
The Genomic Region Encompassing the Nephropathic Cystinosis Gene (CTNS): Complete Sequencing of a 200-kb Segment and Discovery of a Novel Gene within the Common Cystinosis-Causing Deletion
- Jeffrey W. Touchman1,
- Yair Anikster2,
- Nicole L. Dietrich1,
- Valerie V. Braden Maduro3,
- Geraldine McDowell2,
- Vorasuk Shotelersuk2,
- Gerard G. Bouffard1,
- Stephen M. Beckstrom-Sternberg1,
- William A. Gahl2, and
- Eric D. Green1,3,4
- 1NIH Intramural Sequencing Center, National Institutes of Health, Gaithersburg, Maryland 20877; 2Heritable Disorders Branch, National Institute for Child Health and Development and 3Genome Technology Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892
Abstract
Nephropathic cystinosis is an autosomal recessive disorder caused by the defective transport of cystine out of lysosomes. Recently, the causative gene (CTNS) was identified and presumed to encode an integral membrane protein called cystinosin. Many of the disease-associated mutations in CTNS are deletions, including one >55 kb in size that represents the most common cystinosis allele encountered to date. In an effort to determine the precise genomic organization of CTNS and to gain sequence-based insight about the DNA within and flanking cystinosis-associated deletions, we mapped and sequenced the region of human chromosome 17p13 encompassingCTNS. Specifically, a bacterial artificial chromosome (BAC)-based physical map spanning CTNS was constructed by sequence-tagged site (STS)-content mapping. The resulting BAC contig provided the relative order of 43 STSs. Two overlapping BACs, which together contain all of the CTNS exons as well as extensive amounts of flanking DNA, were selected and subjected to shotgun sequencing. A total of 200,237 bp of contiguous, high-accuracy sequence was generated. Analysis of the resulting data revealed a number of interesting features about this genomic region, including the long-range organization of CTNS, insight about the breakpoints and intervening DNA associated with the common cystinosis-causing deletion, and structural information about five genes neighboringCTNS (human ortholog of rat vanilloid receptor subtype 1 gene,CARKL, TIP-1, P2X5, and HUMINAE). In particular, sequence analysis detected the presence of a novel gene (CARKL) residing within the most common cystinosis-causing deletion. This gene encodes a previously unknown protein that is predicted to function as a carbohydrate kinase. Interestingly, bothCTNS and CARKL are absent in nearly half of all cystinosis patients (i.e., those homozygous for the common deletion).
[The sequence data described in this paper have been submitted to the GenBank data library under accession nos.AF168787 and AF163573.]
Nephropathic cystinosis is a rare autosomal recessive, lysosomal storage disease with an incidence estimated at 1 per 100,000–200,000 live births (see http://www.ncbi.nlm.nih.gov/omim; OMIM 219800). The classic disorder is characterized clinically by renal tubular Fanconi syndrome in the first year of life, growth retardation in childhood, renal glomerular failure at ∼10 years of age, hypothyroidism, and a variety of other complications, including photophobia and corneal crystal formation (Gahl 1986; Gahl et al. 1995). After renal transplantation, cystine accumulation continues in nonrenal organs, frequently causing a distal vacuolar myopathy (Charnas et al. 1994), swallowing difficulty (Sonies et al. 1990), or retinal dysfunction (Kaiser-Kupfer et al. 1986), and occasionally causing diabetes mellitus (Fivush et al. 1987), pancreatic exocrine insufficiency (Fivush et al. 1988), or neurological deterioration (Ehrich et al. 1979; Fink et al. 1989). These complications arise because defective lysosomal transport of the disulfide cystine (Gahl et al. 1982a) causes this amino acid to accumulate within the lysosomes of many different cell types, which then triggers cystine crystal formation (Gahl et al. 1982b). The cystine transporter is the first of many lysosomal membrane carriers to be characterized biochemically (Thoene 1992), and cystinosis is the most common of a group of lysosomal transport disorders (Gahl et al. 1995).
The gene altered in patients with cystinosis (CTNS) was recently identified by a positional cloning strategy (Town et al. 1998). CTNS is a 12-exon gene that is transcribed into a ∼2.6-kb mRNA. The encoded protein, named cystinosin, consists of a predicted 367 amino acids, appears to be an integral membrane protein, and most likely functions as a cystine transporter. A number of cystinosis-causing CTNS mutations have now been reported (Shotelersuk et al. 1998a; Town et al. 1998). The most prevalent mutation reported to date is a large (>55-kb) deletion, with 33%–44% of affected patients being homozygous for this deletion (Town et al. 1998; Anikster et al. 1999). In addition, at least 11 other smaller disease-causing deletions have been reported (Shotelersuk et al. 1998a; Forestier et al. 1999), suggesting that this genomic region may be prone to rearrangement.
We sought to establish the long-range organization of the segment of chromosome 17p13 harboring CTNS and to determine the sequence of this clinically important gene and its surrounding DNA. Here we report the assembly of a detailed bacterial artificial chromosome (BAC)-based physical map encompassing CTNS. In addition, two BAC clones spanning >200 kb were sequenced to high accuracy, providing insight into the molecular architecture of the CTNSgene and the genomic segment commonly deleted in cystinosis patients.
RESULTS
Physical Mapping
Our goal was to construct a high-resolution, long-range physical map of the region of chromosome 17p13 containing CTNS. Specifically, we sought to isolate the region in overlapping BAC clones (Shizuya et al. 1992; Birren et al. 1999) and to order a large set of sequence-tagged sites (STSs) across the interval. Although this genomic segment has been isolated in yeast artificial chromosomes (YACs) (McDowell et al. 1996; Stec et al. 1996; Peters et al. 1997), few markers were available for BAC isolation and mapping. Consequently, we generated new STSs across the region using several sources of DNA sequence, including known genes (e.g., ASPA) and genetic markers (e.g., D17S2167, D17S2054, D17S1828), a YAC spanning the interval [CEPH YAC 767F9 (McDowell et al. 1996; Peters et al. 1997)], and BAC insert ends. Available human BAC libraries were screened by PCR- and hybridization-based methods for the available STSs. Following STS-content analysis, nascent contigs were assembled, and clones residing at contig ends were selected and used to derive additional BAC insert-end sequences. New STSs were developed from the latter and used to screen the BAC libraries again. This scheme was repeated in an iterative fashion, eventually allowing assembly of the contig map depicted in Figure 1.

BAC-based STS-content map of the region of chromosome 17p13 containingCTNS. A fully contiguous BAC contig map spanning the genomic segment encompassing CTNS is depicted, oriented with 17pter leftward and 17cen rightward. Shown along the top are the deduced positions of 43 STSs (spaced in an equidistant fashion from one another). Information about the STSs and their corresponding PCR assays is available in GenBank and/or GDB. Genetic markers are indicated by their D17S numbers, the one gene-specific STS by its assigned abbreviation (ASPA), and all the other STSs by their GenBank accession numbers. BACs are depicted as horizontal lines, with the length of each line reflecting the clone's STS content (as opposed to its insert size). The BAC names include the following prefixes reflecting the clone's library of origin: (RG) Research Genetics human BAC library; (GS) Genome Systems human BAC library; and (NH) Roswell Park Cancer Institute human BAC library RPCI-11. (●) The STS was verified to be present in that clone by PCR testing; (◼) the STS was derived from the insert end of that BAC. The two BACs subjected to complete sequencing (see Fig. 2), which together contain the entireCTNS gene, are contained within a dashed box.
The resulting BAC-based STS-content map contains 95 clones and provides ordering information for 43 STSs. The contig is estimated to span >1 Mb based on previous YAC-based mapping of the interval (McDowell et al. 1996; Peters et al. 1997). The average redundancy of BACs per STS is ∼14; such redundancy provides strong support for the indicated BAC overlaps and deduced STS order.
Genomic Sequencing
Two overlapping BACs (RG147P12 and RG87B10; see Fig. 1), which together contain the entire CTNS gene, were sequenced to an estimated accuracy of >99.99% by a shotgun sequencing strategy (Wilson and Mardis 1997). The clone inserts were found to be 68,220 and 138,720 bp in size, respectively, and to overlap by 6703 bp. Thus, a total of 200,237 bp of nonredundant sequence was generated (GenBank accession no. AF168787). Comparison of the sequence with a collection of known human repetitive elements revealed that this genomic region is relatively rich in repeats (constituting 42.6% of the total sequence), in particular short interspersed repetitive elements (SINEs).Alu repeats comprise nearly 30% of the sequence (Table1).
Repetitive Elements in the 200,237-bp Segment Encompassing CTNS
Genomic Organization of the CTNS Gene
Comparison of the CTNS cDNA sequence and the generated genomic sequence allows the precise structure of the gene to be deduced, including details about intron/exon organization (Table 2; Fig. 2). The publishedCTNS cDNA sequence (GenBank accession no. AJ222967) is distributed across 24,816 bp of genomic DNA [positions 72,070–96,885 (GenBank accession no. AF168787)]. This cDNA sequence matches our established genomic sequence throughout, except for a silent A:G substitution at nucleotide position 843 in exon 8 (of the cDNA sequence), the presence of an additional T residue at position 2273 in the 3′-untranslated region (UTR), and a G:A substitution at position 2594 in the 3′ UTR. Furthermore, based on the genomic sequence, intron 1 is 276 bp in length, shorter than that described previously (Town et al. 1998).
Intron/Exon Organization of CTNS andCARKL

Long-range organization of genes within the 200-kb interval encompassing CTNS. The positions and intron/exon organization of five genes detected in the genomic sequence are schematically depicted, with the 17p telomere (Tel) leftward and the centromere (Cen) rightward. In each case, the introns and exons are drawn to scale, with vertical bars reflecting individual exons and arrows indicating the direction of transcription. The general position of a sixth gene, the vanilloid receptor/SIC gene, is also depicted; intron/exon organization is not shown due to the lack of available human cDNA sequence. The positions of the two sequenced BACs (RG147P12 and RG87B10) and the common 57-kb cystinosis-causing deletion are also shown. Additional structural details about this sequence, including the location of human repetitive elements, are provided in GenBank accession no. AF168787.
Deletion Breakpoint Mapping
The breakpoints of the most common cystinosis-causing deletion were identified and sequenced in numerous cystinosis patients and reported previously (Anikster et al. 1999; Forestier et al. 1999). The availability of sequence data for the region encompassing CTNSallowed precise characterization of this deletion. Aligning the breakpoint sequences to the normal genomic sequence reveals that the common deletion spans 57,257 bp, notably smaller than the ∼65-kb estimate reported previously (Town et al. 1998). The 5′ (telomeric) deletion breakpoint occurs after nucleotide position 36,253 (GenBank accession no. AF168787). The 3′ (centromeric) deletion breakpoint occurs before nucleotide position 93,511 and interrupts exon 10 of theCTNS gene. Note that it cannot be determined whether the C nucleotide at the deletion junction originated from the 5′ or 3′ end of the deletion; thus, the breakpoint position at either end may be plus or minus one nucleotide. Whereas the regions immediately surrounding the deletion breakpoints are rich in Alurepetitive elements, the breaks themselves do not occur within these repeats.
Detection of a Novel Gene (CARKL) in the Common Cystinosis-Causing Deletion
Toward the telomeric end of the 57-kb segment commonly deleted in cystinosis is a region matching a series of expressed-sequence tags (ESTs; GenBank accession nos. AA70014, AA553482, AA618422, AA340511,AA331298, AA313538, and AA355260; see Fig. 3), three of which comprise a UniGene cluster (UniGene Hs.190207). These ESTs were derived from various tissues (including colon, fetal kidney, fetal liver/spleen, human embryo, Jurkat T cells, and Schwannoma tumor) and matched the genomic sequence with nearly 100% identity. In addition, gene-prediction programs indicate the presence of a seven-exon gene between CTNS and the matching ESTs (with the 3′ end of the predicted gene residing adjacent to the ESTs; see Fig. 3).

Genomic structure of CARKL. A detailed view of the intron/exon organization of CARKL is provided. Each of the seven exons is depicted to scale, with the hatched regions corresponding to the predicted ORF. Available 3′ and 5′ ESTs from the terminal exon (GenBank accession nos. indicated) are depicted. GRAIL2- and GENSCAN-predicted exons are indicated (top). The asterisks indicate cases where GRAIL2 incorrectly predicts the location of utilized splice sites. The CARKL mRNA sequence is provided in GenBank accession no. AF163573.
In light of its apparent presence within the genomic interval commonly deleted in cystinosis patients, we characterized this putative gene in greater detail. PCR primers were designed from the predicted exons and used in various combinations to amplify human fetal kidney cDNA. The resulting PCR products were sequenced, eventually allowing the assembly of 3838 bp of the mRNA (GenBank accession no. AF163573). Note that the sequence of the most upstream portion of exon 1 has not been determined. These results confirmed the presence of the gene [namedCARKL (carbohydratekinase-like); see below], which contains a 1434-bp ORF encoding a predicted 478 amino-acid protein. Both GENSCAN (Burge and Karlin 1997) and GRAIL2 (Xu et al. 1994) nicely predicted the intron/exon organization of CARKL, the details of which are now known based on the genomic and cDNA sequence data (Table 2). Northern analysis (Fig. 4) of CARKL revealed the strong expression of a ∼3.9-kb transcript in liver, kidney, and pancreas, weaker expression in heart and placenta, and very weak expression in brain and lung. In addition to the ∼3.9-kb mRNA, a ∼2.7-kb transcript was also detected in liver and, to a lesser extent, in heart.
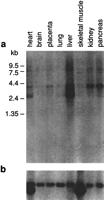
Expression profile of CARKL. A Northern blot containing 2 μg of poly(A)+ RNA from the indicated tissues was hybridized with a 1072-bp CARKL cDNA-specific probe spanning exons 2–7 (a) and then with a human β-actin-specific probe (b). Autoradiography was performed for 24 and 2 hr, respectively.
The predicted amino-acid sequence encoded by CARKL shows 30% identity and 42% similarity over 321 amino acids to the hypotheticalCaenorhabditis elegans protein T25C8.1 (GenBank accession no. Z83241) and 24% identity and 37% similarity across 320 amino acids to a Streptomyces rubiginosus xylulose kinase protein (GenBank accession no. P27156). CARKL has weak homology to several other carbohydrate kinases from a variety of species (data not shown). The predicted protein does not appear to contain a signal sequence, suggesting that it localizes in the cytoplasm. A search for protein motifs identified weak similarity to two domains of the FGGY family of carbohydrate kinases (PROSITE PS00933 and PS00445). Carbohydrate kinases are a class of proteins involved in the phosphorylation of sugars as they enter a cell, inhibiting return across the cell membrane (Worley et al. 1995). In light of the weak similarity to the carbohydrate kinases and the absence of a known substrate for the encoded protein, the gene was named CARKL.
Genes Neighboring CTNS and CARKL
By a combination of sequence database comparisons and computational gene predictions, three additional genes were detected in the 200-kb region immediately surrounding CTNS and CARKL (Fig.2; Table 3). At the telomeric end of this interval is the likely human ortholog of the rat vanilloid receptor subtype 1 gene (Caterina et al. 1997). Most of the gene is contained within the sequenced region. The encoded receptor, which is a cation channel whose ligands include capsaicin, functions as a transducer of pain stimuli. An alternative splicing variant of this gene, called the stretch-inhibitable nonselective cation channel (SIC), has been reported independently (Suzuki et al. 1999). At the centromeric end of the region resides most of the gene encoding the integrin αE precursor (HUMINAE). The mRNA sequence of HUMINAE has been established, with 3647 nucleotides (of 3927 total) identified in the genomic sequence. The integrin αE precursor is a component of a cell adhesion protein complex expressed on a subclass of T lymphocytes known as intraepithelial lymphocytes, which are interspersed among mucosal epithelial cells (Shaw et al. 1994). Also present in the sequenced interval are genes encoding the ionotropic ATP receptor (P2X5), a developmentally regulated gene expressed as two splicing variants (Le et al. 1997), and the Tax interaction protein 1 (TIP-1), a protein containing a PDZ domain that has been found to interact with the HTLV-1 Tax oncoprotein (Rousset et al. 1998).
Genes Within the 200-kb Sequenced Interval
EST T85505, reported previously to reside within the telomeric end of the common 57-kb cystinosis-causing deletion (Town et al. 1998), was analyzed in greater detail. This EST is part of a larger cluster (UniGene Hs.193738). All of the cDNA clones in this cluster were derived from fetal liver/spleen, and the sizes of the corresponding inserts are nearly identical (744–746 bp), based on overlapping the generated 5′ and 3′ ESTs with the genomic sequence. Searches against the public databases failed to identify significant matches to known genes or proteins. The 3′ ESTs in this cluster begin at a polyadenosine stretch located at the end of a partial Alusequence. No polyadenylation signal is found within the 3′ ESTs, and Northern analysis did not detect a transcript in multiple tissues tested (data not shown). Furthermore, GRAIL2 and GENSCAN failed to predict any exons or genes within the 15-kb interval surrounding these ESTs. It seems, therefore, that this T85505-specific sequence in 17p13 likely represents a pseudogene or an artifact of cDNA cloning.
DISCUSSION
The systematic sequencing of large genomic segments represents a powerful tool for revealing the long-range molecular architecture of biologically important chromosomal regions. In the study reported here, we have focused on the segment of human chromosome 17p harboringCTNS, the gene recently implicated in nephropathic cystinosis (Town et al. 1998). Specifically, following the construction of a detailed BAC-based physical map of the region, we generated >200 kb of high-accuracy genomic sequence from two overlapping clones that together contain the entire CTNS gene.
Our sequence data reveal the molecular structure, size, and intron/exon organization of CTNS, as well as insight about the size and sequence context of cystinosis-causing deletions. Our findings reveal that this genomic region is rich in Alu sequences. There is direct and circumstantial evidence that such repetitive motifs may have a role in other chromosomal rearrangements (Luzi et al. 1995; Harteveld et al. 1997; Super et al. 1997; Jeffs et al. 1998; Strout et al. 1998). One might speculate that such instability may have contributed to the genetic event leading to the common 57-kb deletion as well as the other described cystinosis-causing deletions, although a direct involvement of Alu repeats in these deletions has certainly not been established.
The comprehensive sequence data now available for CTNS should facilitate efforts to define the mutational spectrum associated with cystinosis. Already, this sequence has been used to characterize the breakpoints of the common deletion, allowing the development of a PCR assay for diagnosing individuals that are heterozygous or homozygous for that deletion (Anikster et al. 1999). This assay serves as the primary diagnostic tool for cystinosis in the Western Hemisphere, as nearly half of the known cystinosis alleles contain the 57-kb deletion. It should now be straightforward to determine the precise breakpoints in any cystinosis-associated deletion and to design suitable PCR assays for detecting such deletions, such as the second large cystinosis-causing deletion reported by Forestier et al. (1999). For cystinosis patients with splice-site mutations, the intronic sequence will permit the identification of cryptic or alternative splice sites and allow the design of primers for PCR amplification and sequencing of the intronic DNA flanking each exon.
Another use of the sequence has been demonstrated by the discovery of a number of genes flanking CTNS (Fig. 2). In principle, deletions affecting CTNS and any of these flanking genes may lead to more complex phenotypes than those encountered in conventional cystinosis patients; specifically, contiguous gene deletion syndromes may be recognized. In that regard, the most intriguing findings are those associated with the novel gene CARKL, which presumably encodes a carbohydrate kinase. Strikingly, CARKL is fully contained within the 57-kb region commonly deleted in cystinosis patients. Because nearly half of all known cystinosis patients are homozygous for this deletion, these individuals are devoid of both cystinosin and the CARKL-encoded protein. Once the function of the latter protein has been elucidated and its putative substrate(s) identified, it will be important to study the clinical features of cystinosis patients harboring different CTNS deletions (e.g., those with or without the common 57-kb deletion). It is possible that the presence/absence of CARKL may account for the clinical heterogeneity seen in cystinosis patients with respect to distal vacuolar myopathy (Charnas et al. 1994), nephrocalcinosis (Theodoropoulos et al. 1995), and other complications of the disease (Gahl and Kaiser-Kupfer 1987; Gahl et al. 1995). In this regard, we hypothesize that CARKL may be a modifier for the cystinosis phenotype.
The study of patients presumably lacking a carbohydrate kinase may also provide insight about the functional role of this putative enzyme and its associated biochemical pathway. Studies in human biochemical genetics often reveal pathways whose existence and function are elucidated by discovery of individuals lacking a key enzyme; theCARKL gene may provide the latest example.
METHODS
STS Generation
STSs were developed from the following sources of DNA sequence: (1) known genes and genetic markers; (2) plasmid subclones derived from random restriction fragments of CEPH YAC 767F9 [which spans the entire region harboring CTNS (McDowell et al. 1996; Peters et al. 1997)]; and (3) insert ends of isolated BACs. For generating the latter, BAC DNA was purified using an Autogen 740 Automated Nucleic Acid System (Integrated Separation Systems) and concentrated to 200 ng/μl using a Microcon-100 column (Millipore Corp., Bedford, MA). Fluorescent DNA sequencing was performed with the -40M13 universal primer (5′-GTTTTCCCAGTCACGAC-3′) or -28M13 reverse primer (5′-CAGGAAACAGCTATGACC-3′) and BigDye-terminator chemistry (Perkin Elmer/Applied Biosystems Division, Foster City, CA). The 20-μl sequencing reaction contained 11 μl of purified BAC DNA (at 200 ng/μl), 1 μl of primer (at 10 μm), and 8 μl of BigDye-reaction mixture. Thermal cycling was performed as suggested by the manufacturer. The products were then purified on a Centrisep column (Princeton Separations, NJ), dried, suspended in 2 μl of formamide loading buffer, and analyzed on an Applied Biosystems 377XL automated fluorescent sequencing instrument (Perkin Elmer/Applied Biosystems Division, Foster City, CA). For developing suitable STS-specific PCR assays, sequences were analyzed for repetitive elements, and apparently unique sequences were then used to design PCR primers using the computer program OSP (Hillier and Green 1991). PCR assays were optimized essentially as described (Green 1993). Information about the STSs and their corresponding PCR assays is available in GenBank and/or GDB.
BAC Contig Construction
BACs were isolated from the Research Genetics (http://www.resgen.com) and Genome Systems (http://www.genomesystems.com) human BAC libraries by PCR-based screening (according to the suppliers' instructions) and from the Roswell Park Cancer Institute human BAC library RPCI-11 (http://bacpac.med.buffalo.edu) by hybridization-based screening using STS-specific “overgo” probes (Vollrath 1999; seehttp://genome.wustl.edu/gsc). Positive clones were colony purified, and individual colonies were tested by PCR analysis. As nascent BAC contigs were assembled based on the STS content of the BACs, new STSs were developed from insert-end sequences derived from strategically selected BACs and used to isolate additional clones. This process was repeated in an iterative fashion. Our general strategy for constructing human BAC contigs has been reported previously (Ellsworth et al. 1999).
Genomic Sequencing
BAC clones RG147P12 and RG87B10 were sequenced to high accuracy using a shotgun sequencing strategy (Wilson and Mardis 1997). Briefly, purified BAC DNA (http://genome.wustl.edu/gsc/Protocols/BAC.shtml) was kinetically sheared with a nebulizer (CIS-US, Inc., Bedford, MA), and the resulting fragments were end-repaired with T4 DNA polymerase and Klenow and then subcloned into plasmid pBC (Stratagene, La Jolla, CA) and M13mp18 vectors. Randomly selected subclones were sequenced from one (M13mp18) or both (pBC) ends to a final estimated average redundancy of 10-fold. Fluorescent sequencing reactions were performed with BigDye-terminator (Perkin Elmer/Applied Biosystems Division, Foster City, CA) and energy transfer (ET) dye-primer (Amersham-Pharmacia Biotech, Piscataway, NJ) chemistries, and the resulting products analyzed with Applied Biosystems 377XL automated fluorescent sequencing instruments. Individual sequences were edited and assembled using the Phred/Phrap/Consed suite of programs (Gordon et al. 1998; Ewing and Green 1998; Ewing et al. 1998) to a final estimated error frequency of <1 in 104 bp as determined by Phrap and Consed. The validity of each sequence assembly was confirmed by the concordance of forward and reverse sequencing reads from individual plasmid subclones and by alignment with known cDNA sequences.
Sequence Analysis
Genomic sequence was analyzed for the presence of known human repetitive elements using the program RepeatMasker (http://ftp.genome.washington.edu/cgi-bin/RepeatMasker) and Crossmatch (http://www.genome.washington.edu/UWGC/analysistools/swat.htm) (A.F.A. Smit and P. Green, unpubl.). Sequence comparisons with public databases were performed with PowerBLAST (Zhang and Madden 1997) using the following parameters: BLASTN (M = 1, N = −3, S = 40, S2 = 40) and BLASTX (S = 90, S2 = 90, FILTER = SEG). The results from PowerBLAST were collated and viewed using Sequin (Benson et al. 1997). As part of our sequence analysis process, the gene prediction programs GRAIL2 (Xu et al. 1994) and GENSCAN (http://ccr-081.mit.edu/GENSCAN.html) (Burge and Karlin 1997) were used to identify putative genes. Protein motifs were identified using the MOTIF tools (http://www.genome.ad.jp/SIT/MOTIF.html), whereas prediction of signal peptides was performed using the PSORT program (http://psort.nibb.ac.jp).
cDNA Sequencing
Fragments of the CARKL cDNA were generated by PCR amplification of human fetal kidney cDNA (Clontech, Palo Alto, CA) using primers designed from GENSCAN-predicted exons (details available on request). The resulting DNA fragments were sequenced using BigDye-terminator chemistry as described above, eventually allowing assembly of the cDNA.
Northern Analysis
A 1072-bp CARKL-specific DNA probe was generated by PCR from human fetal kidney cDNA (Clontech, Palo Alto, CA) with primers 5′-GAGTAGAATCCTCCAAGCCCTACAC-3′ and 5′-GAAGCATGGAGTGCAGGTTCTG-3′ (see GenBank accession no.AF163573 for corresponding positions within the cDNA sequence). The resulting PCR product was radiolabeled with [α-32P]dCTP (NEN Life Science Products, Boston, MA) and hybridized to a human multiple tissue Northern blot (Clontech, Palo Alto, CA) as described (Shotelersuk et al. 1998b).
Acknowledgments
Y.A. is a Howard Hughes Medical Institute Physician Postdoctoral fellow. We thank M. Furgusson, E. Sorbello, A. Cunningham, A. Gupta, R. Torkzadeh, C. Varner, and M. Walker for excellent technical assistance with DNA sequencing as well as John McPherson and the staff of the Washington University Genome Sequencing Center for assistance in BAC isolation. We also thank Drs. A. Baxevanis, L. Biesecker, L. Everett, W. Gan, and C. Jamison for general advice, assistance, and/or critical review of the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL egreen{at}nhgri.nih.gov; FAX (301) 402-4735.
-
- Received October 21, 1999.
- Accepted December 13, 1999.
- Cold Spring Harbor Laboratory Press








