linked mouse-independent visible phenotypes covering a wide range of traits are reported in the literature (Table 2; Fig.2). Most of these phenotypes have arisen spontaneously in mouse colonies or among the large numbers of mice used in mutagenesis experiments (George et al. 1994). The paucity of induced X-linked mutations is probably due to that fact that most novel mutations have been sought in the F1 progeny of mutagenized males and as a result, only those X-linked mutations that have a phenotypic effect in heterozygous females are identified. Some attempts were made to obtain sex-linked recessive lethals by identifying abnormal sex ratios in the offspring of F1 females produced in classical mutagenesis experiments (Searle et al. 1964). Recessive mutations, such as sex-linked fidget, arose in experiments like these (Lyon et al. 1981b; Phippard et al. 2000).
Spontaneous and Induced X-Linked Mouse Traits
| Symbol | Mutant name | Origin | Map position | No. of alleles | Phenotype details and gene mutated if known | References |
| Bhd | broad-headed | R | 35 | 1 | broad heads and snouts in heterozygous females, males die at birth, have aberrant ossification and may have cleft palate | Phillips and Fisher (1978); Reed and Boyd in prep. (1999) |
| Bn | bent-tail | S | central X | 1 | shortened and kinked tails | Garger (1952) |
| Bpaand Str (NsdhlBpa/Str) | bare patches or striated | SRC | 29 | 4 | hyperkeratosis, punctate calcification in the epiphyses of vertebrae, and long bones in heterozygous females, males die in utero; mutations have been found in Nsdhl[NAD(P)-dependent steroid dehydrogenase], an enzyme involved in sterol biosynthesis | Liu et al. (1999) |
| Bw1 | body weight marker 1 | V | proximal X | 1 | increased body weight in males | Dragani et al. (1995) |
| Bw2 | body weight marker 2 | V | central X | 1 | increased body weight in males | Dragani et al. (1995) |
| Bw3 | body weight marker 3 | V | distal X | 1 | increased body weight in males | Dragani et al. (1995); York et al. (1997) |
| crm | cream | S | distal X | 1 | pale yellow coat that is fluorescent in long-wave UV light, in hemizygous males and homozygous females | Hetherington (1977) |
| Fnld | faint-lined | R | 15 | 1 | faint dorsal stripes on heterozygous females, which are also small, males die in utero | Gormally and Boyd (1998) |
| Gs | greasy | S? | 38 | 1 | shiny fur in homozygous females and males | Larsen et al. (1964) |
| gy | gyro ⧣ | R | 65.4 | 1 | hypophosphatemia, rickets, sterility, and inner ear abnormalities in males; the 5′ end of Phex, the endopeptidase mutated in HYP patients and Sms(spermine synthase) are deleted | Lyon et al. (1986); Strom et al. (1997); Lorenz et al. (1999) |
| Hq | harlequin | S | 17 | 1 | small, almost totally bald, homozygous females and males | Barber (1971) |
| Hst3 | hybrid sterility 3 | V | distal X | 1 | sterility in F1 males from crosses between Mus spretus and laboratory strains of mice | Guenet et al. (1990) |
| hyp | hypophosphatemia ⧣, X-linked | S | 65.4 | 1 | hypophosphatemia and rickets seen in males; the 3′ end of Phex, the endopeptidase mutated in HYP patients is deleted | Eicher et al. (1976); Strom et al. (1997); Strom et al. (1998) |
| le | eye–ear reduction | R | syntenic | 1 | anopthalmia, small external ears in homozygous females and males | Hunsicker et al. (1974) |
| lhpd | interspecific hybrid dysplasia | V | central X | 1 | abnormal development of the spongiotrophoblast, influenced by the sex of the conceptus, with a resultant effect on fetal growth | Zechner et al. (1996) |
| lt | irregular teeth | S? | syntenic | 1 | incisors absent in heterozygous females, males die in utero | Phipps (1969) |
| jp, msd, orrsh (Plpjp/msd/rsh) | jimpy, myelin synthesis deficiency or rumpshaker ⧣ | S | 56 | 3 | tremor of hindquarters and convulsions with early death in males caused by mutations inPlp (myelin proteolipid protein); models for Pelizaeus–Merzbacher disease | Meier and MacPike (1970); Dautigny et al. (1986); Griffiths et al. (1990) |
| Li | lined ⧣ | S? | 65.6 | 1 | transverse dorsal stripes in heterozygous females, males die in utero; ribosomal S6 kinase (Rps6ka3), which is mutated in Coffin–Lowry syndrome, and possibly other genes are deleted | Blair et al. (1998a) |
| mdxor (Dmdmdx) | muscular dystrophy ⧣, X-linked | SC | 32 | 4 | tremors and mild inco-ordination, elevated creatine and pyruvate kinase in males; mutations have been found in Dmd (dystrophin), models for Duchenne muscular dystrophy | Bulfield et al. (1984); Chapman et al. (1989) |
| Mo or (Atp7aMo) | mottled ⧣ | SRC | 44 | >20 | Defective pigmentation, wavy vibrissae/coat, connective tissue, and neurological abnormalities, males have allele-dependent time of death, from in utero to adulthood; mutations have been found in Atp7a (ATPase, Cu2+transporting, type 7a), models for Menkes disease | Hunt (1974); Danks (1986); Das et al. (1995); Cecchi et al. (1997); Reed and Boyd (1997); Cunliffe (1999) |
| Paf | patchy fur | S | 73 | 1 | missing fur with hemizygous males more severely affected than homozygous females, male carriers have delayed disjunction at meiotic metaphase I; associated with rearrangements atMid1 locus (midline 1), which is associated with Opitz syndrome | Lane and Davisson (1990); Quaderi et al. (1998) |
| ptd | palate-tail-digits abnormality | S | central X | 1 | tail malformations, hind feet polydactyly and frequent cleft palate in homozygous females and males | Barra (1990) |
| PyLMP | transgenic insertion of LMP | I | 5.0 | 1 | secondary cleft palate and craniofacial malformation, males die shortly after birth; transgenic insertion in Cask (calcium/calmodulin) dependent serine protein kinase | Laverty and Wilson (1998) |
| Rv3 | Rauscher leukemia virus susceptibility-3 | V | syntenic | 1 | CBA strain variant which results in susceptibility to spleen focus formation by Rauscher leukemia virus in males | Heller and Pluznik (1984) |
| sf | scurfy | S | 2.1 | 1 | scaly and tight skins, hypogonadism, thrombocytopenia with death before weaning in males | Lyon et al. (1990) |
| sla | sex-linked anemia | R? | 36.4 | 1 | severe hypochromic anemia, defective intestinal iron transport; mutation in hephaestin (Heph) a ceruloplasmin homologue | Kingston et al. (1978); Vulpe et al. (1999) |
| slf | sex-linked fidget ⧣ | R | In8H (A7-E1) | 1 | slight vertical head shaking in homozygous females and males; Pou3f4 expression abolished in embryonic ear, model for DFN3 | Phillips and Fisher (1978); Phippard et al. (2000) |
| spf (Otcspf) | sparse-fur ⧣ | S? | 3 | 2 | wrinkled skin, paucity of fur and bladder stones in males; mutations found inOtc (ornithine transcarbamylase) | Rosenberg et al. (1983); Veres et al. (1987) |
| Stpy | stripey | S | 65.6 | 1 | transverse dorsal stripes in heterozygous females, males die in utero; ribosomal S kinase (Rps6ka3, also known as Rsk2) and pyruvate dehydrogenase E1α (Pdha1), which is mutated in lactic acidosis, and possibly other genes are deleted | Blair et al. (1998a) |
| Ta (EdaTa) | tabby ⧣ | SRC | 37 | >15 | hair defects, tooth abnormalities and aberrant eccrine sweat glands in males and heterozygous females; mutations found in Eda(ectodysplasin-A), model for anhidrotic ectodermal dysplasia | Srivasta et al. (1997) |
| Td (EbpTd) | tattered ⧣ | R | 2.1 | 2 | hyperkeratosis and craniofacial malformations in heterozygous females, males die in utero with skeletal and developmental defects; mutation in the sterol isomerase Ebp (emopamil-binding protein); model for chondrodysplasia punctata | Derry et al. (1999) |
| Tfm (ArTfm) | testicular feminization ⧣ | S | 36 | 2 | external female genitalia and insensitivity to androgens in males; mutations in Ar(androgen receptor), model for testicular feminization | Lyon et al. (1981a); Charest et al. (1991); Gaspar et al. (1991); Tanaka et al. (1993) |
| Tyl | trembly-like | S | central X | 1 | tremors, seizures and paralysis with death before weaning in males | Sweet et al. (1990) |
| wf | wide-faced | S | 12.7 | 1 | craniofacial malformations in homozygous females and males, males have an increased body weight with age | Boyd et al., (in prep.) |
| Xcat | X-linked cataract | R | 68 | 1 | total lens opacity in homozygous females and males, suggested model for Nance-Horan syndrome | Favor and Pretsch (1990); Zhou et al. (1995) |
| Xid (Btkxid) | X-linked immunodeficiency ⧣ | S | 51 | 1 | defective and impaired immune responses; mutations in Btk (Bruton agammaglobulinemia tyrosine kinase) | Rawlings et al. (1993); Thomas et al. (1993) |
| Xpl | X-linked polydactyly | S | distal X | 1 | preaxial polydactyly and tibial hemimelia in carrier and homozygous females and males | Sweet and Lane (1980) |
| Ym | yellow mottled | S? | 44 | 1 | yellowish mottling of heterozygous females, males die in utero, lies close to, but recombines withMo | Hunsicker (1969) |
[i] Mutants that carry lesions at genes associated with human genetics disease are indicated by a pound sign (⋕) after the mutant name. Mouse mutants are generally given a name and symbol when they are first identified; these are used throughout this review for clarity, and the mutants described here are arranged in the alphabetical order of the symbol. Once the gene responsible for a mutant has been cloned, current nomenclature rules dictate that the official name for the mutant is (gene symbolmutant name ) and this is given in parentheses beside the original symbol, where appropriate. Mutants with small deletions that remove several genes, e.g., gy, which has a deletion that removes both Phex and Sms,retain their original mutant symbol. The origin of the mutation, if known, is indicated : (C) chemical mutagenesis; (I) insertional mutagenesis; (R) radiation mutagenesis; (S) spontaneous; (V) natural variant. Several reports of X-linked loci affecting various aspects of immune responsive have also been published. These include regulation of the secondary immune response to LDH-C4 (Marsh et al. 1977), to thymus-independent antigens (Zeicher et al. 1977), to spleen focus formation by Rauscher leukemia virus (Heller and Pluznik 1984), and to denatured DNA (Mozes and Fuchs 1974). These have not been included here, as their relationship to each other and to X-linked loci known to be involved in immune regulation, e.g., xid, is unclear. They are indicated by Ir below the mouse X chromosome on Fig. 1.
The comparative phenotype map of the human and mouse X chromosomes. Each conserved segment, in which the order of loci is the same in mouse and man, is indicated by a colored rectangle. The loci that define each segment are given in Table 1; note that the order of segments1, 2, and 3 has not been established and is arbitrary. The segments are numbered from 1–12 from the centromere to the telomere on the mouse X chromosome and the order of loci indicated by an arrow alongside each block. The hatched region within segment 9 represents the 600-kb region that is inverted around the Xist locus (see text). The centromeric regions are indicated by black and white hatched rectangles; there is, as yet, no known evidence for evolutionary conservation of centromeric sequences between mouse and man. The Xp pseudoautosomal region, which has a complex evolutionary history (Blaschke and Rappold 1997), is not included because it is outside the scope of this review, as loci in this region do not exhibit X-linked inheritance. Classical mutants carrrying spontaneous and induced mutations and variants (Table 2) are indicated on the mouse X chromosome, and targeted mutations (Table 3) are positioned on the human X chromosome for clarity. Names in black text are (1) known to have an X-linked inheritance pattern but have not been positioned on the X (e.g., Ie, It), or (2) have not been subjected to high-resolution mapping (e.g., Bw1, the black line indicates the probable region in which the locus lies), or (3) have been mapped to evolutionary breakpoint positions and therefore the position of the human homolog cannot be defined (e.g.,exma, Bhd, wf). For those traits marked with an asterisk (*), the gene responsible is not known; for all other traits the underlying lesion has been defined.
a(Ir) Immune response genes that are X linked (see Table 2 footnote).
Examples of spontaneous and induced X-linked traits in the mouse. (A,B) Males carrying the greasy (Gs) (A) and harlequin (Hq) (B) mutations; neither of the genes responsible has been cloned. (C) A heterozygous female carrying the broad-headed (Bhd) mutation, which is associated with a craniofacial anomaly, note the unusually short and broad snout. Comparative mapping has shown that Bhdcannot be a model for FGD1 or ATRX, but it remains an interesting skeletal mutant in which males have multiple ossification anomalies and die shortly after birth (V. Reed and Y. Boyd, in prep.). (D) A heterozygous female carrying a mutation at one of the mottled (Mo) alleles associated with death between birth and weaning of affected males (Atp7aMo-10H ). The mosaic coat can be clearly seen, in which hypopigmented areas represent populations of cells carrying the mutant allele and the normally pigmented (brown) areas represent areas of cells carrying the wild-type allele. Details of the phenotypes and references describing each of these mutants are given in Table 2.
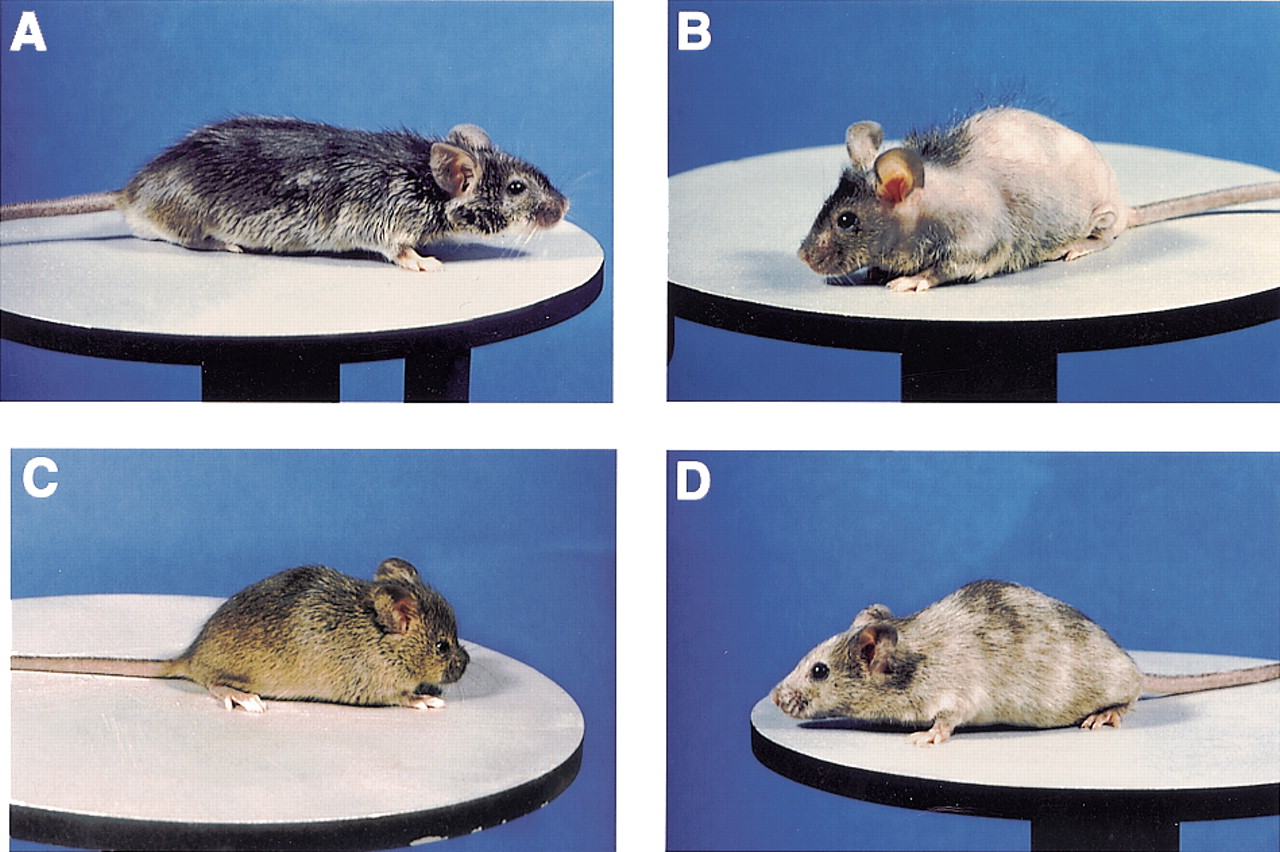
For most of the recovered mutations, only a single mutant allele is available for analysis, limiting the value of the mouse as a model for X-linked human disease, which is often associated with a high number of different new mutations (Rossiter and Caskey 1991). However, where several alleles exist, the mouse provides an ideal tool for studying the phenotypic effects of different mutations in the same gene on an identical genetic background. Over 20 alleles, associated with differing phenotypes have been recovered at the mouse mottled locus and these provide a useful paradigm for using the mouse to study human disease (Cunliffe 1999).
Mottled: A Paradigm for Mouse Models for Human X-Linked Disease
For many years, the mottled mouse has been recognized as a model for Menkes disease (MD), and in both species, a range of mutations has been found in the gene encoding the copper transporter ATP7A(Cecchi et al. 1997; Reed and Boyd 1997; Tümer et al. 1997). Affected mice and human patients show a similar and variable course of disease with the main features being growth retardation, neurological and connective tissue abnormalities, peculiar hair and hypopigmentation (Danks 1986; Tümer and Horn 1997; Fig. 2D). Mutations that have a mild phenotypic effect in the mouse, such as the splice-site lesion leading to the production of both normal and aberrant transcripts in mottled blotchy, seem to be of a similar type to those associated with occipital horn syndrome (OHS, a mild allele of MD). In both man and mouse, this type of mutation is associated mainly with connective tissue problems (Das et al. 1995). However, there are significant differences between man and mice in the type of molecular lesions associated with the more severe phenotypes. The classical MD phenotype is mainly associated with nonsense or frameshift mutations and there is also a substantial proportion (∼20%) of patients with gross deletions covering varying proportions of the ATP7A-coding region. In the mouse, the largest known deletion, which covers exons 11–14 (Cunliffe 1999), is in frame, and no nonsense or frameshift mutations have been reported to date. This fact demonstrates that caution must be taken when assessing potential therapies with mottled mice as animal models. The most striking phenotypic difference is that MD patients, with what might be expected to be null mutations at ATP7A, survive until birth, whereas over half of the mottled alleles are associated with prenatal lethality of males sometime after mid gestation. It has long been thought that the mottled mutations causing early postnatal death are the most appropriate model for classical MD in which affected boys die in the first few years of life. However, recent evidence has shown that in the mouse, Atp7a mutations most similar to those seen in classical MD cause prenatal death (Cunliffe 1999; P. Cunliffe, V. Reed, and Y. Boyd, in prep.). Therefore, at the level of cellular copper processing, mottled alleles associated with prenatal lethality may provide a better model. This is particularly important in light of the varied response of MD patients to copper histidine treatment (Tümer and Horn 1997).
Using the Mouse to Identify the Genes Responsible in X-Linked Dominants Associated with Intrauterine Male Lethality
A further interesting picture has emerged from studies on mouse mutants as possible models for X-linked dominant disease, which, because they are associated with prenatal lethality of males, are difficult to position accurately on the human X chromosome. Disorders associated with the lethality of males in utero are probably caused by lesions in genes that have important developmental roles and that may be conserved in man and mouse. In the mouse, the relative ease of high-resolution mapping renders the genes responsible amenable to positional cloning. Recently, an interesting association between sterol biosynthesis and skeletal defects has been revealed by the identification of the underlying molecular lesions in the Bpa,Str, and Td mutants, which are all associated with skeletal and skin anomalies in heterozygous females (Table 2). Mutations in the 3β-hydroxysteroid dehydrogenase Nsdhlwere first found in several independently derived Bpa andStr mutants, which were shown to be allelic, although there are differences in the severity of the phenotype (Liu et al. 1999). TheBpa mouse was suggested originally as a mouse homolog of CPDX2 as both display-striated hyperkeratosis and skeletal abnormalities including short stature, rhizomelic shortening of the limbs, epipyhseal stippling, and craniofacial anomalies (Happle 1983). However, a more detailed phenotypic analysis of Td has revealed that it also has many of these features and Td was discovered to be a model for CPDX2 when mutations in the sterol isomerase Ebp were discovered inTd and CPDX2 patients (Braverman et al. 1999; Derry et al. 1999). Thus, the Bpa/Str alleles and Td mice provide tools to investigate the relationship between sterol biosynthesis, intrauterine death of males, and the skin and skeletal defects seen in heterozygous females.
X-Linked Phenotypes Associated with Mutations Introduced by Gene Targeting
There are now as many mouse X-linked phenotypes introduced deliberately by gene targeting than have been recovered over the years in mouse colonies. Approximately one-half of the targeted genes are implicated in overt human disease and have been ablated to create models for understanding gene function and disease pathogenesis (Table3). Some of these genes have shown comparable phenotypes in the mouse, such as the targeted disruption of genes encoding factor VIII and IX, which have provided excellent mouse models for studies on haemophilia A and B (Bi et al. 1995; Wang et al. 1997). Others exhibit some, but not all, aspects of the corresponding human disease; for example, there is an impaired humoral immune response in CD40 ligand-deficient mice, but they do not develop full-blown hyperIgM syndrome (Renshaw et al. 1994). In the future, mouse models carrying defined molecular lesions identical to those found in human disease can be provided by gene-targeting technology. In conjunction with the production of a series of different mutations in the same target gene, as has been done at the autosomal Fgf8locus by Meyers et al. (1998), the availability of engineered mouse models relevant to human disease can only increase.
Targeted Mutations at Mouse X-Linked Loci
| Symbol | Targeted gene | Phenotype details | References |
| Ags | α-galactosidase, mutated in Fabry disease # | phenotypically normal at 10 weeks, liver and kidney pathophysiology similar to human Fabry disease | Ohshima et al. (1997) |
| Agtr2 | angiotensin II receptor, type 2 | blood pressure increase, increased sensitivity to pressor action of angiotensin II, lowered body temperature, reduced exploratory behavior, anxiety-like behavior | Hein et al. (1995); Ichiki et al. (1995); Okuyama et al. (1999) |
| Abcd1 | ATP-binding cassette, subfamily D, member 1, mutated in adrenoleukodystrophy # | reduced β-oxidation of very long chain fatty acids (VLCFAs) with consequent elevation of saturated VLCFAs in total lipids of all tissues and cholesterol esters in adrenocortical cells, no neurological involvement seen in mice up to 6 months | ForssPetter et al. (1997); Lu et al. (1997) |
| Bgn | biglycan | skeletal phenotype marked by progressive lowering of bone mass, suggested model for role of ECM proteins in osteoporosis | Xu et al. (1998) |
| Brs3 | Bombesin receptor subtype-3 | mild obesity, hypertension, impaired glucose metabolism, reduced metabolic rate, increased feeding efficiency and subsequent hyperphagia | Ohki-Hamazaki et al. (1997) |
| Btk | Bruton agammaglobulinemia tyrosine kinase # | mild X-linked immunodeficiency, with additional compromise of B cell precursor expansion | Kerner et al. (1995); Khan et al. (1997) |
| CD40I | CD40 antigen ligand, mutated in hyper-IgM syndrome # | failure to undergo isotype switching to T-cell-dependent antigens, normal response to T-cell-independent antigens | Renshaw et al. (1994); Xu et al. (1994); Castigli et al. (1995) |
| Cf8 | coagulation factor VIII, mutated in hemophilia A # | <1% factor VIII clotting activity, significant bleeding after tail biopsy, which may be lethal, no spontaneous bleeding | Bi et al. (1995) |
| Cf9 | coagulation factor IX, mutated in hemophilia B # | absence of factor IX antigen in plasma, <5% factor IX clotting activity | Wang et al. (1997) |
| Cybb | subunit of NADPH-oxidase complex, mutated in chronic granulomatous disease # | chronic granulomatous disease, lack of phagocyte superoxide production, increased susceptibility to infection and altered inflammatory response to thioglycollate peritonitis | Pollock et al. (1995); Morgenstern et al. (1997) |
| Dmd | dystrophin, mutated in Duchenne muscular dystrophy # | hypertrophic skeletal muscles, fibre size variations with necrosis and regeneration | Araki et al. (1997) |
| Fmr1 | fragile X mental retardation syndrome 1 homolog # | macro-orchidism, learning deficits | Bakker et al. (1994); Oostra and Hoogeveen et al. (1997) |
| G6pdx | glucose-6-phosphate dehydrogenase | at ES cell level only, clones with undetectable levels of the enzyme are extremely sensitive to hydrogen peroxide and diamide | Pandolfi et al. (1995) |
| Gata1 | GATA-binding protein 1 | male neonatal lethal, mid-gestation embryos pallid with arrest of erythroid development | Pevny et al. (1991); Fujiwara et al. (1996); Takahashi et al. (1997) |
| Gjb1 | gap junction protein connexin32, mutated in X-linked CharcotMarie-Tooth disease # | from 3 months progressive demyelinating neuropathy, motor fibers more affected than sensory fibers | Scherer et al. (1998) |
| Gpc3 | glypican 3, mutated in, Simpson– Golabi–Behmel syndrome # | overgrowth, cystic kidneys | Li et al. (1998) |
| Grpr | gastrin releasing peptide receptor | no gross phenotypic abnormalities, loss of bombesin-induced feeding suppression | Hampton et al. (1998) |
| Gyk | glycerol kinase # | males normal at birth but exhibit growth retardation, altered fat metabolism with profound hypoglycerolemia and elevated free fatty acids, autonomous glucocorticoid synthesis and death by 3–4 days; heterozygous females are healthy and biochemically normal | Huq et al. (1997) |
| Hprt | hypoxanthine phosphoribosyl transferase # | overgrooming in older mice, increased locomotor activity after amphetamine administration; self-injurious behavior only when inhibitor of Aprt also present (Wu and Melton 1993), model for Lesch–Nyhan disease | Hooper et al. (1987); Kuehn et al. (1987); Nehls et al. (1994); Tsuda et al. (1997) |
| Htr2c | 5-hydroxytryptamine (serotonin) receptor 2C | overweight because of abnormal feeding behavior, spontaneous death from seizures | Tecott et al. (1995) |
| II2rg | interleukin 2 receptor, γ chain, mutated in X-linked severe combined immunodeficiency # | similar phenotype to human XSCID, decrease in lymphocyte numbers but increase in monocytes, few T cells in young mice and no natural killer cells | Leonard et al. (1995); Ohbo et al. (1996); Sugamura et al. (1996) |
| L1cam | L1 cell adhesion molecule, mutated in CRASH syndrome # | smaller than wild type animals, uncoordinated hind legs, hypoplasia of corticospinal tract, abnormal brain pathology, and impaired exploratory behavior | Dahme et al. (1997); Fransen et al. (1998) |
| Maoa | monoamine oxidase a # | pups have elevated serotonin levels, trembling, difficulty in righting, fearfulness; adults have distinct behavioral syndrome with enhanced aggression in males | Cases et al. (1995) |
| Maob | monoamine oxidase b | increased reactivity to stress, increased levels of beta- phenylethylamine, resistance to neurodegenerative effects of MPTP toxin (which induces a Parkinson's- like condition) | Grimsby et al. (1997) |
| Mecp2 | methyl CpG binding protein 2, mutated in Rett syndrome # | chimeric embryos exhibit developmental defects with severity proportional to mutant cell contributions | Tate et al. (1996); Amir et al. (1999) |
| Ndph | Norrie disease homolog # | development of retrolental structures in vitreous body, disorganization of retinal ganglion cell layer, occasional loss of outer plexiform layer with resultant interchange of inner/outer nuclear layer, absence of outer segments of photoreceptor cell layer | Berger et al. (1996) |
| Ocrl | oculocerebrorenal syndrome of Lowe # | no abnormal phenotype, with postulated compensation by the autosomal gene inositol polyphosphate 5-phosphatase (lnpp5b) as explanation | Janne et al. (1998) |
| Piga | phosphatidylinositol glycan, class A | wrinkled and scaly skin, death a few days after birth | Tarutani et al. (1997) |
| Plp | myelin proteolipid protein, mutated in Pelizaeus– Merzbacher disease # | no gross effect, assembly and maintenance of normal amounts of myelin, progressive tract-specific axonopathy | Boison and Stoffel (1994);Griffiths et al. (1995); Klugmann et al. (1997); Griffiths et al. (1998) |
| Pou3f4 | Pou domain, class 3, transcription factor 4, mutated in DFN3 # | vertical head bobbing and hearing loss, dysplastic bony compartment of the inner ear | Minowa et al. (1999); Phippard et al. (1999) |
| Pou4f2 | POU domain, class 4, transcription factor 2 | selective loss of 70% of retinal ganglion cells, other neurons in the retina and brain essentially unaffected | Erkman et al. (1996); Gan et al. (1996) |
| Rep1 | Rab escort protein 1, mutated in choroideremia # | embryonic male lethal; heterozygous females and chimeras have a variable number of photoreceptor cells | van den Hurk et al. (1997) |
| Syn1 | synapsin 2 | no gross abnormalities, mossy fibre giant terminals reduced, fewer synaptic vesicles, presynaptic structures altered | Rosahl et al. (1993); Takei et al (1995) |
| Syp | synaptophysin | indistinguishable from normal littermates, predendritic neurites and axon outgrowth retarded in hippocampal neurons, with delayed synapse formation; homozygotes die prior to 10.5 dpc | Chin et al. (1995); Erhkind and Leube (1995); Arrandale et al. (1996); McMahon et al. (1996) |
| Timp | tissue inhibitor of metalloproteinase | no effect on steroidogenesis, reduced ovarian TIMP2 and TIMP3, at ES cell level only, more invasive than normal cells | Alexander and Werb (1992); Nothnick et al. (1997) |
| Wasp | Wiskott–Aldrich syndrome protein # | decreased peripheral blood lymphocyte and platelet numbers, chronic colitis | Snapper et al. (1998) |
| Xist | inactive X specific transcripts | mutant males unaffected, females inheriting mutant paternal X chromosome severely retarded and die in utero | Penny et al. (1996); Marahens et al. (1997) |
| Xnp | X-linked nuclear protein, mutated in α-thalassemia mental retardation syndrome # | at cellular level, increased sensitivity to ionizing radiation, mitomycin C, and MMS | Essers et al. (1997) |
| Zfx | X-linked zinc finger protein | male and female mutants smaller, with lower viability and fewer germ cells, hemizygotes had reduced sperm count and homozygotes a reduced number of oocytes | Luoh et al. (1997) |
[i] Genes associated with known human disease are indicated by a pound sign (#) after the gene name; where the disease has a different name, it is given in the same column. Targeted genes are given in alphabetical order of gene symbol. Note that a meeting abstract also mentions the targeting of the mouse p55 gene Mpp1 (A.C. Kim, C.D. Southgate, B.J. Mitchell, and A.H. Chistiti, unpubl.), but as no details of the resulting phenotype are provided this locus is not included in the table.
Other mutants have been created to investigate the potential functions and the phenotypic consequence of a deficiency in the targeted gene. Some of these studies have revealed interesting clinical effects in mice with potential applications for studying complex human disorders, for example the demonstration that a lack of biglycan leads to osteoporosis (Xu et al. 1998). Other gene disruptions have also proven to be highly informative in understanding gene function, for example, the targeting of the Xist locus demonstrated its vital role in the X-inactivation process (Penny et al. 1996; Marahens et al. 1997).
Until stable female embryonic stem cell lines are widely available and can be transmitted to the germ line with a high frequency, one problem that remains with the targeted disruption of X-linked genes is the study of those genes that are potentially lethal in males. Genes may be dispensable in stem cells but essential for embryonic development, as is the case with the methyl-CpG-binding protein Mecp2 (Tate et al. 1996). The Mecp2 knockout mice are particularly interesting in light of the recent revelation that the X-linked dominant neurological disorder Rett syndrome is associated with MECP2 mutations (Amir et al. 1999). The problem of early lethality preventing the recovery of mice carrying targeted mutations of X-linked genes can be partially circumvented by the production of conditional knockouts, in which ablation or modification of the gene of interest is engineered on a temporal or tissue-specific basis (for review, see Müller 1999;Roths et al. 1999). Tarutani et al. (1997) used this approach to demonstrate that the X-linked Piga gene plays an important role in skin development.
X Inactivation and Analysis of the Phenotypic Variation Associated with Heterozygous Females
Mice carrying X-linked mutations can also be used as tools to investigate the influence of cellular mosaicism associated with X-inactivation patterns in different tissues on the phenotype. X inactivation occurs early in mammalian female development and results in the transcriptional silencing of one of the two X chromosomes, at random, in every somatic cell (Lyon 1999). As a consequence, females heterozygous for X-linked coat mutations such as mottled have a variegated coat pattern resulting from the two cell populations, one expressing the mutant allele and the other expressing the wild-type allele (Fig. 2D). Because the number of cells at the time of X inactivation is small, and the choice of which X chromosome is inactivated is usually random, considerable differences are seen between females in the relative sizes of the two cell populations; this manifests as a variation in the severity of the phenotype between female mice carrying the same mutation. For some X-linked mutations, cellular mosaicism is a benefit with the population of cells expressing the normal allele, providing enough of the normal gene product to permit a normal phenotype to develop, for example, in the sfor spf mutations (DeMars et al. 1976; Lyon et al. 1990). Another possibility is that growth competition between the two cell populations may result in the virtual elimination of the cells expressing the mutant allele (Ogura et al. 1998). In this situation, the resultant skewed X-inactivation pattern provides heterozygous females with a normal, or mild, phenotype.
Traditionally, X-inactivation patterns in mice have been investigated in the coat by the observation of doubly heterozygous female mice produced when the mutation is bred to well-characterized coat mutants, such as the mottled blotchy or tabby mutants. In humans, approaches that exploit the differences in DNA methylation between the inactive and active X chromosomes are most widely used to study X-inactivation patterns in heterozygous females (Belmont 1996). In the mouse, extensive studies of the X-inactivation patterns can be achieved because of the availability of tissues from several replica heterozygotes at a range of times in development on a constant genetic background. Techniques such as the single nucleotide primer extension (SNuPE) assay have been used to quantitatively measure the relative expression from the two X chromosomes in females heterozygous for X-linked mutations (Greenwood et al. 1997; Ogura et al. 1998). Further, the X-linked LacZ transgenic mice created by Tam and Tan (1992) have great potential for the study of X-inactivation patterns in heterozygous females as the transgene is subject to the inactivation process. When the lacZ reporter is present on only one of the X chromosomes of a female heterozygous for an X-linked mutation, the β-galactosidase activity is limited to cells with only that X chromosome active. Therefore, the distribution of cells expressing the mutant allele in a heterozygous female can be studied and insights into the mode of action of the normal X-linked gene product can be provided by the analysis of the phenotype and the X-inactivation pattern of heterozygous females. In our laboratory, such studies are under way on the Li, Stpy, and Td mutants.
Future Progress
The 80 X-linked phenotypes that have been reported in the laboratory mouse correspond to mutations in approximately half of the cloned X chromosomal genes. Most of these traits are associated with mutations in known genes and are of immediate value as animal models for human X-linked diseases. However, they represent <5% of the probable number of X-linked genes and the phenotype map of the mouse X chromosome is still sparse. Alternative methods will be needed to provide the detailed phenotype maps worthy of complementing the encyclopedias of mouse genes that are being developed (Marra et al. 1999). Although several laboratories are attempting to increase the size of the mouse mutant resource (Justice et al. 1997; You et al. 1997; deAngelis and Balling 1998; Schimenti and Bucan 1998; Zheng et al. 1999), no plans appear to have been made to screen systematically the progeny of F1 females carrying mutagenized X chromosomes for X-linked traits. Without such a specific effort, it will be interesting to see whether the number of X-linked mutant stocks will increase to meet the needs and interests of groups trying to understand the roles of X-linked genes in health and disease.
We thank Kevin Glover for help in producing Figure 2. This work was supported by the MRC of Great Britain.
Notes
[4] Present addresses: 1Institute for Animal Health, Compton, Newbury RG20 7NN, UK; 2Department of Physiological Sciences, Medical School, Framlington Place, University of Newcastle, Newcastle Upon Tyne NE2 4HH, UK; 3Department of Medicine, Manchester Royal Infirmary, Oxford Road, Manchester M13 9WL, UK.
[5] Corresponding author.
Notes
[6] E-MAIL [email protected]; FAX 00 44 1635 577237.
[7] A comprehensive list of international mouse strain resources, developed jointly by the UK MRC laboratory at Harwell and the USA Jackson Laboratory (Eppig and Strivens 1999), and a list of strains available at the newly established European Mouse Mutant Archive (EMMA) in Italy, can be accessed via the World Wide Web athttp://ismr.har.mrc.ac.uk; http://www.emma.cnr.it.
REFERENCES
- ↵C.M. AlexanderZ. Werb(1992) Targeted disruption of the tissue inhibitor of metalloproteinases gene increases invasive behaviour of primitive mesenchymal cells derived from embryonic cells in vitro. J. Cell Biol. 118:727–739.
- ↵L.C. AmarL. DandaloA. HanauerA. Ryder-CookD. ArnaudJ-L. MandelP. Avner(1988) Conservation and re-organisation of loci on the mammalian X chromosome; a molecular framework for the identification of homologous regions in man and mouse. Genomics 22:220–230.
- ↵R.E. AmirI.B. Van den VeyverM. WanmC.Q. TranU. FranckeH.Y. Zhogbi(1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23:185–188.
- ↵E. ArakiK. NakamuraK. NakaoS. KameyaO. KobayashiI. NonakaT. KobayashiM. Katsuki(1997) Targeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in Duchenne muscular dystrophy. Biochem. Biophys. Res. Comm. 238:492–497.
- ↵J.M. ArrandaleA. Gore-WillseS. RocksJ.M. RenJ. ZhuA. DavisJ.N. LivingstonD.U. Rabin(1996) Insulin signaling in mice expressing reduced levels of Syp. J. Biol. Chem. 271:21353–21358.
- ↵C.E. BakkerC. VerheijR. WillemsenR. van der HelmF. OerlemansM. VermeyA. BygraveA.T. HoogeveenB.A. OostraE. Reyniers(1994) FMR1 knockout mice—A model to study fragile-X mental retardation. Cell 78:23–33.
- ↵B.R. Barber(1971) Research News. Mouse NewsLett. 45:34–35.
- ↵J. Barra(1990) An X-linked recessive mutation producing cleft palate, crooked tail, and polydactyly in mice. J. Hered. 81:388–392.
- ↵M.A. BedellN.A. JenkinsN.G. Copeland(1997) Mouse models of human disease. Genes & Dev. 11:1–43.
- ↵J.W. Belmont(1996) Genetic control of X inactivation and processes leading to X-inactivation skewing. Am. J. Hum. Genet. 58:1101–1108.
- ↵W. BergerD. van de PolD. BachnerF. OerlemansH. WinkensH. HameisterB. WieringaW. HendriksH.-H. Ropers(1996) An animal model for Norrie disease (ND); gene targeting of the mouse ND gene. Hum. Mol. Genet. 5:51–59.
- ↵L. BiA.M. LawlerS.E. AntonarakisK.A. HighJ.D. GearhartH.H. Kazazian(1995) Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat. Genet. 10:119–121.
- ↵H.J. BlairV. ReedS.H. LavalY. Boyd(1994) New insights into the man-mouse comparative map of the X chromosome. Genomics 19:215–220.
- ↵H.J. BlairM. HoA.P. MonacoS. FisherI.W. CraigY. Boyd(1995) High resolution comparative mapping of the proximal region of the mouse X chromosome. Genomics 28:305–310.
- ↵H.J. BlairE. GormallyI.C. UwechueY. Boyd(1998a) Mouse mutants carrying deletions that encompass the genes deleted in Coffin-Lowry syndrome and lactic acidosis. Hum. Mol. Genet. 7:549–555.
- ↵H.J. BlairI.C. UwechueG.S. BarshP.S.N. RoweY. Boyd(1998b) An integrated genetic and man-mouse comparative map of the DXHXS674-Pdha1 region of the mouse X chromosome. Genomics 48:128–131.
- ↵R.J. BlaschkeG.A. Rappold(1997) Man to mouse—lessons learned from the distal end of the human X chromosome. Genome Res. 7:1114–1117.
- ↵D. BoisonW. Stoffel(1994) Disruption of the compacted myelin sheath of axons of the central nervous system in proteolipid-deficient mice. Proc. Natl. Acad. Sci. 91:11709–11713.
- ↵Y. BoydH.J. BlairP. CunliffeP. DennyE. GormallyG.E. Herman(1998) Mouse Chromosome X. Mamm. Genome 8:S361–S377.
- ↵Y. BoydP. DennyW.K. MassonV. ReedR. Elliott(1999) Mouse X chromosome. Mamm. Genome 10:961.
- ↵N. BravermannP. LinF.F. MoebiusC. ObieA. MoserH. GlossmanW.R. WilcoxD.L. RimoinM. SmithL. Kratz(1999) Mutations in the gene encoding 3β-hydroxysteroid-Δ8,Δ7-isomerase cause X-linked dominant Conradi-Hünerman syndrome. Nat. Genet. 22:291–294.
- ↵G. BulfieldW.G. SillerP.A.L. WightK.J. Moore(1984) X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. 81:1189–1192.
- ↵O. CasesI. SeifJ. GrimsbyP. GasparK. ChenS. PourninU. MüllerM. AuguetC. BabinetJ.C. Shih(1995) Aggressive behaviour and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science 268:1763–1766.
- ↵E. CastigliR. FuleihanN. RameshE. TsitsikovA. TsytsykovR.S. Geha(1995) CD40 ligand/CD40 deficiency. Int. Arch. Allergy Immunol. 107:37–39.
- ↵C. CecchiM. BiasottoM. TosiP. Avner(1997) The mottled mouse as a model for human Menkes disease: Identification of mutations in the Atp7a gene. Hum. Mol. Genet. 6:425–433.
- ↵V.M. ChapmanD.R. MillerD. ArmstrongC.T. Caskey(1989) Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl. Acad. Sci. 86:1292–1296.
- ↵N.J. CharestZ. ZhouD.B. LubahnK.E. OlsenE.M. WilsonF.S. French(1991) A frameshift mutation destablizes androgen receptor RNA in the Tfm mouse. Mol. Endocrinol. 5:573–581.
- ↵L.S. ChinL. LiA. FerreiraK.S. KosikP. Greengard(1995) Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc. Natl. Acad. Sci. 92:9230–9234.
- ↵P. Cunliffe(1999) “Molecular genetic analysis of mottled mice”, Ph. D. thesis. (University of Oxford, Oxford, UK).
- ↵M. DahmeU. BartschR. MartiniB. AnlikerM. SchachnerN. Mantei(1997) Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat. Genet. 17:346–349.
- ↵D.M. Danks(1986) Of mice and men, metals and mutations. J. Med .Genet. 23:99–106.
- ↵S. DasB. LevinsonC. VulpeS. WhitneyJ. GitschierS. Packman(1995) Similar splicing mutations of the Menkes/mottled copper-transporting ATPase gene in occipital horn syndrome and the blotchy mouse. Am. J. Hum. Genet. 56:570–576.
- ↵A. DautignyM.-G. MatteiD. MorelloP. M. AllielD. Pham-DinhL. AmarD. ArnaudD. SimonJ.-F. MatteiJ.-L. Guenet(1986) The structural gene coding for mylein-associated proteolipid protein is mutated in jimpy mice. Nature 321:867–868.
- ↵M.T. Davisson(1987) X-linked homologies between mouse and man. Genomics 1:213–227.
- ↵M.H. De AngelisR. Balling(1998) Large scale ENU screens in the mouse: Genetics meets genomics. Mutat. Res. 400:25–32.
- ↵R. De MarsS.L. LeVanB.L. TrendL.B. Russell(1976) Abnormal ornithine carbamoyltransferase in mice having the sparse-fur mutation. Proc. Natl. Acad. Sci. 73:1693–1697.
- ↵J.M.J. DerryE. GormallyG.D. MeansW. ZhaoA. MeindlR.I. KelleyY. BoydG.E. Herman(1999) Mutations in a Δ8-Δ7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat. Genet. 22:286–290.
- ↵M. D'EspositoM.R. MatarazzoA. CiccodicolaM. StrazzulloR. MazzarellaN.A. QuaderiH. FujiwaraM.S.H. KoL.B. RoweA. Ricco(1997) Differential expression pattern of XqPAR-linked genes SYBL1 and IL9R correlates with the structure and evolution of the region. Hum. Mol. Genet. 6:1917–1923.
- ↵M.B. DinulosM.T. BassiE.I. RugarliV. ChapmanA. BallabioC. Disteche(1996) A new region of conservation is defined between the human and mouse X chromosomes. Genomics 35:244–247.
- ↵C.M. DistecheM.B. DinulosM.T. BassiR.W. ElliottE.I. Rugarli(1998) Mapping of the murine Tbl1 gene reveals a new rearrangement between mouse and human X chromosomes. Mamm. Genome 9:1062–1064.
- ↵T.A. DraganiZ.-B. ZengF. CanzianM. GaribaldiM.T. GhilarducciG. ManentoM.A. Pierotti(1995) Mapping of body weight loci on mouse chromosome X. Mamm. Genome 6:778–781.
- ↵I.E. EhrmanP.S. EllisS. MazeyratS. DuthieN. BrockdorffM.G. MatteiM.A. GavinN.A. AffaraG.M. BrownE. Simpson(1998) Characterization of genes encoding translation initiation factor eIF-2γ in mouse and human: Sex chromosome localization, escape from X-inactivation and evolution. Hum. Mol. Genet. 7:1725–1737.
- ↵E.M. EicherJ.L. SouthardC.R. ScriverF.H. Glorieux(1976) Hypophosphatemia: Mouse model for human familial hyposphosphatemic (vitamin D-resistant) rickets. Proc. Natl. Acad. Sci. 73:4667–4671.
- ↵J.T. EppigM. Strivens(1999) Finding a mouse: The International Mouse Strain Resource (IMSR). Trends Genet. 15:81–82.
- ↵L. ErkmanR.J. McEvillyL. LuoA.K. RyanF. HooshmandS.M. O'ConnellE.M. KeithleyD.H. RapaportA.F. RyanM.G. Rosenfeld(1996) Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature 381:603–606.
- ↵L.G. EshkindR.E. Leube(1995) Mice lacking synaptophysin reproduce and form typical synaptic vesicles. Cell Tiss. Res. 3:423–433.
- ↵J. EssersR.W. HendriksS.M.A. SwagemakersC. TroelstraJ. de WitD. BootsmaJ.H.J. HoeijmakersR. Kanaar(1997) Disruption of mouse RAD54 reduces ionizing radiation resistance. Cell 89:195–204.
- ↵J. FavorW. Pretsch(1990) Genetic localization and phenotypic expression of X-linked cataract (Xcat) in Mus musculus. Genet. Res. 56:157–162.
- ↵E.M.C. Fisher(1997) The contribution of the mouse to advances in human genetics. Adv. Genet. 35:155–205.
- ↵S. ForssPetterH. WernerJ. BergerH. LassmannB. MolzerM.H. SchwabH. BernheimerF. ZimmermannK.A. Nave(1997) Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J. Neurosci. Res. 50:829–843.
- ↵E. FransenR. DhoogeG. van CampM. VerhoyeJ. SijbersE. ReyniersP. SorianoH. KamiguchiR. WillemsenS.K.E. Koekkoek(1998) L1 knockout mice show dilated ventricles, vernis hypoplasia and impaired exploration patterns. Hum. Mol. Genet. 7:999–1009.
- ↵Y. FujiwaraC.P. BrowneK. CunniffS.C. GoffS.H. Orkin(1996) Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc. Natl. Acad. Sci. 93:12355–12358.
- ↵L. GanM. XiangL. ZhouD.S. WagnerW.H. KleinJ. Nathans(1996) POU domain factor Brn-3b is required for the development of a large set of retinal ganglion cells. Proc. Natl. Acad. Sci. 93:3920–3925.
- ↵E.D. Garber(1952) “Bent-tail”, a dominant sex-linked mutation in the mouse. Proc. Natl. Acad. Sci. 38:8786–8789.
- ↵M.-L. GasparT. MeoP. BourgarelJ.-L. GuenetM. Tosi(1991) A single base deletion in the Tfm androgen receptor gene creates a short-lived messenger RNA that directs internal translation initiation. Proc. Natl. Acad. Sci. 88:8606–8610.
- ↵A.M. GeorgeV. ReedP. GlenisterZ. TümerN. HornJ. ChellyA.P. MonacoY. Boyd(1994) Analysis of Mnk, the murine homologue of the locus for Menkes disease, in normal and mottled (Mo) mice. Genomics 37:96–104.
- ↵E. GormallyY. Boyd(1998) Faint lined (Fnl): A novel X-linked coat mutant in the mouse. Genet. Res. 72:211–216.
- ↵A.D. GreenwoodE.M. Southard-SmithA.T.J. GaleckiD.T. Burke(1997) Coordinate control and variation in X-linked gene expression among female mice. Mamm. Genome 8:818–822.
- ↵I.R. GriffithsI. ScottM.C. McCullochJ.A. BarrieK. McPhilemyB.M. Cattanach(1990) Rumpshaker mouse: A new X-linked mutation affecting myelination: Evidence for a defect in PLP expression. J. Neurocytol. 19:273–283.
- ↵I.R. GriffithsA. SchneiderJ. AndersonK.A. Knave(1995) Transgenic and natural mouse models of proteolipid (PLP)-related dysmyelination and demyelination. Brain Pathol. 5:275–281.
- ↵I.R. GriffithsM. KlugmannT. AndersonC. ThomsonD. VouyiouklisK.A. Nave(1998) Current concepts of PLP and its role in the nervous system. Microscop. Res. Technique 41:344–358.
- ↵J. GrimsbyM. TothK. ChenT. KumazawaL. KlaidmanJ.D. AdamsF. KaroumJ. GalJ.C. Shih(1997) Increased stress response and β-phenylethylamine in MAOB-deficient mice. Nat. Genet. 17:206–210.
- ↵J.-L. GuenetC. NagamineD. Simon-ChazettesX. MontagutelliF. Bonhomme(1990) Hst-3: And X-linked hybrid sterility gene. Genet. Res. 56:163–165.
- ↵L.L. HamptonE.E. LadenheimM. AkesonJ.M. WayH.C. WeberV.E. SutliffR.T. JensenL.J. WineH. ArnheiterJ.F. Battey(1998) Loss of bombesin-induced feeding suppression in gastrin-releasing peptide receptor-deficient mice. Proc. Natl. Acad. Sci. 6:3188–3192.
- ↵R. Happle(1983) Homologous genes for X-linked dominant chondrodysplasia punctata in man and mouse. Hum. Genet. 63:24–27.
- ↵L. HeinG.S. BarshR.E. PrattV.J. DzauB.K. Kobilka(1995) Behavioural and cardiovascular effects of disrupting the angiotensin II type-2 receptor in mice. Nature 377:744–747.
- ↵E. HellerD.H. Pluznik(1984) Chromosomal assignment of two murine genes controlling susceptibility to spleen focus formation by Rauscher leukemia virus. Exp. Hematol. 12:645–649.
- ↵C. Hetherington(1977) Research News. Mouse NewsLett. 56:35.
- ↵M. HooperK. HardyA. HandysideS. HunterM. Monk(1987) HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature 326:292–295.
- ↵P.R. Hunsicker(1969) Research News. Mouse NewsLett. 40:42.
- ↵P.R. Hunsicker(1974) New mutants: Eye-ear reduction Ie. Mouse NewsLett. 50:51–52.
- ↵D.M. Hunt(1974) Primary defect in copper transport underlies mottled mutants in the mouse. Nature 249:852–854.
- ↵A.H.M.M. HuqR.S. LovellC.N. OuA.L. BeaudetW.J. Craigen(1997) X-linked glycerol kinase deficiency in the mouse leads to growth retardation, altered fat metabolism, autonomous glucocorticoid secretion and neonatal death. Hum. Mol. Genet. 6:1803–1809.
- ↵T. IchikiP.A. LaboskyC. ShiotaS. OkuyamaY. ImagawaA. FogoF. NiimuraI. IchikawaB.L.M. HoganT. Inagami(1995) Effects on blood pressure and exploratory behaviour of mice lacking angiotensin II type-2 receptor. Nature 377:748–750.
- ↵P.A. JanneS.F. SuchyD. BernardM. MacDonaldJ. CrawleyA. GrinbergA. Wynshaw-BorisH. WestphalR.L. Nussbaum(1998) Functional overlap between murine Inpp5b and Ocrl1 may explain why deficiency of the murine ortholog for OCRL1 does not cause Lowe syndrome in mice. J. Clin. Invest. 101:2042–2053.
- ↵M.J. JusticeB. ZhengR.P. WoychikA. Bradley(1997) Using targeted large deletions and high efficiency N-ethyl-N-nitrosourea mutagenesis for functional analyses of the mammalian genome. Methods: Companion Methods Enzymol. 13:423–436.
- ↵J.D. KernerM.W. ApplebyR.N. MohrS. ChienD.J. RawlingsC.R. MaliszewskiO.N. WitteR.M. Perlmutter(1995) Impaired expansion of mouse B cell progenitors lacking Btk. Immunity 3:301–312.
- ↵W.N. KhanA. NilssonE. MizoguchiE. CastigliJ. ForsellA.K. BhanR. GehaP. SiderasF.W. Alt(1997) Impaired B cell maturation in mice lacking Bruton's tyrosine kinase (Btk) and CD40. Int. Immunol. 9:395–405.
- ↵P.J. KingstonC.E.M. BannermanR.M. Bannerman(1978) Iron deficiency anaemia in newborn sla mice: A genetic defect of placental iron transport. Br. J. Haematol. 40:265–276.
- ↵M. KlugmanM.H. SchwabA. PulhoferA. SchneiderF. ZimmermanI.R. GriffithsK.A. Knave(1997) Assembly of CNS melin in the absence of proteolipid protein. Neuron 18:59–70.
- ↵M.R. KuehnA. BradleyE.J. RobertsonM.J. Evans(1987) A potential animal model for Lesch-Nyhan syndrome through introduction of HPRT mutations into mice. Nature 326:295–298.
- ↵P.W. LaneM.T. Davisson(1990) Patchy fur (Paf), a semidominant X-linked gene associated with a high level of X-Y nondisjunction in male mice. J. Hered. 81:43–50.
- ↵M.M. Larsen(1964) Research news: Greasy. Mouse NewsLett. 30:47.
- ↵S.H. LavalY. Boyd(1993) Partial inversion of gene order within an homologous segment on the X chromosome. Mamm. Genome 4:119–123.
- ↵H.G. LavertyJ.B. Wilson(1998) Murine CASK is disrupted in a sex-linked cleft palate mouse mutant. Genomics 53:29–41.
- ↵W.J. LeonardE.W. ShoresP.E. Love(1995) Role of common cytokine receptor gamma chain in cytokine signaling and lymphoid development. Immunol. Rev. 148:97–114.
- ↵M. LiJ.A. SquireR. Weksberg(1998) Overgrowth syndromes and genomic imprinting: From mouse to man. Clin. Genet. 53:165–170.
- ↵X.Y. LiuA.W. DangelR.I. KelleyW. ZheoP. DennyM. BotcherbyB. CattanachJ. PetersP.R. HunsickerA.-M. Mallon(1999) The mouse bare patches and striated gene encodes a novel 3β-hydroxysteroid dehydrogenase. Nat. Genet. 22:182–187.
- ↵B. LorenzF. FrancisK. GempelA. BoddrichM. JostenW. SchmahlJ. SchmidtH. LehrachT. MeitingerT.M. Strom(1999) Spermine deficiency in Gy mice caused by deletion of the spermine synthase gene. Hum. Mol. Genet. 7:541–547.
- ↵J.F. LuA.M. LawlerP.A. WatkinsJ.M. PowersA.B. MoserH.W. MoserK.D. Smith(1997) A mouse model of X-linked adrenoleukodystrophy. Proc. Natl. Acad. Sci. 94:9366–9371.
- ↵S.W. LuohP.A. BainR.D. PolakiewiczM.L. GoodheartH. GardnerR. JaenischD.C. Page(1997) ZFX mutation results in small animal size and reduced germ cell number in male and female mice. Development 124:2275–2284.
- ↵M.F. Lyon(1999) X-inactivation. Curr. Biol. 9:R235–R237.
- ↵M.F. LyonB.M. CattanachH.M. Charlton(1981a) Genes affecting sex differentiation in mammals. in Mechanisms of sex differentiation in animals and man, eds C.R. AustinR.G. Edwards(Academic Press, New York, NY), pp 329–386.
- ↵M.F. LyonR.J.S. PhillipsG. Fisher(1981b) Use of an inversion to test for induced X-linked lethals in mice. Mutat. Res. 92:217–228.
- ↵M.F. LyonC.R. ScriverL.R.I. BakerH.S. TenenhouseJ. KronickS. Mandla(1986) The Gy mutation: Another cause of X-linked hypophosphatemia in mouse. Proc. Natl. Acad. Sci. 43:4899–4903.
- ↵M.F. LyonJ. PetersP.H. GlenisterS. BallE. Wright(1990) The scurfy mouse has previously unrecognized hematological abnormalities and resembles Wiskott-Aldrich syndrome. Proc. Natl. Acad. Sci. 87:2433–2437.
- ↵M. MarraL.-D. HillierT. KucabaM. AllenR. BarsteadC. BeckA. BlistainM. BonaldiY. BowersL. Bowles(1999) An encylopedia of mouse genes. Nat. Genet. 21:191–194.
- ↵Y. MarahensB. PanningJ. DausmanW. StraussR. Jaenisch(1997) Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes & Dev. 11:156–166.
- ↵J.A. MarshT.E. WheatE. Goldberg(1977) Temporal regulation of the immune response to LDH-C4 by an X-linked gene in C3H/HeJ and SJl/J mice. J. Immunol. 118:2293–2295.
- ↵H.T. McMahonV.Y. BolshakovR. JanzR.E. HammerS.A. SiegelbaumT.C. Sudhof(1996) Synaptophysin, a major synaptic vesicle protein is not essential for neurotransmitter release. Proc. Natl. Acad. Sci. 10:4760–4764.
- ↵H. MeierA.D. MacPike(1970) A neurological mutation (msd) of the mouse causing a deficiency of myelin synthesis. Exp. Brain Res. 10:510–525.
- ↵E.N. MeyersM. LewandoskiG.R. Martin(1998) An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat. Genet. 18:136–141.
- ↵J.R. Miller(1990) X-linked traits: A catalog of loci in non-human mammals. (Cambridge University Press, Cambridge, UK).
- ↵O. MinowaK. IkedaY. SugitaniT. OshimaS. NakaiY. KatoriM. SuzukiM. FurukawaT. KawaseY. Zheng(1999) Altered cochlear fibrocytes in a mouse model of DFN3 nonsyndromic deafness. Science 285:1408–1411.
- ↵D.E. MorgensternM.A. GiffordL.L. LiC.M. DoerschukM.C. Dinauer(1997) Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defence and inflammatory response to Aspergillus fumigatus. J. Exp. Med. 185:207–218.
- ↵E. MozesS. Fuchs(1974) Linkage between iummune response potential to DNA and X chromosome. Nature 249:167–168.
- ↵U. Müller(1999) Ten years of gene targeting: Targeted mouse mutants, from vector design to phenotype analysis. Mech. Dev. 82:3–21.
- ↵M. NehlsM. MesserleA. SirulnikA.J. SmithT. Boehm(1994) Two large insert vectors lambda PS and lambda Ko, facilitate rapid mapping and targeted disruption of mammalian genes. BioTechniques 17:770–775.
- ↵W.B. NothnickP. SolowayT.E. Curry(1997) Assessment of the role of tissue inhibitor of metalloproteinase-1 (TIMP-1) during the periovulatory period in female mice lacking a functional TIMP-1 gene. Biol. Reprod. 56:1181–1188.
- ↵H. OguraS. TakadaN. MiseM. SugimotoS.-S. TanN. Takagi(1998) Translocation breakpoint possibly predisposes to nonrandom X-chromosome inactivation in mouse embryos bearing Searle's T(X;16)16H translocation. Cytogenet. Cell Genet. 80:173–178.
- ↵K. OhboT. SudaM. HashiyamaA. MantaniM. IkebeK. MiyakawaM. MoriyamaM. NakamuraM. KatsukiK. Takahashi(1996) Modulation of haematopoiesis in mice with a truncated mutant of the interleukin-2 receptor gamma chain. Blood 87:956–967.
- ↵H. Ohki-HamazakiE. WadaK. MatsuiK. Wada(1997) Cloning and expression of the neuromedin B receptor and the third type of bombesin receptor genes in the mouse. Brain Res. 762:165–172.
- ↵S. Ohno(1973) Ancient linkage groups and frozen accidents. Nature 244:259–262.
- ↵T. OhshimaG.J. MurrayW.D. SwaimG. LongeneckerJ.M. QuirkC.O. CardarelliY. SugimotoI. PastanM.M. GottesmanR.O. Brady(1997) Alpha-galactosidase deficient mice: A model of Fabry disease. Proc. Natl. Acad. Sci. 94:2540–2544.
- ↵S. OkuyamaT. SakagawaS. ChakiY. ImagawiT. Ichiki(1999) Anxiety-like behaviour in mice lacking the angiotensin II type-2 receptor. Brain Res. 821:150–159.
- ↵B.A. OostraA.T. Hoogeveen(1997) Animal model for fragile X syndrome. Ann. Med. 29:563–567.
- ↵K. Paigen(1995) A miracle enough: The power of mice. Nat. Med. 1:215–220.
- ↵P.P. PandolfiF. SonatiR. RiviP. MasonF. GrosveldL. Luzzatto(1995) Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defence against oxidative stress. EMBO J. 14:5209–5215.
- ↵G.D. PennyG.F. KayS.A. SheardownS. RastanN. Brockdorff(1996) Requirement for Xist in X chromosome inactivation. Nature 379:131–137.
- ↵L. PevnyM.C. SimonE. RobertsonW.H. KleinS.F. TsaiV. D'AgatiS.H. OrkinF. Constantini(1991) Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 349:257–260.
- ↵R.J.S. PhillipsG. Fisher(1978) New mutants. Mouse NewsLett. 58:43–44.
- ↵D. PhippardL. LuD. LeeJ.C. SaundersE.B. Crenshaw(1999) Targeted mutagenesis of the POU domain gene Brn4/Pou3f4 causes developmental defects in the inner ear. J. Neurosci. 19:5980–5989.
- ↵D. PhippardY. BoydV. ReedG. FisherW.K. MassonE.P. EvansJ.C. SaundersE.B Crenshaw(2000) The sex-linked fidget mutation abolishes Brn4/Pou3f4 expression in the embryonic inner ear. Hum. Mol. Genet. 9:79–85.
- ↵E.L. Phipps(1969) New mutants: Irregular teeth It. Mouse NewsLett. 40:41–42.
- ↵J.D. PollockD.A. WilliamsM.A. GiffordL.L. LiX. DuJ. FishermanS.H. OrkinC.M. DoerschukM.C. Dinauer(1995) Mouse model of X-linked chronic granulomatous disease, an inherited defect in superoxide production. Nat. Genet. 9:202–209.
- ↵N.A. QuaderiS. MessaliS. CainarcaA. BallabioG. MeroniE. Rugarli(1998) Possible involvement of Mid1 in the patchy fur (Paf) mutant. Twelfth International Mouse Genome Conference (Garmisch-Partenkirchen, Bavaria).
- ↵D.J. RawlingsD.C. SaffranS. TsukadaD.A. LargasepadaJ.C. GrimaldiL. CohenR.N. MohrJ.F. BazanM. HowardN.G. Copeland(1993) Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science 261:358–361.
- ↵V. ReedY. Boyd(1997) Mutation analysis provides additional proof that mottled is the mouse homologue of Menkes disease. Hum. Mol. Genet. 6:417–423.
- ↵B.R. RenshawW.C. FanslowR.J. ArmitageK.A. CampbellD. LiggittB. WrightB.L. DavisonC.R. Maliszewski(1994) Humoral responses in CD40-ligand deficient mice. J. Exp. Med. 180:1889–1900.
- ↵T.W. RosahlM. GeppertD. SpillaneJ. HerzR.E. HammerR.C. MalenkaT.C. Sudhof(1993) Short-term plasticity is altered in mice lacking synapsin-1. Cell 75:661–670.
- ↵L.E. RosenbergF. KalousekM.D. Orsulak(1983) Biogenesis of ornithine transcarbamylase in spf ash mutant mice: Two cytoplasmic precursors, one mitochondrial enzyme. Science 222:426–428.
- ↵B.J.F. RossiterC.T. Caskey(1991) Molecular studies of human genetic disease. FASEB J. 5:21–27.
- ↵J.B. RothsW.B. FoxworthM.J. McArthurC.A. MontgomeryA.B. Kier(1999) Spontaneous and engineered mutant mice as models for experimental and comparative pathology: History, comparison, and developmental technology. Lab. Anim. Sci. 49:12–29.
- ↵C. RougelleP. Avner(1996) Cloning and characterisation of a murine brain specific gene Bpx and its human homologue lying within the Xic candidate region. Hum. Mol. Genet. 5:41–49.
- ↵S.S. SchererY.T. XuE. NellesK. FischbeckK. WillekeL.J. Bone(1998) Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24:8–20.
- ↵J. SchimentiM. Bucan(1998) Functional genomics in the mouse: Phenotype based mutagenesis screens. Genome Res. 8:698–710.
- ↵A.G. Searle(1964) Genetic effects of spermatogonial X-irradiation on the productivity of F1 female mice. Mutat. Res. 1:99–108.
- ↵A.G. SearleJ. PetersM.F. LyonE.P. EvansJ.H. EdwardsV.J. Buckle(1987) Chromosome maps of man and mouse III. Genomics 1:3–18.
- ↵A.G. SearleJ. PetersM.F. LyonJ.G. HallE.P. EvansJ.H. EdwardsV.J. Buckle(1989) Chromosome maps of man and mouse IV. Ann. Hum. Genet. 53:89–140.
- ↵S.B. SnapperF.S. RosenE. MizoguchiP. CohenW. KhanC.H. LiuT.L. HagemannS.P. KwanmR. FerriniL. Davidson(1998) Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity 9:81–91.
- ↵A.K. SrivastavaJ. PispaA.J. HartungY. DuS. EzerT. JenksT. ShimadaM. PekkanenM.L. MikkolaM.S.H. Ko(1997) The Tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin-A) with collagenous domains. Proc. Natl. Acad. Sci. 94:13069–13074.
- ↵T.M. StromF. FrancisB. LorenzA. BöddrichM.J. EconsH. LehrachT. Meitinger(1997) Pex gene deletions in Gy and Hyp mice provide mouse models for X-linked hypophosphatemia. Hum. Mol. Genet. 6:165–171.
- ↵K. SugamuraH. AsaoM. KondoN. TanakaN. IshiiK. OhboM. NakamuraT. Takeshita(1996) The interleukin-2 receptor gamma chain: Its role in the multiple cytokine receptor complexes and T cell development in XSCID. Annu. Rev. Immunol. 14:179–205.
- ↵H.O. SweetP.W. Lane(1980) X-linked polydactyly (Xpl), a new mutation in the mouse. J. Hered. 71:207–209.
- ↵H.O. SweetR.T. BronsonB.S. HarrisM.T. Davisson(1990) Trembly-like (Tyl)—a new X-linked mutation in the mouse. Mouse Genome 87:112.
- ↵S. TakahashiK. OnoderaH. MotohashiN. SuwabeN. HayashiN. YanaiY. NabesimaM. Yamamoto(1997) Arrest in primitive erythroid cell development caused by promoter-specific disruption of the GATA-1 gene. J. Biol. Chem. 272:12611–12615.
- ↵Y. TakeiA. HaradaS. TakedaK. KobayashiS. TeradaT. NodaT. TakahashiN. Hirokawa(1995) Synapsin I deficiency results in the structural change in the presynaptic terminals in the murine nervous system. J. Cell Biol. 131:1789–17880.
- ↵P.L.P. TamS.-S. Tan(1992) The somitogenic potential of cells in the primitive streak and the tail bud of the organogenesis-stage mouse embryo. Development 115:703–715.
- ↵S. TanakaT. KanekoT. MoriyamaA. Matsuzawa(1993) A new testicular feminization mutation found in C57BL/6J mice. Lab. Anim. 28:262–264.
- ↵M. TarutaniS. ItamiM. OkabeM. IkawaT. TezukaK. YoshikawaT. KinoshitaJ. Takeda(1997) Tissue-specific knockout of the mouse Pig-a gene reveals important roles for GPI-anchored proteins in skin development. Proc. Natl. Acad. Sci. 94:7400–7405.
- ↵P. TateW. SkarnesA. Bird(1996) The methyl-CpG binding protein MeCP2 is essential for embryonic development in the mouse. Nat. Genet. 12:205–208.
- ↵L.H. TecottL.M. SunS.F. AkanaA.M. StrackD.H. LowensteinM.F. DallmanD. Julius(1995) Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature 374:542–546.
- ↵J.D. ThomasP. SiderasC.I.E. SmithI. VorechovskýV. ChapmanW.E. Paul(1993) Colocalization of X-linked agammalobulinemia and X-linked immunodeficiency genes. Science 261:355–358.
- ↵H. TsudaC.E. Maynard-CurrieL.H. ReidT. YoshidaK. EdamuraN. MaedaO. SmithiesA. Jakobivits(1997) Inactivation of the mouse HPRT locus by a 203-bp retroposon insertion and a 55-kb gene-targeted deletion: Establishment of new HPRT-deficient mouse embryonic stem cell lines. Genomics 42:413–421.
- ↵Z. TümerN. Horn(1997) Menkes disease: Recent advances and new aspects. J. Med. Genet. 34:265–274.
- ↵Z. TümerC. LundJ. TolshaveB. VuralT. TønnesenN. Horn(1997) Identification of point mutations in 41 unrelated patients affected with Menkes disease. Am. J. Hum. Genet. 60:63–71.
- ↵J.A. van den HurkW. HendriksD.J. van de PolF. OerlemansG. JaissleK. RutherK. KohlerJ. HartmannE. ZrennerH. van Bokhoven(1997) Mouse choroideremia gene mutation causes photoreceptor cell degeneration and is not transmitted through the female germline. Hum. Mol. Genet. 6:851–858.
- ↵G. VeresR.A. GibbsS.E. SchereC.T. Caskey(1987) The molecular basis of the sparse fur mutation. Science 237:415–417.
- ↵C.D. VulpeY-M. KuoT.I. MurphyL. CowleyC. AskwithN. LibinaJ. GitschierG.J. Anderson(1999) Hephaestin, a ceruloplasmin homologue implicated in intestinal ion transport, is defective in the sla mouse. Nat. Genet. 21:195–199.
- ↵L. WangM. ZoppeT.M. HackengJ.H. GriffinK.F. LeeI.M. Verma(1997) A factor IX deficient mouse model for hemophila B gene therapy. Proc. Natl. Acad. Sci. 94:11563–115636.
- ↵J. XuT.M. FoyJ.D. lamanE.A. ElliottJ.J. DunnT.J. WaldschmidtJ. ElsemoreR.J. NoelleR.A. Flavell(1994) Mice deficient for the CD40l ligand immunity. Immunity 1:423–431.
- ↵T.S. XuP. BiancoL.W. FisherG. LongneckerE. SmithS. GoldsteinJ. BonadioA. BoskeyA.M. HeegardB. Sommer(1998) Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat. Genet. 20:78–82.
- ↵B. YorkK. LeiD.B. West(1997) Inherited non-autosomal effects on body fat in F2 mice derived from am AKR/J x SWR/J cross. Mamm. Genome 8:726–730.
- ↵Y. YouR. BergstromM. KlemmB. LedermanH. NelsonC. TicknorR. JaenischJ. Schimenti(1997) Chromosomal deletion complexes in mice by radiation of embryonic stem cells. Nat. Genet. 15:285–288.
- ↵U. ZechnerM. ReuleA. OrthF. BonhommeB. StrackJ.-L. GuenetH. HameisterR. Fundele(1996) An X-chromosome linked locus contributes to abnormal placental development in the mouse. Nat. Genet. 4:398–403.
- ↵M. ZeicherE. MozesP. Lonai(1977) Lymphocyte alloantigens associated with X-chromosome-linked immune response genes. Proc. Natl. Acad. Sci. 74:721–724.
- ↵B.H. ZhengA.A. MillsA. Bradley(1999) A system for rapid generation of coat color-tagged knock-outs and defined chromosomal rearrangements in mice. Nucleic Acids Res. 27:2354–2360.
- ↵E. ZhouJ. FavorW. DilversD. Stamboulian(1995) Exclusion of the candidate genes, Grpr, Cxn33 and Pdha1, for X-linked cataract gene on the distal region of mouse chromosome X. Mamm. Genome 6:357–359.