
Multiple LTR-Retrotransposon Families in the Asexual Yeast Candida albicans
Abstract
We have begun a characterization of the long terminal repeat (LTR) retrotransposons in the asexual yeast Candida albicans. A database of assembled C. albicans genomic sequence at Stanford University, which represents 14.9 Mb of the 16-Mb haploid genome, was screened and >350 distinct retrotransposon insertions were identified. The majority of these insertions represent previously unrecognized retrotransposons. The various elements were classified into 34 distinct families, each family being similar, in terms of the range of sequences that it represents, to a typical Ty element family of the related yeast Saccharomyces cerevisiae. These C. albicans retrotransposon families are generally of low copy number and vary widely in coding capacity. For only three families, was a full-length and apparently intact retrotransposon identified. For many families, only solo LTRs and LTR fragments remain. Several families of highly degenerate elements appear to be still capable of transposition, presumably via trans-activation. The overall structure of the retrotransposon population in C. albicans differs considerably from that of S. cerevisiae. In that species, retrotransposon insertions can be assigned to just five families. Most of these families still retain functional examples, and they generally appear at higher copy numbers than the C. albicans families. The possibility that these differences between the two species are attributable to the nonstandard genetic code of C. albicans or the asexual nature of its genome is discussed. A region rich in retrotransposon fragments, that lies adjacent to many of theCARE-2/Rel-2 sub-telomeric repeats, and which appears to have arisen through multiple rounds of duplication and recombination, is also described.
[The sequence data described in this paper have been submitted to the GenBank data library. Accession numbers are listed in Table 1 and in the Materials and Methods section.]
Candida albicans is a major fungal pathogen of humans and C. albicansinfections have become more of a problem in recent years with the spread of AIDS and the increased use of invasive surgical techniques (Odds 1988). In addition to its medical importance, C. albicans is of interest because it is diploid and asexual (Scherer and Magee 1990), yet often displays a high level of interstrain variation—by various criteria including pathogenicity (Cutler 1991), karyotype (Iwaguchi et al. 1990), and ability to utilize various carbon sources (Rustchenko et al. 1997). These factors are contributing to its use as a model system for studying both the means by which genetic variation can be introduced in the absence of meiotic recombination, and the long-term effects of asexuality.
Several laboratories have devoted considerable effort over recent years toward understanding the genomic organization of C. albicansand how this varies among strains. Important results to date include the construction of an SfiI restriction map of the complete genome (Chu et al. 1993); a detailed physical map of chromosome 7 (Chibana et al. 1998); the finding that, despite extensive karyotypic variation, the underlying genetic map may be largely similar in most strains, with the karyotypic variation being introduced by a relatively small number of large-scale chromosomal translocations (Thrash-Bingham and Gorman 1992); and the finding (Chu et al. 1993) that these exchanges of material between chromosomes are likely to occur within a large array of repetitive sequence, the major repeat sequence (MRS), which seems to be present, at least in part, on all chromosomes (Chindamporn et al. 1998).
Retrotransposons are a significant component of many eukaryote genomes. They often make up a large proportion of the genome, for instance, the L1 retrotransposon comprises ∼15% of the human genome (Kazazian and Moran 1998), and are known to cause mutations and promote genomic alterations (e.g., Zou et al. 1996b). The yeast Saccharomyces cerevisiae has five families of retrotransposons, which are collectively referred to as Ty elements (Boeke and Sandmeyer 1991). These elements are all of the long terminal repeat (LTR) class, and as such, they resemble the vertebrate retroviruses in their genomic organization and replication cycle. The most well-characterized Ty element is Ty1, which is composed of two ∼330-bp LTRs (delta elements) flanking an ∼5.3-kb internal domain. The element replicates via the reverse transcription of a genomic mRNA into a double-stranded DNA copy, followed by the insertion of this DNA into a new site within the host genome. The proteins required for the reverse transcription and integration reactions are encoded by two long ORFs in the internal region, whereas the LTRs contain the promoter and polyadenylation signals that direct the synthesis of the full-length transcript.
Full-length retrotransposons seem to be quite unstable structures and are often lost as a result of recombination between their two LTRs. This results in single, isolated LTRs, termed solo LTRs, remaining at the original sites of insertion. Full-length retrotransposons, and also the solo LTRs, are often found flanked by short (4- or 5-bp) direct repeats. These are duplications of the target-site sequence, which are formed during the insertion process.
Of the S. cerevisiae Ty elements, Ty1, Ty2, and Ty3 are known to be functional (Curcio et al. 1988; Hansen and Sandmeyer 1990). Full-length, apparently uncorrupted, Ty4 elements are present, but they have not yet been shown to be active (Hug and Feldmann 1996). No functional Ty5 elements are known in S. cerevisiae, although some retain significant amounts of internal coding sequence, and functional Ty5s have been found in the closely related species S. paradoxus (Voytas and Boeke 1992; Zou et al. 1996a). A recent survey of the complete genome sequence of S. cerevisiaeidentified a total of 331 Ty element insertions, which together comprise 3.1% of the 12-Mb genome (Kim et al. 1998). A large proportion (85%) of these were solo LTRs or LTR fragments. Evidence was also found for recombination between Ty elements at different sites and it was suggested that rearrangements associated with Ty elements may have played a significant role in shaping the yeast genome (Kim et al. 1998).
Most LTR retrotransposons can be classified into one of two distinct groups, the copia group or the gypsy group, on the basis of their reverse transcriptase sequences and other structural features (Xiong and Eickbush 1990). Ty1, Ty2, Ty4, and Ty5 are allcopia-like elements, whereas Ty3 is a member of thegypsy group.
Five retrotransposons or retrotransposon-like elements have been identified in C. albicans to date. The first of these is Tca1 (Chen and Fonzi 1992; Chen et al. 1998), which consists of 388-bp LTRs (alpha elements) flanking an ∼5-kb internal domain. No long ORFs are present in the internal region, suggesting that Tca1 is not capable of autonomous transposition. Tca1 is present at just a low copy number, 0–3 per cell, whereas its LTR is more abundant at 5–10 copies per cell. The second element, beta, is known only as some 395-bp solo LTRs, which are present at 5–10 copies per cell (Perreau et al. 1997). Third, is the unusual Tca2 (or pCal) retrotransposon (Matthews et al. 1997). Tca2 consists of 280-bp LTRs (gammaelements) flanking an ∼6-kb internal domain. The internal domain contains two long ORFs resembling the gag and polORFs of other retrotransposons. It differs from all other known retrotransposons, however, in that it produces an abundance of extrachromosomal DNA copies. The fourth described element is a 280-bp LTR, kappa (Goodwin and Poulter 1998), present at 10–15 copies per cell. Some kappa elements can be found in association with internal fragments resembling the internal regions of Tca2, suggesting that the two are related. The fifth element, LRT2, is a non-LTR retrotransposon reverse transcriptase identified by Chibana et al. (1998) during their mapping project.
Here we describe a more thorough characterization of the retrotransposons in the C. albicans genome. This characterization was made possible by a C. albicans genome sequencing project at Stanford University. Our results show that the structure of the retrotransposon population in C. albicansdiffers considerably from that of S. cerevisiae. The differences suggest that the forces shaping retrotransposon evolution have differed in the two species. Further analyses and comparisons may yield interesting insights into more general aspects of genome structure, function, and evolution.
RESULTS
Identification of New C. albicansRetrotransposon Sequences
The genome of C. albicans strain SC5314 (Gillum et al. 1984) is currently being sequenced by the Stanford DNA Sequencing and Technology Center. A website is provided (http://www-sequence.stanford.edu/group/candida), in which the data is made publicly available in a searchable form soon after it is obtained. At present, there are two sequence databases at the Stanford site. The first has been operating since soon after the project's inception and contains individual sequence reads together representing a 1.5-fold coverage of the haploid C. albicans genome (∼16 Mb, Chu et al. 1993). Each entry is annotated with the top three hits from a BLASTX search of the sequence against the Genpept database and contains links to the sequence itself and the trace data. There is also a link provided for directly submitting the sequence as a query in a BLASTN search of the entire Stanford database. The second database has been available since March, 1999 and contains sequence assembled into high-quality contigs. Assembly 4, which was used in this work, consists of 1631 contigs, all >2 kb in length, containing 14.9 Mb.
These sequence databases were used to identify new families of C. albicans retrotransposons. The major focus of this paper is the identification and characterization of the LTRs of the new retrotransposon families, although a number of new full-length elements were identified. These full-length retrotransposons are described briefly here but will be analyzed in greater depth elsewhere. In this paper we first describe how the new LTRs were discovered, then how they were divided into families, before presenting a more in-depth characterization of the various families.
The criteria for assigning a C. albicans sequence as a new retrotransposon LTR were that such a sequence should (1) have distinct termini, most likely 5′-TG...CA-3′; (2) be present at multiple locations in the genome, as evidenced by multiple different flanking sequences; (3) sometimes be found flanked by short (4- or 5-bp) direct repeats, representing duplications of the target sequence; and (4) be within a limited size range. Initially, we considered elements ∼200–600 bp in length, a range that encompasses the size distribution of previously described fungal LTRs (251–388 bp,Lauermann et al. 1997). It soon became apparent, however, that there were LTRs both larger and smaller than this. We therefore relaxed these limits somewhat and eventually have considered elements anywhere between 127 and 780 bp in length. All of the elements we describe here fulfill all of the above criteria, although several have the terminal dinucleotides 5′-TG...TA-3′, as described previously for beta(Perreau et al. 1997).
The new LTRs were identified in a variety of ways. A few were identified simply as a result of their association with recognizable retrotransposon internal regions. An example is the san LTR. One sequence carrying part of a san LTR also carries part of an ORF predicting a protein similar in sequence to the carboxy-terminal end of the Tca2 Pol protein (Fig. 1A). This similarity was recognized in the BLASTX search against the Genpept database and was noted in the sequence's annotation. Closer examination of this sequence revealed a region downstream of the ORF closely resembling the polypurine tracts (PPTs) of other C. albicans retrotransposons (Fig. 1B), and adjacent to this PPT was a new LTR-like repetitive sequence—san.

Identification of the san LTR. (A) A translation of a partial ORF found upstream of the san LTR in sequence 396001F03s (san ORF) is compared with the predicted carboxy-terminal sequence of the Tca2 RNase H. (B) A comparison of the PPTs found upstream of four C. albicansretrotransposon LTRs. The PPTs are in italics and LTR sequences are in boldface. The name of the associated retrotransposon (see text) is shown at left. (C) Comparison of san andalpha. The alignment was produced by the FASTA program of the GCG package. The percent identity is also shown.
The next method involved screening the database containing the individual sequences (the database containing the assembled data was not available at the time) for repetitive elements by performing sequential BLAST searches of individual sequences against the database. With the level of coverage of the genome in the database at the time, most single-copy sequences produced just a single hit (themselves), or, occasionally, two or three. Repetitive sequences, on the other hand, usually had multiple high-quality matches. Most of the repetitive sequences identified in this manner appeared to originate from the mitochondrial DNA, the ribosomal DNA repeats, the MRS (Chibana et al. 1998), or from CARE-2/Rel-2 (Lasker et al. 1992;Thrash-Bingham and Gorman 1993). Occasionally, however, other repetitive sequences were found and these were examined to see if distinct ends to the repetitive DNA, and the other features expected of LTRs, were apparent. At least 2000 sequences were screened in this manner and five or six new LTRs, for example zeta andsampi, were identified. This was a rather time-consuming method, but was useful as a unbiased way of detecting completely new LTRs.
Further LTRs were identified as a result of the apparent pattern of evolution of retrotransposons in C. albicans (see below), which has resulted in the appearance of related families of elements that still retain significant sequence similarity. For instance, thesan element mentioned above and the alpha LTR of Tca1 share ∼60% sequence identity. These two elements are shown aligned in Figure 1C. They can be seen to share very similar terminal sequences and scattered regions of similarity in the internal regions. These areas of similarity may contain sequences important for LTR function, such as promoters, terminators, or recognition sites for integrase, etc. An example of a new LTR being detected as a result of its similarity to other elements is xi, which is related to thesan and alpha LTRs. This element was detected following a BLASTN search of the assembled sequence data with asan LTR as a query. As expected, the search detected the contigs containing san LTRs with the most significant scores, followed by the contigs containing alpha LTRs. However, several other contigs were also detected with scores well above background. Close examination of these contigs revealed that they all contain a similar element, which bears all of the characteristics of an LTR. This new element was named xi.
The final method by which new LTRs were discovered was as inserts within, or near to, previously recognized elements. For instance, while characterizing the members of one retrotransposon family, we would frequently find truncated examples or elements with large insertions. Analysis of the sequences causing the disruptions often revealed new LTRs. An example of an LTR identified in this manner is tui. This LTR was originally detected as a 193-bp element, present within a kappa LTR, with the terminal dinucleotides 5′-TG...TA-3′ and flanked by a 5-bp duplication of the target site (Fig. 2). Further analysis of this putative LTR revealed multiple similar sequences in the genome with a variety of different flanking sequences.
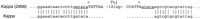
Identification of the tui LTR. The region of thekappa LTR in contig 2956 that contains the tuiinsertion is compared with a typical kappa LTR. Thetui LTR is in capitals and its target-site duplication is underlined.
By these four methods, we have identified a total of 355 LTRs or LTR fragments in assembly 4 of the Stanford database, which represents 14.9 Mb from a haploid genome of size 16 Mb. It is worth noting, however, that our methods were not exhaustive, and it is possible that there are other families of LTR sequences in the SC5314 genome that we have failed to detect. In addition, it is likely, given that C. albicans is asexual and that many strains have thus been evolving independently for a long time, that there are retrotransposons present in other C. albicans strains that are not present in SC5314 at all. Furthermore, there are a few other LTR-like sequences present, which we haven't included in this analysis because they don't fulfill all of the above criteria, such as having two distinct termini.
Division of the New Elements into Families
Many of the new LTRs that we have identified are simple to group into distinct families as they are highly similar within a family, bearing, for example, >80% sequence identity to all other members of the family, yet they bear no detectable similarity to any other LTR at all (apart from the terminal dinucleotides). For others, however, it is not so simple, as there are quite a few cases of pairs of elements sharing 60%–70% sequence identity. It is not immediately clear whether such elements should be classified in the same family, or into related, but distinct, families. To allow comparisons between our results and the findings from S. cerevisiae, we have tried to keep the concept of a family consistent between these two species. Although the term family has not been defined precisely for retrotransposons in S. cerevisiae, the extent of diversity of sequences within a family has been fairly well documented (Kim et al. 1998 ; Jordan and McDonald 1999b). For instance, Kim et al. (1998) have reported that, of the 28 full-length Ty3 LTRs present in the genome sequence, 21 share >98% identity with a reference LTR, whereas the most divergent LTR was 85% identical. For Ty4, 17 of 19 full-length LTRs were >96% identical to the reference LTR, with the other two being 73% and 75% identical. Ty5 LTRs were found to share between 80% and 95% identity to a reference LTR. The delta LTRs of Ty1 and Ty2 seem to be a more heterogenous group, with some having <70% identity to a reference LTR. These divergent deltaelements have, however, been considered to be degenerate and most of the delta elements that are associated with full-length Ty elements have >85% identity to a reference LTR (Kim et al. 1998). The greater diversity of delta elements may in part be explained by it being considered the LTR of two distinct elements. Recently, it has been recognized that the LTRs of Ty2 can, in fact, be distinguished from Ty1 LTRs and it has been proposed that the large group of delta elements be subdivided into two families,delta and phi, for Ty1 and Ty2, respectively (Jordan and McDonald 1999a).
With these findings from S. cerevisiae in mind, we decided on a working definition of a family of C. albicansretrotransposon LTRs as being a monophyletic group of sequences, having all the characteristics of LTRs, that typically share ≥70% nucleotide identity. Single sequences that share ∼55%–70% identity with the members of a family were considered to represent diverged or degenerate members of that family, so that a family could contain members with <70% identity to other members. In some cases, there are multiple LTR sequences (as evidenced by multiple different flanking sequences) that have 55%–70% identity to a particular family, but which have ≥70% identity to each other. Such elements were considered to belong to related, but distinct, families. In borderline cases, we tended to be conservative, preferring to make a family larger and more diverse rather than creating an additional family.
Phylogenetic trees were constructed for all families to allow the diversity of sequences among and within families to be visualized. Examples of these are shown, drawn to the same horizontal scale, in Figure 3A. These trees are useful to illustrate some borderline cases and to compare them with well-defined examples of related, but distinct, families. An example of the latter is provided by alpha, san, and xi. Alpha and xieach have ≥90% identity within a family and san has ≥98%. The elements share ≤62% identity in between-family comparisons. The tree separated the three families into three groups, each receiving 100% bootstrap support. The elements within each family can be seen to be very closely related, but well separated from elements of each of the other families. In marked contrast, however, is the tree for pi elements, which represents one of the borderline cases. The tree divides the pi elements into two broad groups, each receiving >90% bootstrap support. These two groups might be considered as two families; however, some elements in each group still share 73% identity with members of the other group. In addition, the two groups still appear to be capable of recombination. This was suggested by an apparent chimaeric element, the most divergent element in the upper group in Figure 3A, which associated with the lower group when bases 1–80 of the alignment were used to construct a tree, but with the upper group when the remainder of the alignment was used (not shown). For these reasons, it was decided to assign all of these elements to the one family, which then becomes a diverse family, as some members share as little as 58% identity with others.
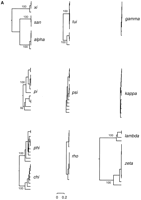

Phylogenetic trees of full-length LTRs. (A) C. albicans LTRs. (B) S. cerevisiae LTRs. The trees were constructed by the UPGMA method with PHYLIP (Felsenstein 1989) and are shown drawn to the same horizontal scale. The distance is Kimura's (1980) two-parameter distance. The level of bootstrap support (%) for the major branches is indicated.
Another borderline case is the phi and chi LTRs. These two families fall into two monophyletic groups with 100% bootstrap support (Fig. 3A), but some phi LTRs share 71% identity with particular chi LTRs, suggesting that perhaps they should all be assigned to the one family. If, however, this were to be done, then this would become a very diverse family, with some elements sharing as little as 56% identity and many failing to align full-length at all. In addition, no evidence for recombination between the two families was detected. For the above reasons, these elements were assigned to two separate families. The somewhat greater divergence between phi and chi LTRs than between the two halves of the pi family can be seen by the longer branch length separating phi and chi than that separating the two halves of the pi family (Fig. 3A). A final borderline case was the tui LTRs. These elements fall into two groups receiving 100% bootstrap support, with the elements from one group typically sharing 67%–69% identity with the other group. However, all of the elements in the upper group, as shown in Figure 3A, have very similar flanking sequences and appear to have been duplicated by a mechanism other than autonomous retrotransposition (see below), suggesting that they are not worthy of a separate family designation. These elements were therefore grouped together with the related LTRs into one family.
On the basis of the above family definition, designed to be broadly comparable to S. cerevisiae, and the resolution of borderline cases as described, the C. albicans LTRs fall into 34 distinct families. To be consistent with the naming of previous C. albicans and S. cerevisiae LTRs, the new elements have been named after letters of the Greek alphabet, although we tended to avoid using letters already assigned to S. cerevisiae LTRs (delta, omega, sigma, and tau). When we ran out of classical Greek letters, we started naming them after archaic Greek letters (sampi, san, etc.) and phonetically similar names of New Zealand birds (moa,tara, weka, etc.). The various families are listed in Table 1. Related families of LTRs are listed in Table2.
Properties of C. albicans Retrotransposon LTR Families
Related Families of LTRs
The number of distinct families we have identified in C. albicans is considerably more than the number found in S. cerevisiae. This raises the question of whether a C. albicans retrotransposon family, as we have defined it, is really equivalent to an S. cerevisiae Ty family. We tested this in two ways. Firstly, we constructed phylogenetic trees of thedelta, sigma, and tau Ty LTRs, using as a dataset all of the full-length LTRs identified in the yeast genome (available from the Voytas laboratory web site:http://www.public.iastate.edu/∼voytas/ltrstuff/ltrtables/yeast.html). These trees are shown, drawn to the same horizontal scale as theC. albicans trees (Fig. 3B). As discussed by Kim et al. (1998), the trees reveal that the tau elements are very similar to one another, with the exception of two quite divergent ones. Similarly, sigma LTRs can be seen to be a homogeneous group with a small number of diverged examples. In contrast, thedelta LTRs are revealed to be a fairly heterogeneous group (Fig. 3B). The diversity of tau elements, as revealed by this method, is very similar to, for example, that of the zeta LTR of C. albicans, whereas the sigma tree is similar to that of rho and psi. The delta LTRs appear to represent a slightly more diverse range of sequences than the most diverse C. albicans family illustrated in Figure 3A. They are not as diverse as the phi and chi families combined, however, and are substantially less diverse than the more typical groups of related elements, such asalpha-san-xi andlambda-zeta. Overall, the trees indicate that C. albicans retrotransposon families, as defined above, and families of Ty elements, are broadly similar in terms of the diversity of sequences that they represent.
As a second means of comparing the concept of a family in C. albicans and S. cerevisiae, we calculated the level of nucleotide diversity (π, Nei and Li 1979) for each family. For a set of aligned sequences, π is the average number of differences per site between each pair of sequences in the alignment. For instance, π = 0.02 means that any two sequences differ on average at 2% of the sites. Jordan and McDonald (1999b) have calculated π for the LTRs of full-length Ty elements and found that it ranged from 0 for Ty3, to 0.027 for Ty1, with Ty2 (0.023) and Ty4 (0.011) in between. Unfortunately, many of the C. albicans retrotransposon families appear not to have any full-length examples remaining (see below), so we were forced to use LTRs that weren't solely from full-length elements. Therefore, to allow the results to be compared, we recalculated π for delta, sigma, andtau using all of the full-length LTRs found in the genome (i.e., including solo LTRs), and did the same for each C. albicans family. The results are listed in Table 1. As might be expected, π is considerably higher for the S. cerevisiaeLTRs when the solo LTRs are included, for example, eight times higher for delta. The calculated values of π again suggest thatC. albicans and Ty element families are similar in the diversity of sequences that they represent. There are six or sevenC. albicans element families for which π is similar to or greater than that for delta, including the pi andtui families mentioned above. The values of π forsigma and tau are lower than for delta, but again similar to a variety of C. albicans families. The average π for all C. albicans families (0.134) is very similar to that for S. cerevisiae (0.126). Together, the results from the trees and nucleotide diversity calculations suggest that a family of retrotransposons in C. albicans, as defined above, and a Ty element family are approximately equivalent in terms of the range of sequences they represent. Thus, the figure of at least 34 distinct families in C. albicans is directly comparable with the 5 distinct families present in S. cerevisiae. This great difference in the number of retrotransposon families suggests that the forces directing element evolution in these two species have varied considerably.
General Characteristics of the LTR Families
Having established that C. albicans retrotransposon families are comparable to S. cerevisiae families, we went on to characterize the LTRs of each family in more detail. Some of our findings are summarized in Table 1. The elements range in length from 127 to 780 bp, with an average length of 359 bp. This is a somewhat wider range than that found in previously identified yeast retrotransposon LTRs—251–388 bp (Lauermann et al. 1997), which probably reflects the large number of C. albicans elements that have been identified. Most of the elements have the terminal dinucleotides 5′-TG...CA-3′ although a few, such astara and tui, have 5′-TG...TA-3′. These dinucleotides often form part of larger terminal inverted repeats. For example, the termini of the theta element are 5′-TGTTACGA...TCGTAACA-3′. The copy number of each C. albicans LTR was estimated by determining the number of LTRs with distinct flanking regions present in assembly 4 of the Stanford database. These estimates are likely to be close to the true figure, as this database represents 14.9 Mb of assembled sequence from a haploid genome of 16 Mb. The copy numbers of most families are similar to the copy number of the Ty5 LTR in S. cerevisiae, but consideraby lower than that of delta, sigma, and tau. The total number of LTRs (355) is, however, very similar to the number found in S. cerevisiae (331). An independent estimate of copy number by Southern blotting has been obtained previously foralpha (Chen and Fonzi 1992), beta (Perreau et al. 1997), and kappa (Goodwin and Poulter 1998). We analyzed another four elements by Southern blotting here (examples in Fig.4). The estimates from the blots (figures in parentheses in Table 1) and from the sequence are quite similar, although the blots often give slightly lower figures. This is likely due to partial or diverged elements not being detected or distinct elements comigrating in the gels. Comparison of several different strains on the blots reveals that the number of hybridizing bands, and their sizes, varies among strains. This is consistent with these new LTRs being part of transposable elements and suggests that they have moved since the divergence of the strains. The extent of difference in the banding pattern between any two strains is likely a function of the time since those strains diverged. DNA from C. albicans'close relatives C. maltosa, C. parapsilosis, andC. tropicalis, and a more distantly related species C. pseudotropicalis (Cai et al. 1996), was also included on the blots. None of the elements tested hybridized to DNA from these species, suggesting that they are specific to C. albicans.

Hybridization patterns of zeta and san. (A) A Southern blot of EcoRI-digested DNA from six C. albicans strains (lanes 1–6) and four otherCandida species (lanes 7–10) was probed with a radio-labeled zeta element. (Lane 1) hOG1042; (lane 2) SGY269; (lane 3) SC5314; (lane 4) ATCC10261; (lane 5) SA40; (lane 6) F16932; (lane 7) C. maltosa; (lane 8) C. parapsilosis; (lane 9) C. tropicalis; (lane 10) C. pseudotropicalis. Sizes (in kb) are indicated at left. (B) The blot inA was stripped, checked for complete removal of the probe, then reprobed with the san element.
For each LTR family, the sequences flanking all of the copies were recorded and compared. Examples of such comparisons are given for thepi and san elements in Figure 5. More than half of the full-length LTRs were found to be flanked by short direct repeats representing target-site duplications (TSDs). For the majority of elements, such TSDs were 5 bp in length, as is the case with the S. cerevisiae Ty elements. Five elements, however, were commonly found flanked by 4-bp TSDs. To the best of our knowledge, 4-bp TSDs have not been reported previously for fungal elements, but are common among gypsy-class elements of insects (for a summary, see Table 6 of Boeke and Stoye 1997). For one of these elements, pi (Fig. 5A), 10 of the 14 full-length LTRs are flanked by 4-bp direct repeats, but two are flanked by 5-bp direct repeats. One of these 5-bp repeats was confirmed as a TSD, as we were able to compare the flanking regions with a similar sequence lacking the pi insertion. Similarly, weka has a confirmed 7-bp TSD, but can also be found flanked by more typical 5-bp repeats.

Immediate flanking regions of pi LTRs (A) andsan LTRs (B). The data are taken from assembly 4 of the Stanford database. The name of the contig on which the element lies is given at left. Flanking direct repeats are underlined. LTR sequences are in boldface. Putative PBSs and PPTs are in italics.
Kim et al. (1998) analyzed the sequences of all the perfect TSDs of Ty elements and observed a strong preference for A and T in the three internal positions. A similar preference is apparent for the perfect 5-bp TSDs of the C. albicans elements (Table3). For the elements with 4-bp TSDs, a bias toward a purine in the first position and a pyrimidine in the fourth was observed (Table 4). Close examination of the 5-bp TSDs of both the C. albicans and S. cerevisiaeelements (Table 3; Kim et al. 1998), also reveals a bias toward a purine in the first position and a pyrimidine in the last.
Five-base-pair TSD Sequences
Four-base-pair TSD Sequences
Analysis of the regions flanking each C. albicans LTR suggested that recombination between LTRs at different locations has occured. For instance, just one of the san LTRs in Figure 5B (2986) is flanked by a 5-bp direct repeat. However, the 3′ target site of the san LTR in 2584 is identical to the 5′ target site of that in 3083, and similarly its 5′-flank is the same as the 3′ flank of the san LTR in 2757. The san LTRs in 2757 and 3083 likely represent the left and right LTRs of a single full-length element (T. Goodwin, unpubl.). This apparent swapping of flanking sequences suggests that one LTR of this retrotransposon has undergone a recombination with a san LTR at some other location. There are several other examples of pairs of LTRs that appear to have swapped flanking sequences, and there are many examples of elements flanked by sequences, bearing no resemblance to a direct repeat, which are probably the result of either recombination or frequent mutation of the TSD.
Identification of Retrotransposons Retaining Internal Sequences
The finding that many of the new LTRs are flanked by direct repeats suggests that these elements are solo LTRs. It is of interest to know whether the actual retrotransposons, corresponding to the many LTR families we have identified, are still present and to know what form such retrotransposons might take. We have therefore searched the databases for retrotransposon internal regions. Sixteen different families were identified, including the previously described Tca1 and Tca2. The new elements have been named Tca3 through Tca16 and are listed in Table 5.
C. albicans Retrotransposons with Internal Regions
For some elements, such as nu, the identification of a corresponding retrotransposon was relatively straightforward—anu LTR is present on a particular contig alongside of a large region with significant homology to the ORFs of gypsy-type retrotransposons and retroviruses. A short distance downstream of this apparent retrotransposon coding region lies a polypurine tract immediately followed by the nu LTR.
Retrotransposons that have highly degenerate or divergent coding regions, or for which only fragments remain, are unlikely to be detected by homology to other elements at the protein level. Therefore, as a possible means of detecting such elements, we scanned the regions flanking those LTRs that lack TSDs for possible minus-strand primer-binding sites (PBSs). In most retrotransposons, the PBS is a 10–20-nucleotide sequence just downstream of the left LTR that is complementary to part of a cytoplasmic tRNA. As examples, the Ty1 PBS consists of a 10-nucleotide sequence, immediately adjacent to the LTR, that is complementary to the 3′ end of the initiator methionine tRNA (tRNAiMet), whereas the Ty5 PBS is a 13-nucleotide sequence, adjacent to the LTR, that is complementary to the anticodon stem-loop of the tRNAiMet (Voytas and Boeke 1992).
New PBSs were detected in a variety of ways. Some were found as a result of their similarity to previously identified C. albicans PBSs. For instance, both the previously described full-length retrotransposons, Tca1 (Chen and Fonzi 1992) and Tca2 (Matthews et al. 1997), have PBSs complementary to an internal fragment of the C. albicans tRNAArg(UCU) (GenBank accession no. AF041470), as does the kappa-carrying retrotransposon (Goodwin and Poulter 1998). A very similar sequence was observed adjacent to a whio LTR when its flanking sequences were being analyzed, and this was recognized as a PBS. Other PBSs were detected because they are complementary to the tRNAiMet that is very commonly used by copia-type elements and also occasionally bygypsy-group elements. We extracted the C. albicanstRNAiMet gene sequence from the database, using the C. utilis tRNAiMet sequence as query, and then compared it with the sequences adjacent to the LTRs. Several LTR families were thus found to have associated PBSs. For instance, some beta LTRs were found in association with PBSs complementary to up to 27 nucleotides at the 3′ end of the tRNAiMet.
In some cases, two LTRs of a family were found repeated in a direct orientation on a contig, but it wasn't clear whether they were two independent insertions or the two LTRs of a single retrotransposon. In such cases, we looked for sequences near to the LTR termini that began 5′-TGG-3′. This sequence is complementary to the CCA trinucleotide present at the 3′ end of all tRNAs and is a feature of all PBSs that utilize the 3′ end of a tRNA as a primer. Such sequences were visually compared with the C. albicans tRNAs in GenBank or to a database of all the S. cerevisiae tRNA sequences (http://biochimica.unipr.it/yeast/all_trna.txt). In the case in which a matching S. cerevisiae tRNA was found, its sequence was used to screen the C. albicans databases and identify the homologous gene, which was then compared with the putative PBS.
By these methods, 14 different families of C. albicans LTRs were found with associated PBSs. The PBSs could be divided into seven different classes (Fig. 6); those with an extensive region of homology to a tRNAArg(UCU) internal region (e.g., gamma), those with a short region of homology to the same tRNA (e.g., whio), those homologous to an internal region of the tRNAiMet (e.g., zeta), and those homologous to the 3′ ends of various tRNAs including the iMet, Ile, Gln, and Ala tRNAs. In several cases, the PBS was not immediately adjacent to the LTR, but between 2 and 10 bp downstream. A short gap between the LTR and the PBS is a common feature of gypsy-class retrotransposons (see, e.g., Table 1 of Chavanne et al. 1998).

The diversity of PBSs in C. albicans retrotransposons. The names of the retrotransposons and associated LTRs are shown atleft. LTR sequences are underlined. The GenBank accession nos. of the tRNAs are as follows: tRNAArg, AF041470; tRNAiMet, AF069449; tRNAIle, Y08492; tRNAGln, AF180282; and tRNAAla, Y08493.
The 16 families of elements that we have found to retain some internal sequences vary considerably in their coding capacity (Table 5). A few are apparently intact, bearing all the characteristic features of functional retrotransposons (Tca2, Tca4, and Tca5). Others, such as Tca3 and Tca8, still have long, uninterrupted ORFs with homology to other retroelements, but we haven't yet detected a full-length element. One, Tca6, has no long ORFs in the internal region but has ORF fragments that still bear detectable similarity to related retrotransposons. Some, such as Tca9 and Tca13, can be found as composite elements, with identical LTRs, intact PBSs and PPTs, and flanked by 5-bp direct repeats. Between the LTRs, however, lies 4–5 kb of sequence with no apparent coding capacity at all, nor any detectable similarity to other retroelements. A further element, Tca10, consists of a composite element, flanked by a 5-bp direct repeat, whose LTRs share 99.5% identity. The internal region is ∼2 kb long and contains a PBS and PPT adjacent to the left and right LTRs, respectively, and some extensive ORFs. The predicted products of these ORFs, however, bear no significant similarity to any protein sequence in the databases.
Most of the elements could be assigned to either the copia orgypsy family (Table 5). For some elements without extensive ORFs, this assignment is tentative as it is based on the nature of their PBSs. For instance, a small gap between the LTR and the PBS is common among gypsy-class elements, but to the best of our knowledge, has not been found in copia-type elements. Conversely, the use of tRNA fragments as primers appears to be restricted to copia-like elements.
None of the other 18 newly identified LTR families were found associated with sequences resembling the internal regions of retrotransposons. Solo LTRs and LTR fragments may be the only remnants of these retrotransposon families. However, it is possible that internal retrotransposon sequences corresponding to some of these LTRs may have escaped detection, given that the coverage of the SC5314 genome in the Stanford database, although extensive, is not complete. It is also possible that other C. albicans strains harbor full-length retrotransposons that are absent from SC5314.
Retrotransposons in the C. albicans subtelomeric regions
We have reported previously that some kappa LTRs are associated with the C. albicans repetitive elementsCARE-2 (Lasker et al. 1992) and Rel-2 (Thrash-Bingham and Gorman 1993) and presented evidence suggesting that such LTRs have been subjected to some form of rearrangement (Goodwin and Poulter 1998). Subsequently, it was reported that CARE-2- and Rel-2-like sequences are present near the ends of several chromosomes, and it was proposed that they are subtelomeric repeats (Chibana et al. 1998). The availability of extensive assembled sequence data allowed us to analyze in greater detail the relationship between retrotransposon LTRs and these putative subtelomeric sequences.
As might be expected for repetitive elements, there are a relatively large number of contigs bearing CARE-2- and Rel-2-like sequences in the database. More than half of these contigs were found to contain a similar and LTR-rich region neighboring theCARE-2/Rel-2-like sequences. A few other contigs also have a similar LTR-rich region but lack CARE-2/Rel-2. The LTR-rich regions of all of these contigs are depicted in Figure7A.
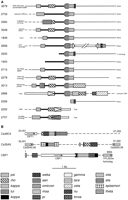
(A) LTR sequences adjacent to the putative C. albicans subtelomeric repeats. LTRs are represented by the boxes with different fill patterns. kappa LTRs that are truncated just prior to the 3′ end are indicated by the thin, vertical lines. Sequences to the right of the LTRs with some homology toCARE-2/Rel-2 are represented by the thin, continuous lines. Broken lines represent different sequences. A DNA transposon is depicted as a narrow box flanked by inward-pointing triangles. The direction of transcription of LTRs with associated internal regions is indicated. The thick black line represents sequences similar to theC. albicans CTA2 gene (GenBank accession no. AJ006637). The nearest recognizable upstream and downstream genes are indicated. The sequences of some contigs end within the region shown. (B) LTR groups in intergenic regions. LTRs are again represented by the filled boxes. ORFs are indicated by the boxed arrows, the arrows indicating the orientation of the ORF. The accession nos. of the sequences areas follows: Ca49C4, AL033503; Ca35A5, AL033396; and CBP1, L08824. The common scale for A and B is also shown. Note that Ca35A5 also contains MRS (Chibana et al. 1998) sequences but that these are >15 kb away from the sequences shown here.
To ascertain whether these areas are of subtelomeric origin, the distribution of genes and repeated sequences in the surrounding areas was studied. We found that upstream of the LTRs (Fig. 7A, left) the sequences soon diverge. These different upstream flanking sequences contain a variety of different genes. For example, contig 3048 has a tRNALeu gene, contig 1846, part of a TUP1 gene, and contig 2757, a homolog of the S. cerevisiae YJL004c gene, each within 500 bp of the psi LTR fragment. The other contigs, apart from 2250, each have a gene within 1 kb of the psi LTR. Contig 2250 has a degenerate non-LTR retrotransposon starting ∼300 bp upstream. Several of the contigs contain sequences extending 20 kb or more upstream of the LTR-rich region. In contrast, the contigs withCARE-2/Rel-2-like sequences downstream of the LTRs generally extend only 2–6 kb downstream and these sequences contain no recognizable genes. An exception is contig 2935, which extends >8 kb downstream and contains a long ORF with similarity (not shown) to the Y′ subtelomeric elements of S. cerevisae (Louis and Haber 1992). For the contigs that have CARE-2- and Rel-2-like sequences, but that don't have the LTR-rich region, theCARE-2/Rel-2-like sequences were also found to lie at the end of the contig with no genes downstream, apart from another couple of Y′ homologs. These findings that the CARE-2- and Rel-2-like sequences are bounded on one side by variable and gene-rich regions, but that the opposite flank could not be identified, because in each case the available sequence ends, together with their association with homologs of the Y′ subtelomeric elements ofS. cerevisiae, strongly supports the proposal of Chibana et al. (1998) that CARE-2 and Rel-2 are subtelomeric repeats. [Note, however, that none of the above contigs extend into the 23-bp telomeric repeats (McEachern and Hicks 1993), although no contigs in assembly 4 do]. In addition, it is likely that CARE-2 and Rel-2 are subtelomeric repeats on the majority of C. albicanschromosomes as they are both present on most, if not all, chromosomes (Lasker et al. 1992; Thrash-Bingham and Gorman 1993). It is also noteworthy that about half of these contigs also carry theCARE-1 (Lasker et al. 1991) and Rel-1 (Thrash-Bingham and Gorman 1993) repetitive elements, and that these represent the vast majority of contigs on which these elements appear. This suggests thatCARE-1 and Rel-1 are subtelomeric sequences as well.
The LTR-rich regions that are present at the boundary between the subtelomeric repeats and the centromere-proximal unique sequences appear to have been subjected to high levels of sequence rearrangement (Fig. 7A). Elements that are common to a majority of these regions arekappa LTRs, each bearing a tui LTR insertion, and, a short distance upstream, partial psi LTRs. The tuiLTRs are inserted into the kappa LTRs at the same position and in the same orientation in all of the various contigs, although many of them have suffered small deletions. The immediate upstream flanks of the kappa LTRs are identical in all of the contigs, but there are a variety of distinct downstream flanks. For instance, some are immediately flanked by CARE-2/Rel-2-like sequences, others have an intervening partial weka LTR, whereas others are truncated, etc. Upstream of the corrupted kappa LTRs, the partial psi LTRs vary on the different contigs, some suffering more widespread deletions than others. The upstream flanks of thepsi fragments mark the boundary between the repeated sequences and the upstream unique sequence in some contigs, although other contigs share similar sequence for up to several hundred base pairs further upstream. Several contigs have a rho LTR between thepsi and kappa LTRs. Again, these rho LTRs have the same flanking sequences and the same orientation. Some contigs carry different combinations of these common features. For instance, contig 3079 has the rho LTR between psi andkappa and the downstream flank of kappa is within aweka LTR. Contig 3048, however, has the rho LTR, but not the partial weka, whereas contig 2956 has the partialweka but not the rho LTR. Other contigs carry unique variations—for instance, an additional tui LTR between thepsi fragment and rho in contig 3013 and a large (4.4-kb) inverted-repeat DNA transposon in the kappa LTR of contig 2956.
The arrangement of these sequences suggests that they are the result of multiple rounds of sequence duplication and recombination, interspersed with a variety of transposition events. Presumably, the ancestral structure consisted of a kappa LTR, with a tuiinsertion, lying downstream of a partial psi LTR. For some reason, this sequence was the subject of multiple rounds of duplication. At some stage during the duplication process, arho LTR likely became inserted between the kappa LTR and psi fragment of one copy and subsequently became part of the duplicated sequences. Recombinations, at various stages amongkappa LTRs with different flanking sequences is also likely. In addition, a variety of deletions occurring in the tui andpsi LTRs during the duplication process is suggested.
The sequences of some of these contigs are suggestive of some quite dramatic rearrangements, involving areas of the genome not closely associated with the subtelomeres. For instance, contig 2898 has not only suffered a Tca2 insert within the rho LTR, but atara insert within the kappa LTR and anotherrho insert within the tara element. The tarainsert in this contig is not flanked by a direct repeat, and its downstream flanking sequence, in which the remainder of thekappa LTR might be expected to lie, bears no resemblance to akappa LTR. The sequences downstream of tara also bear no resemblance to the subtelomeric CARE-2/Rel-2 sequences. Instead, following an additional zeta LTR and a truncated toroa LTR, there are several apparent genes. This suggests that the LTR-rich region in this contig, although clearly related to the others, is no longer closely associated with the subtelomeres.
LTRs that lie in close proximity to one another are not restricted to the subtelomeric regions. Several examples of LTRs grouped together in intergenic regions are shown in Figure 7B. The arrangement of LTRs in the CBP1 (corticosteroid-binding protein) gene is of particular interest, as LTRs have inserted within the ORF and now supply its last 13 codons and presumably the transcriptional termination signals as well. The protein encoded by this ORF is still functional, however, with demonstrated high-affinity corticosteroid-binding activity (Malloy et al. 1993). Another LTR is present a short distance (150 bp) upstream of the CBP1 ORF. Given that LTRs often carry strong promoter elements, it is possible that LTRs that lie so close to genes could alter the regulation of those genes. LTRs in intergenic regions can also be found as truncated forms and without flanking direct repeats, suggesting that rearrangements associated with LTRs can occur in these regions. However, the only LTR we could find in association with the major repeat sequence (Chibana et al. 1998), in which chromosomal translocations may be most common (Chu et al. 1993), was zeta. A degenerate and truncated copy of this LTR comprises ∼400 bp of the MRS (not shown).
DISCUSSION
Retrotransposons are an abundant and ubiquitous component of the eukaryote genome, and, as such, are a common source of genetic variation. The analysis of the retrotransposon complements of different species is of interest, as it should further our understanding of the role played by these elements in host evolution and may reveal the various strategies by which the hosts have attempted to prevent the overproliferation of these elements. Conversely, the various strategies used by the elements to try to avoid any host-driven elimination mechanisms may also become apparent.
We have presented here an analysis of the retrotransposons in the genome of the asexual yeast C. albicans. The results of such an analysis are of special interest as the retrotransposons of the related yeast S. cerevisiae have been analyzed in depth and thus provide an excellent reference for comparison. Here, we have described in some detail the methods by which a wide variety of newC. albicans elements were identified and classified into families. We have presented a working definition of a retrotransposon family for C. albicans that may assist in the classification of any further C. albicans retrotransposons. We compared the concept of a retrotransposon family in C. albicans with the recognized families of Ty elements and concluded that they are roughly equivalent in that they represent a similar diversity of sequences. An initial characteriza-tion of the various families was also undertaken. A more in-depth analysis of the several relatively intact retrotransposons that we identified will be presented elsewhere.
Perhaps the most interesting finding to emerge from this work is that the number of distinct retrotransposon families is much higher inC. albicans than it is in S. cerevisiae. Even if the families of C. albicans elements that show some sequence similarity (Table 2) were to be combined into superfamilies of related elements, there would be 20 such groups, still considerably more than in S. cerevisiae. Another difference is that the majority of the C. albicans families appear to be nonfunctional and of low copy number. In contrast, the Ty elements are largely intact and are present at higher copy numbers. Furthermore, C. albicans has non-LTR retrotransposons and DNA transposons (Chibana et al. 1998; this work), neither of which are found in S. cerevisiae. These differences suggest that the forces driving the evolution of transposable elements have differed quite considerably between the two species. It is of interest to consider the possible origin of such differences.
One major difference between C. albicans and S. cerevisiae is that C. albicans has a nonstandard genetic code—the standard CUG-leucine codon has been reassigned to serine (Santos et al. 1997). Several lines of evidence suggest that the reassignment is not complete (Suzuki et al. 1997), so that CUG is actually an ambiguous codon. Santos et al. (1999) have recently attempted to reconstruct the early stages of this reassignment by introducing a single copy of the C. albicans CUG-decoding tRNA into S. cerevisiae. Interestingly, they found that ambiguous CUG-decoding induced the general stress response and resulted inS. cerevisiae cells that exhibited a high level of stress tolerance, similar to that of normal C. albicans cells. It was postulated that a constitutive induction of the general stress response, induced by ambiguous decoding, may have been important in the evolution of pathogenesis in Candida species. Induction of retrotransposons in response to stress has been well documented (Anaya and Roncero 1996; Wessler 1996; Vasil'eva et al. 1998). It is possible that a constitutive stress response over millions of years may have played a role in generating the diversity of retrotransposons inC. albicans.
Another obvious difference between the two species is that S. cerevisiae can reproduce sexually, whereas C. albicansappears to be strictly asexual (Scherer and Magee 1990). This could have influenced the diversity of transposable elements in several ways. For instance, the process of meiotic recombination may allow S. cerevisiae to remove any ill-adapted elements, for example, those whose integration isn't directed away from coding regions, much more rapidly than can any alternative process in C. albicans. Alternatively, the genetic diversity generated by transposable elements may be advantageous to C. albicans, leading to more elements being retained. A large number of transposable elements could even promote the appearance of asexuality as recombination between elements at different loci could lead to large-scale chromosomal rearrangements and subsequent difficulties in chromosome pairing during meiosis. Indeed, Rachidi et al. (1999) have described recently an S. cerevisiae strain that harbors a number of Ty-mediated chromosomal translocations which is no longer able to sporulate, possibly due to difficulties in chromosome pairing. The above proposals, however, require that the asexual genome has a greater absolute number of transposable elements, whereas, in fact, the total number of insertions appears to be similar in C. albicans and S. cerevisiae. Wild isolates of S. cerevisiae generally have fewer Ty elements than strains that have been subjected to extended periods of laboratory culture (Wilke et al. 1992), although the same may be true of C. albicans. Comparison of element diversity and copy number in recently isolated strains may be required to fully evaluate the role of sexuality/asexuality in the different patterns of transposable element evolution. However, consistent with the sexual state of the genome having an influence on transposable element diversity, only two retrotransposons (Levin et al. 1990; Weaver et al. 1993), and no DNA transposons, have been found in the well-characterized sexual yeast Schizosaccharomyces pombe, whereas many transposable elements have been found in the asexual filamentous fungus Fusarium oxysporum—at present there are at least 15 known families of F. oxysporum transposable elements, including four retrotransposons (Daboussi and Langin 1994; a search of sequences in GenBank).
Several of the full-length retrotransposons in C. albicanshave highly degenerate internal regions, suggesting that they are nonfunctional, yet have the identical LTRs and perfect TSDs characteristic of recently transposed elements. What could explain these apparent contradictions? Frequent gene conversion among the LTRs of these families is one possibility. If this were the case, however, the expectation would then be that most or all of the LTRs of these families would be highly similar, whereas, in fact, they are fairly heterogeneous groups. Gene conversion also doesn't account for the fact that these elements all have intact PBSs and PPTs as well. Rather, it seems likely that these elements have indeed transposed fairly recently. Presumably, this would occur via the mRNAs of these elements being processed by the products of other retrotransposons intrans. The minimum requirements fortrans-activation are that an element be transcribed and have an intact PBS, PPT, and mRNA-packaging signal, and that these be recognized by the trans-activating element. We know nothing about the mRNA-packaging signals of C. albicansretrotransposons, but all of the elements that appear to move viatrans-activation do have intact PBSs and PPTs, and Tca1, at least, is known to be transcribed (Chen and Fonzi 1992). We know of no other species in which retrotransposons that have degenerated to the extent of these C. albicans elements still appear to be capable of transposition. This trans-activation may go some way toward explaining the large number of retrotransposon families inC. albicans as it would allow dead elements, such as Tca1, to persist for much longer than would otherwise be expected.
The various families of elements in C. albicans appear to represent the full range of stages of retroelement speciation (Fig.3A). For some families, such as san or gamma, the members are all nearly identical. Other families, such as psi, include several elements that are very similar, but also some more divergent ones. Further families, such as pi, represent quite a diverse range of sequences and the members clearly fall into two distinct subfamilies. Then, there are closely related families of elements, such as phi and chi, followed by families such as lambda and zeta, which are related but probably diverged a long time ago. Finally, there are families, such assan and gamma, for which the LTRs are very different, but the PBSs and PPTs are similar in the full-length versions, suggesting that they are also related. These elements thus provide snap shots of retrotransposon speciation in action. Further analysis of such sequences may yield interesting insights into the speciation process. For example, the role of recombination, in particular, gene conversion, in maintaining sequence homogeneity and thus inhibiting speciation, may become apparent. Another example could be in determining whether positive selection ever has a role, for instance, in helping related elements to avoid competition for limited host factors, such as tRNA primers.
All of the families for which we have identified full-length and apparently intact members, gamma (Tca2), san (Tca4), and omega (Tca5), have very low levels of sequence diversity (Table 1), suggesting that they have arisen only recently. These elements are unlikely to have arrived via horizontal transmission, however, as they are clearly related to other C. albicanselements. Therefore, they must have arisen as a result of divergence from some progenitor element. These findings are consistent with the retrotransposons of C. albicans being in a state of flux with new elements being continually generated and diverging, whereas older elements become nonfunctional as a result of either random mutation or deletion via inter-LTR recombinations. The abundance of nonfunctional families suggests that the remnants of ancient elements are not efficiently removed from the genome, but rather persist and gradually diverge as random mutations accumulate.
Each of the Ty element families of S. cerevisiae has a preference for inserting at particular sites within the genome (Kim et al. 1998). These targeting mechanisms appear to direct the Ty elements to areas of the genome in which they are unlikely to corrupt an essential gene. It is of interest to determine whether elements inC. albicans also target particular sites in the genome. Unfortunately, the unfinished state of the C. albicans genome project with many genes, in particular tRNA genes, unrecognized, precludes an in-depth analysis of the target sites at this time. In a preliminary analysis of about one-half of the C. albicanselements, with just short (100- to 200-bp) flanking sequences, the only element for which a preference was apparent was beta. This element, like Ty3 (Hansen et al. 1988), appears to target the immediate upstream flanks of tRNA genes, as described previously (Perreau et al. 1997).
At the junction between the C. albicans subtelomeric repeats and the centromere-proximal unique sequences, there is often a region that is rich in LTRs and other transposable elements (Fig. 7A). The frequency of this region in the database suggests that it is likely to be present on half or even more of the chromosome ends. The different copies of this region share a similar underlying pattern of LTR insertions, suggesting that they all have been derived from some ancestral sequence via multiple rounds of duplication. Recombinations and additional transpositions during the duplication process have served to make each copy distinct. The frequent occurrence of this LTR-rich region at C. albicans subtelomeres prompts the question of whether it has any adaptive significance or whether theC. albicans chromosome ends are in a continual state of flux, and this just happens to be a prevalent structure at this time. Analysis of the subtelomeric regions of other strains and relatedCandida species may reveal the significance of such structures. Of interest, a recent report (Morschhauser et al. 1999) reveals that Candida dubliniensis, a very close relative of C. albicans, lacks sequences hybridizing toCARE-2, suggesting that the subtelomeric regions of these two species have diverged rapidly.
The arrangement of sequences in the subtelomeres suggests that recombinations between LTRs are a common occurrence in C. albicans. For instance, the subtelomeric kappa LTRs all the have the same upstream flanking sequence, but a variety of downstream flanks, suggesting several recombination events. Recombinations between LTRs at different locations in the genome has also apparently occurred. For instance, the arrangement of LTRs and genes in contig 2898 (Fig. 7A) is most easily explained by a recombination between a tara insert within a subtelomerickappa LTR, and a tara element at an intergenic location. The result of such an exchange would be that subtelomeric sequences located upstream of the recombination would subsequently be in an intergenic region and vice versa for the other region involved. Similarly, groups of corrupted and rearranged elements in other locations (Fig. 7B) bear witness to past genomic rearrangements involving retrotransposon sequences.
Our results suggest that the pattern of retrotransposon evolution inC. albicans has differed markedly from that of S. cerevisiae. We hope that further analyses of retrotransposon populations in these and related species will contribute to our understanding of more general aspects of genome evolution. We also hope that our description of the methods by which we identified and classified a wide variety of C. albicans retrotransposons will assist in the study of retrotransposons in other species.
METHODS
Candida albicans Sequence Analysis
Sequence data for C. albicans strain SC5314 was obtained from the Stanford DNA Sequencing and Technology Center website athttp://www-sequence.stanford.edu/group/candida. Sequencing of C. albicans at Stanford was accomplished with the support of the NIDR and the Burroughs Wellcome Fund. The Stanford C. albicanssequence databases were screened for retrotransposon sequences, as described in the Results section. LTRs found in the assembled data were labeled with a family name, for example sampi, the number of the contig on which they were found, for example, 1749, and a number to distinguish all of the elements of a family on a particular contig, usually 1. General sequence analysis and manipulation was performed with version 8.1 of the University of Wisconsin Genetics Computer Group sequence analysis package (Devereux et al. 1984). Chromatograms of sequences of interest were examined with EditView 1.0.1 and the sequences were edited where necessary. Phylogenetic trees were constructed either with the GCG programs Distances and Growtree, or the programs of the PHYLIP package (Felsenstein 1989). The complete set of full-length delta, sigma, and tau elements in the S. cerevisiae genome was retrieved from the Voytas laboratory website available athttp://www.public.iastate.edu/∼voytas/ltrstuff/ltrtables/yeast.html. Nucleotide diversity (π, Nei and Li 1979) for a set of aligned sequences is the average number of differences per site between each pair of sequences in the alignment and was calculated with Arlequin (Schneider et al. 1997). Gaps were scored as a fifth nucleotide state. Elements containing large deletions (>50 bp) were excluded from the analysis and similar large inserts were removed from those elements that had them. Only full-length LTRs were included in the trees and in the nucleotide diversity calculations. In determining the number of LTRs present in assembly 4, LTRs bisected by another element were counted as a single copy and partial elements were included. LTRs with identical flanking sequences could arise in a number of ways; they could be homozygous on the two homologs of a chromosome; they could have resulted from duplication of the chromosomal region on which they lie; they could be two independent insertions into a similar target; or they could be the result of the same region being present twice in the assembly. Unless there was evidence suggesting that the latter was not the case, such as substantial differences in the broader flanking regions, such elements were counted as a single insertion. The degenerate zeta LTRs present in the MRS were not included in the copy number estimates. Percent nucleotide identities are as scored by the alignment procedure of Chao et al. (1992) as implemented by the FASTA program within the GCG package. The C. albicanstRNAiMet and tRNAGln(UUG) genes were discovered by searching the C. albicans databases with the corresponding tRNA sequences from Candida utilis (Genbank accession no.K00323) and S. cerevisiae (Z00038), respectively. The C. albicans sequences detected in these searches were confirmed as tRNAs by use of the tRNAscan-SE program of Lowe and Eddy (1997). As the assembled sequence database at Stanford is updated, the numbering of the contigs changes. Therefore, to allow easy future access to the sequences described in this report, we have a complete copy of assembly 4 in our laboratory which can be made available upon request.
Representative LTRs from several families were cloned from SC5314 by PCR with the Expand high-fidelity PCR system (Boehringer Mannheim) and custom-designed oligonucleotide primers. Each primer pair gave a single major PCR product of the expected size. The identity of the cloned PCR products was confirmed by sequencing.
Southern Analysis
Candida genomic DNA was isolated by the method ofPhilippsen et al. (1991) from cells grown at 27°C in YPD medium (1% yeast extract, 2% peptone, 2% glucose). The DNAs were digested withEcoRI (Boehringer Mannheim), separated in a 0.8% agarose gel, then capillary transferred to a Hybond-N+ nylon membrane (Amersham) with 0.4 m NaOH. Probes were prepared by random-primed labeling of purified fragments with [α-32P]dCTP. Hybridization was performed in 0.36 mNa2HPO4, 0.12 mNaH2PO4, 1 m EDTA, and 7% SDS at 65°C overnight. Post-hybridization washes consisted of two 5 min washes in 2× SSC at room temperature and two 15 min washes in 0.2× SSC, 0.1% SDS at 65°C. The membrane was then rinsed in 2×SSC and exposed to X-ray film at −80°C. The membrane was stripped by rinsing in sterile water for 1 min, then washing twice, 10 min per wash, in 0.2 m NaOH, 0.1% SDS at 37°C.
Candida Strains
C. albicans strains used were hOG1042 (Matthews et al. 1997), SGY269 (Kelly et al. 1987), SC5314 (Gillum et al. 1984), ATCC10261 (Mackinnon and Artagaveytia-Allende 1945), SA40 (Agatensi et al. 1991), and F16932 (Goodwin and Poulter 1998). OtherCandida species used were C. parapsilosis (CDC MCC 499), C. tropicalis (CDC B397), and C. pseudotropicalis (CDC B2455), provided by the National Health Institute, Porirua, New Zealand, and C. maltosa (CHAU1, Ohkuma et al. 1993).
Accession Numbers
The GenBank accession numbers of each of the new LTRs are listed in Table 1. The accession numbers of the C. albicanstRNAiMet and tRNAGln(UUG) genes are AF069449 andAF180282, respectively.
Acknowledgments
Sequence data for C. albicans was obtained from the Stanford DNA Sequencing and Technology Center website athttp://www-sequence.stanford.edu/group/candida. Sequencing of C. albicans was accomplished with the support of the NIDR and the Burroughs Wellcome Fund.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL timg{at}sanger.otago.ac.nz; FAX 64 3 479 7866.
-
- Received September 28, 1999.
- Accepted December 9, 1999.
- Cold Spring Harbor Laboratory Press








