
Abstract
Isoprenoid compounds are ubiquitous in living species and diverse in biological function. Isoprenoid side chains of the membrane lipids are biochemical markers distinguishing archaea from the rest of living forms. The mevalonate pathway of isoprenoid biosynthesis has been defined completely in yeast, while the alternative, deoxy-D-xylulose phosphate synthase pathway is found in many bacteria. In archaea, some enzymes of the mevalonate pathway are found, but the orthologs of three yeast proteins, accounting for the route from phosphomevalonate to geranyl pyrophosphate, are missing, as are the enzymes from the alternative pathway. To understand the evolution of isoprenoid biosynthesis, as well as the mechanism of lipid biosynthesis in archaea, sequence motifs in the known enzymes of the two pathways of isoprenoid biosynthesis were analyzed. New sequence relationships were detected, including similarities between diphosphomevalonate decarboxylase and kinases of the galactokinase superfamily, between the metazoan phosphomevalonate kinase and the nucleoside monophosphate kinase superfamily, and between isopentenyl pyrophosphate isomerases and MutT pyrophosphohydrolases. Based on these findings, orphan members of the galactokinase, nucleoside monophosphate kinase, and pyrophosphohydrolase families in archaeal genomes were evaluated as candidate enzymes for the three missing steps. Alternative methods of finding these missing links were explored, including physical linkage of open reading frames and patterns of ortholog distribution in different species. Combining these approaches resulted in the generation of a short list of 13 candidate genes for the three missing functions in archaea, whose participation in isoprenoid biosynthesis is amenable to biochemical and genetic investigation.
A challenge for computational biology in the “post-genomic” era is to reconstruct cellular metabolic pathways, in as much detail as possible and appropriate, by analysis of genome sequences. If genes in a given pathway have been characterized in one species, and a newly sequenced species has the complement of the orthologous genes, characterized by high sequence similarity and consistent position in the phylogenetic tree (Tatusov et al. 1996;Eisen 1998; Yuan et al. 1998), then modeling of the unknown metabolism is straightforward. However, many instances of gaps in metabolic pathways have been reported, especially when arbitrary similarity cutoffs were imposed in the database searches. Comparative protein sequence analysis, with the attention to both close and more distant similarities, remains the principal method of function prediction and pathway reconstruction (Tatusov et al. 1996; Koonin et al. 1997; Bork et al. 1998). In the attempt to break through the “similarity barrier”, complementary approaches have been proposed, including identification of genes that are physically close, and presumably coordinately regulated, in multiple genomes (Overbeek et al. 1999), and the use of phyletic profiles, i.e., the occurrence in some genomes of sets of genes that are missing in others (Tatusov et al. 1997;Pellegrini et al. 1999). We are interested in applying all available methods to reconstruct pathways in poorly characterized species, in order to develop a highly automated strategy facilitating this process and to get an insight in general and specific trends in the evolution of biochemical pathways.
More than 25,000 naturally occurring isoprenoid derivatives are known, including such important classes of bioactive compounds as vitamin A and related light-capturing and ultraviolet-protecting carotenoids, steroids that modify lipid membranes and participate in signal transduction in eukaryotes, phytols that form the membrane-anchoring side chains of chlorophyll, and plant protective isoprenoids (Eisenreich et al. 1998). Pathways of isoprenoid biosynthesis are perturbed in such human diseases as mevalonic aciduria (OMIM Entry 251170) and hyperimmunoglobulinaemia D with periodic fever syndrome (OMIM Entry 260920; Houten et al.1999a,b), but can be modified to the advantage of human health, as in the case of inhibition of HMG-CoA reductase by cholesterol-reducing drugs (Moghadasian 1999). From an evolutionary perspective, isoprenoids are notable as a biochemical marker that distinguishes archaeal lipids from bacterial and eukaryotic lipids, which have fatty acids as the side chains (Kates 1993).
The mevalonate pathway of isoprenoid biosynthesis provides isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP), the essential five-carbon building blocks from which the more complex isoprenoids are formed. Genetic determinants for the complete mevalonate pathway for IPP synthesis have been defined in yeast (Saccharomyces cerevisiae), followed by the biochemical analysis of the individual gene products. An overview of the mevalonate pathway and its genetic control in yeast is shown in Figure1.
The mevalonate and deoxy-D-xylulose (DXP) pathways of isoprenoid biosynthesis. Gene names and GenBank accession nos. for the prototype yeast proteins of the mevalonate pathway are shown. Green shading indicates genes orthologous to the yeast prototypes. Gene displacements are shown in yellow or, when the replacing enzymes have not been characterized, in red. Blue shading indicates the enzymes of the DXP pathway. No shading indicates that these functions are more likely to be absent in a given genus. Compounds are indicated by Roman numerals: I, acetyl-CoA; II, acetoacetyl-CoA; III, hydroxy-3-methylglutaryl-CoA; IV, mevalonate; V, phosphomevalonate; VI, diphosphomevalonate; VII, isopentenyl pyrophosphate; VIII, dimethylallyl pyrophosphate; IX, geranylpyrophosphate; X, pyruvate; XI, glyceraldehyde 3-phosphate; XII, 2-deoxy-D-xylulose 5-phosphate; XIII, 2C-methyl-D-erythritol 4-phosphate; XIV, 4-diphosphocytidyl-2C-methyl-D-erythritol; XV, 4-diphosphocytidyl-2C-methyl-D-erythritol 2-phosphate; XVI, 2C-methyl-D-erythritol 2,4-cyclodiphosphate; XVII, isopentenyl monophosphate. Arabic numerals indicate enzymes. Mevalonate pathway: 1, acetoacyl-CoA synthetase; 2, hydroxy-3-methylglutaryl-CoA synthase; 3, hydroxy-3-methylglutaryl-CoA reductase; 4, mevalonate kinase; 5, phosphomevalonate kinase; 6, diphosphomevalonate decarboxylase; 7, isopentenyl pyrophosphate delta-isomerase; 8, geranyl pyrophosphate synthase family (the † sign indicates that orthologs and paralogs are not well distinguished in this family, which is compatible with the observation that substrate specificity of these enzymes is modulated easily by small number of point mutations). DXP pathway: 9, deoxy-D-xylulose phosphate synthase; 10, deoxy-D-xylulose phosphate reductoisomerase; 11, 2C-methyl-D-erythritol 4-phosphate cytidylyltransferase (YgbP); 12, isopentenyl monophosphate kinase; 13, 2C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (YgbB).
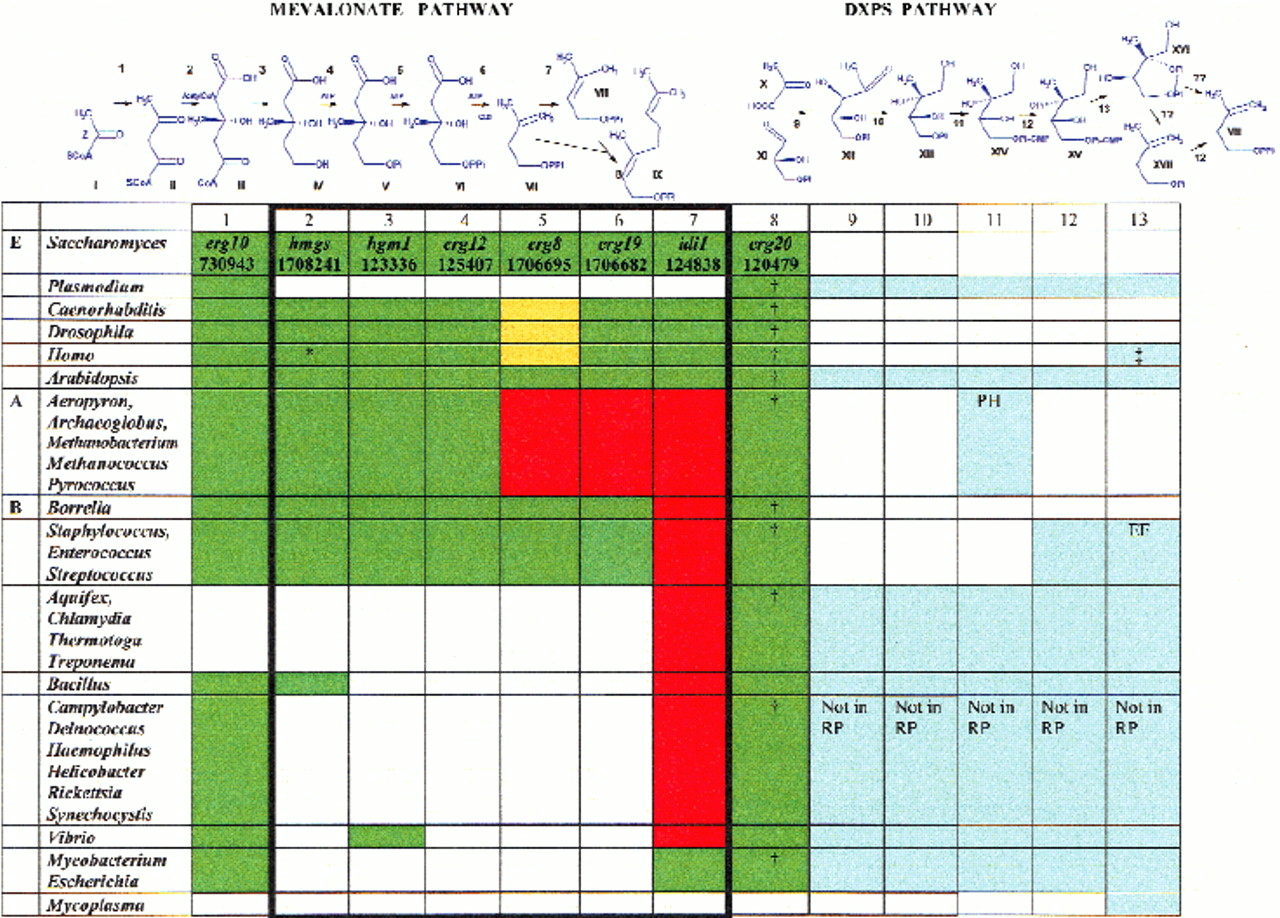
The central role of isoprenoids in living cells has prompted the search for the enzymes of the mevalonate pathway in different species. Biochemical evidence has suggested the occurrence of this pathway in various eukaryotes and in archaea, and its apparent substitution in bacteria by an alternative route involving biosynthesis and reductive isomerization of deoxy-D-xylulose phosphate (DXP) (Eisenreich et al. 1998; Fig. 1).
We used comparative sequence analysis to show that most of the species synthesizing isoprenoids lack one or more of the orthologs of the yeast mevalonate pathway genes. In particular, the route from phosphomevalonate to geranyl pyrophosphate, requiring three separate enzymes in yeast, has not been found in archaea. In an attempt to find the missing links in archaea and to reconstruct the evolution of isoprenoid biosynthesis, we analyzed the sequences of the known mevalonate pathway proteins, positional linkage of relevant genes, and patterns of ortholog distribution in completely sequenced genomes. Frequent functional takeovers, i.e., gene replacements by a gene with the same function but distantly related or unrelated sequence, appear to be a major problem in reconstructing metabolism of a poorly studied species. In such cases, combination of computational approaches enables significant reduction of the space of gene candidates, providing short lists of targets for biochemical characterization.
RESULTS AND DISCUSSION
Defining the Orthologs: Only Higher Plants Have the Full Complement of Yeast Mevalonate Pathway Enzymes
We searched DNA and protein sequence databases for candidate orthologs of yeast mevalonate pathway enzymes and Escherichia coli DXP pathway enzymes in completely sequenced or extensively covered genomes. The results of the ortholog definition are shown in Figure 1.
Inspection of the committed portion of the mevalonate pathway, leading from acetoacetyl-CoA to the two main isoprenoid precursors IPP and DMAPP (columns 2–7), revealed substantial differences between evolutionary lineages. Higher plants contain orthologs of all relevant yeast genes. Genes coding for the enzymes comprising the alternative DXP pathway also are present in higher plants, in agreement with the biochemical evidence of both pathways in green algae and in dicots (Disch et al. 1998; Lange et al. 1998; Lange and Croteau 1999).
In most completely sequenced bacterial genomes, the mevalonate pathway is missing and is substituted by the DXP pathway. However, in some bacteria, such as the completely sequenced spirochete Borrelia burgdorferi and the almost completely covered gram-positive cocci, most of the mevalonate pathway genes are found, with the exception of isopentenyl diphosphate isomerase. Another case of a “one-bit” difference with the yeast pathway is presented by Metazoa, including the completely sequenced Caenorhabditis elegans andDrosophila melanogaster, both of which have a single phosphomevalonate kinase unrelated to the yeast enzyme. In archaea, the orthologs for only three of six committed enzymes could be found, leaving phosphomevalonate kinase (PMK), diphosphomevalonate decarboxylase, and isopentenyl diphosphate isomerase (IPPI) unaccounted for. Given the biochemical evidence that biosynthesis of the side chains of archaeal lipids proceeds via the mevalonate route, at least in the case of halophiles (Kates 1993; Tachibana et al. 1996), this gap in the downstream portion of the trunk pathway needs to be explained.
Detailing the Motifs: New Sequence Relationships for Three Enzymes in the Mevalonate Pathway
It has been shown that mevalonate kinases and the yeast PMK belong to a large superfamily, which also includes galactokinases and homoserine kinases (Bork et al. 1993). Recently, a role in the DXP pathway has been proposed for another member of this superfamily, called YchB in E. coli (indicated by 12 in Fig. 1), which is capable of forming IPP by phosphorylation of an isopentenyl monophosphate (IMP) precursor, and also phosphorylates the essential intermediate phosphocytidyl-2-C-methylerythritol (Lange and Croteau 1999; Luttgen et al. 2000). We found that related sequence motifs are present in diphosphomevalonate decarboxylases. A PSI-BLAST search initiated by a putative mevalonate kinase sequence fromEnterococcus faecalis, retrieved, at the second iteration, a diphosphomevalonate decarboxylase sequence from Arabidopsiswith a probability of matching by chance of 10–4. Other members of the galactokinase family also were observed in these searches. All kinases (EC 2.7) in the superfamily transfer a phosphate to a hydroxyl group on a substituted tetra-, penta-, or hexacarbon linear scaffold. Diphosphomevalonate decarboxylases work on a similar substrate, but belong to a different enzyme class (EC 4.1.1.33). An explanation for a common origin of these decarboxylases and kinases is provided by the understanding of the decarboxylation mechanism, in which the 3′- position of diphosphomevalonate undergoes ATP-dependent phosphorylation, followed by elimination of phosphate and CO2 (Dhe-Paganon et al. 1994). Thus, affinities for ATP and a substituted aliphatic chain are the common denominators for the whole superfamily.
Multiple sequence alignment (Fig.2A) revealed four conserved sequence motifs in the galactokinase superfamily. A role in binding of ATP gamma-phosphate has been suggested for the lysine-13 residue of rat mevalonate kinase, based on affinity mapping and site-directed mutagenesis (Potter et al. 1997a). This residue, found within motif I (marked by an asterisk in Fig. 2A), is conserved in many kinases but replaced in diphosphomevalonate decarboxylases and in a subset of uncharacterized archaeal proteins (Fig. 2A). Another residue in rat mevalonate kinase, aspartate-204, is thought to serve as the base that facilitates proton extraction from the substrate (Potter et al. 1997b). This amino acid (asterisk in motif 3, Fig. 2A) is substituted by a similarly nucleophilic serine in decarboxylases. Glycine-rich loops commonly form the phosphate-binding sites in ATP-dependent enzymes (e.g., Bork and Koonin 1994), and motifs 2 and 4 are notable candidates for such a function. Verification of the specific roles of the four conserved motifs awaits the site-directed mutagenesis of additional representatives of this family and crystallographic studies.
Conserved sequence motifs in mevalonate pathway enzymes. Blocks of high-sequence similarity are shown. Unique identifiers in SWISSPROT or GenBank are given for each sequence. Yellow shading indicates conserved bulky hydrophobic residues (I, L, F, M, V, Y, and W), red type indicates conserved small side chain residues (A, G, and S), and blue type indicates other conserved residues. Secondary structures predicted with reliability of eight or higher (PHD program) are shown; h indicates a helix, and s indicates a strand. (A) Diphosphomevalonate kinase belongs to the galactokinase superfamily. Secondary structures for yeast phosphomevalonate kinase (ERG8) and diphosphomevalonate decarboxylase (ERG19) predicted with reliability of eight or higher are shown. (B) Conserved ATP-binding motifs of nucleotide monophosphate kinase type in metazoan phosphomevalonate kinases. Secondary structure elements observed in the T4 bacteriophage deoxynucleoside monophosphate kinase (pdb code 1DEL) and predicted for human phosphomevalonate kinase (PMKA_HUMAN) are shown. Green shading indicates residues located within 3å distance from the bound ADP. (C) MutT-like pyrophosphate-binding motifs in isopentenyl pyrophosphate delta-isomerases. Secondary structure elements observed in Escherichia coli MutT protein (pdb code 1TUM) and predicted for yeast IPPI (IDI1_YEAST) are shown.

The known PMK enzymes encoded by metazoan genomes are unrelated to the galactokinase family, and their evolutionary origin was unclear. We found that metazoan PMK are distantly related to nucleoside monophosphate kinases found in all superkingdoms of life and in viruses. The database searches revealed moderate similarity (p = 10–3 upon first-time passing the cutoff in PSI-BLAST analysis) between animal PMK, uncharacterized proteins encoded by the catfish herpes virus, and the deoxynucleoside monophosphate (NMP) kinase of bacteriophage T4. The latter protein shares significant sequence similarity and the same three-dimensional fold with a number of nucleoside monophosphate kinases (Teplyakov et al. 1996). The region of highest sequence similarity in nucleoside kinases (motif I in Fig.2B) also is found in many other ATP-binding proteins where it corresponds to a Walker-type P-loop (Koonin 1993). It is modified in the bacteriophage kinase and in metazoan phosphomevalonate kinases, but the essential lysine residue is conserved (Fig. 2B). The C-terminal motif II in T4 deoxynucleoside monophosphate kinase consists of a strand, a turn, and a helix, which comprise the grasp holding the adenine moiety of the bound nucleoside phosphate (Teplyakov et al. 1996). Only marginal sequence similarity between nucleoside monophosphate kinases and PMK was detected in the middle, substrate-binding regions of these proteins, reflecting the difference in the phosphorylation targets.
Another new sequence relationship was observed in the case of IPPI. At the second PSI-BLAST iteration, statistically significant matches (p <10–6) were detected between the C-terminal halves of IPPI and the MutT family of nucleoside pyrophosphatases (Fig. 2C). Multiple sequence alignment of MutT-like proteins and IPPI, and analysis of the known solution structure of the E. coli MutT protein indicate that the conserved motifs correspond to the alpha helix I and connected loops (Mildvan et al. 1999). In the MutT protein, these structural elements are involved in interactions with the bound nucleotide and with divalent metal cations essential for the nucleophilic attack on the pyrophosphate bond. Although a pyrophosphate bond is present in isopentenyl pyrophosphate, its hydrolysis would be unexpected of and not sufficient for the isomerase reaction. Possibly, MutT-like motifs in the IPPI enzymes are catalytically inefficient and are used to bind the pyrophosphate moiety of the substrate, while the isomerization requires the aid of the unique N-terminal domain.
Reconstructing the Pathway in Archaea: Paralogous Candidates for Missing Functions
Archaeal genomes encode members of each of the three newly delineated superfamilies. For example, the genome of Methanococcus jannaschii codes for at least five members of the galactokinase superfamily, six members of the nucleoside monophosphate kinase family (as well as a large number of remotely related kinases and ATPases with Walker-type P-loops), and one member of the MutT/IPPI superfamily. The biochemical functions of these proteins cannot be established by database searches alone; nevertheless, for a subset of archaeal proteins, orthologous relationships with biochemically characterized proteins from other species could be reliably inferred by analysis of best matches. In many cases, however, ortholog definition was complicated by nontransitive relationships and/or domain rearrangements between proteins. Therefore, blocks of high-sequence similarity were collected from the alignments of each of the three superfamilies, and phylogenetic trees were constructed from four concatenated blocks, in order to trace the evolutionary relationships of some of the “orphan” members of the families of interest.
Using this approach, we were able to reliably resolve most branches in the galactokinase superfamily tree (bootstrap support 75% or higher). The known galactokinases, homoserine kinases, mevalonate kinases, phosphomevalonate kinases, isopentenyl monophosphate kinases, and diphosphomevalonate decarboxylases each form a distinct cluster on the tree. In the case of the five M. jannaschii proteins in this superfamily, the initial assignments of gi 2497515 to the homoserine kinase family and gi 2497517 to the mevalonate kinase family1 were confirmed. The three remaining paralogs (gi 2128859, gi 3024949, and gi 3183371) belong to ancient, archaea-specific clusters. Distribution of orphan paralogs in other archaea is very similar to what is observed in M. jannaschii(Fig. 3).
(A) Phylogenetic tree of the galactokinase superfamily. The results of neighbor joining analysis are shown, which fully correspond to the maximum likelihood data. The tree was built with 116 galactokinase superfamily members, all < 90% identical to one another. Forty-seven less-informative proteins have been weeded out afterwards. Thick lines indicate a bootstrap value >75% for the corresponding node. Eukaryotic branches are in blue, eubacterial in green, and archaeal in red lines. Three orphan archaeal-specific families within the superfamily stand out, and may include phosphomevalonate kinases, isopentenyl monophosphate kinases, and, less likely, diphosphomevalonate decarboxylases (see text).(B) Blowup of the central region of the tree in Fig. 3A. Numbers indicate the bootstrap support, out of 1000 replicates, for the deep branches in the tree.

The exact biochemical function of any member of these clusters is unknown, and it is possible that one of them is the missing PMK (or, in a convergence with the DXP pathway, the IMP kinase). None of the paralogs groups together with either the PMK or IMP kinase (IMPK) families; in fact, two of the paralogs are slightly closer related to homoserine kinases. Thus, if one of these proteins indeed performs either PMK or IPMK function, this would mean that members of two different families within the same superfamily have been recruited to perform the same biochemical reaction on at least two independent occasions. However, this assumption is plausible, as similar scenarios have been documented for other enzymes, including sugar kinases from the unrelated families (Bork et al. 1993) and aminoacyl-tRNA synthetases (Koonin and Aravind 1998). The identifiers of these archaeal candidates for a missing kinase activity are listed in Table1.
Short List of Candidates that May Replace the “Missing” Enzymes of Mevalonate Pathway in Archaea, Borrelia, and Gram-Positive Cocci
| Missing function | Suggested candidates | Criteria for selection | Comments |
| Phosphomevalonate kinase (archaea) | Family of proteins related to uridylate and acetyl glutamate kinases 3258052_Pyrho Y044_METJA 2621082_Metth 2648231_Arcfu 5105458_Aerpe | Linked to mevalonate kinase in all archaea, similarly to the linkage observed between mevalonate kinase and phosphomevalonate kinase in Borellia and cocci; isolated phylogenetic position. | A more likely candidate. |
| Family of orthologs within galactokinase superfamily 3258263_Pyrho 3024949_Metja 2621655_Metth 2648904_Arcfu 5105835_Aerpe | Sequence similarity and isolated phylogenetic position within the superfamily. | ||
| Family of orthologs within galactokinase superfamily 2128859_Metja 2621895_Metth 2850101_Arcfu 5105835_Aerpe | Sequence similarity and isolated phylogenetic position within the superfamily. | No orthologs in two of three Pyrococcus species. | |
| Family of orthologs within galactokinase superfamily YE27_METJA 3256616_Pyrho 3257645_Pyrho Y830_METTH YK89_ARCFU 5106179_Aerpe 5105197_Aerpe | Sequence similarity and isolated phylogenetic position within the superfamily. | P. horikoshii andAeropyrum have a set of paralogs, generally not seen for mevalonate pathway enzymes. | |
| Family of orthologs within nucleoside monophosphate kinase superfamily 3257422_Pyrho 2128835_Metja 2621499_Metth 2649179_Arcfu | Sequence similarity and isolated phylogenetic position within the superfamily. | Closer to CMP/UMP kinases and may be required for the nucleotide kinase function (judged from the numbers of paralogs with such specificity in bacterial genomes). | |
| Diphosphomevalonate decarboxylase (archaea) | Homologs (probably paralogs) of SAM decarboxylase from E. coli 5103467_Aerpe 2648951_Arcfu 2495908_Metja 3258436_Pyrho | Elimination of other decarboxylases in archaea, either as misannotations, or because they are predicted to have a clearly unrelated biological function. | Related genes in some bacteria, but an internal deletion in archaeal genes suggests that the substrate in archaea may be different. No homolog in M. thermoautotrophicum. |
| Isopentenyl pyrophosphate delta-isomerase (archaea, Borrelia,cocci) | FMN-dependent dehydrogenases, including glycolate oxidase (NCBI COG1304) 2688617_Borbu 5105455_Aerpe 2648236_Arcfu 1591547_Metja 2621084_Metth 3257619_Pyrho | In Borrelia,found within an “operon” with all other enzymes of mevalonate pathway; neighbors with relevant functions in some archaea. Consistent phyletic pattern, although paralogs are widespread. | Oxidoreductase activity predicted by sequence similarity. In plants, some oxidoreductases are known to have a desaturase activity, which, in this case, could be exploited to isomerize the double bonds in IPP. |
| Uncharacterized family (NCBI COG1916) 2688316_Borbu 2650316_Arcfu 2129184_Metja 2622291_Metth 3257569_Pyrho | Exceptionally consistent phyletic pattern in microorganisms (Borrelia, Enterococcus, only one additional bacterium, Treponema, and archaea except forAeropyrum). | In Enterococcus, plasmid-borne copy is involved in response to peptide pheromone, which controls conjugation. Orthologs in plants, C. elegans and humans. | |
| Uncharacterized family (NCBI COG0327) 2688374_Borbu 2648773_Arcfu Y927_METJA 726074_Metth 3257033_Pyrho | Phyletic pattern (archaea+ Borrelia), although orthologs are found in many bacteria. | Presence of several invariant histidines suggests that this may be a metalloenzyme. | |
| Uncharacterized family (NCBI COG0061) 2688218_Borbu 5104774_Aerpe 2650718_Arcfu 2621965_Metth 2826350_Metja 3257490_Pyrho | Phyletic pattern (archaea+ Borrelia), although orthologs are found in many bacteria. | Glycine-rich (phosphate-binding?) motif shared with diacylglycerol kinases and 6- phosphofructokinases. | |
| Undecaprenyl diphosphate synthase (NCBI COG0020) UPPS_BORBU 5105068_Aerpe UPPS_ARCFU UPPS_METJA UPPS_PYRHO | Phyletic pattern (archaea+ Bacteria except for Mycoplasmae), note that the DXPS-pathway bacteria may need such activity, too (see text). | Multiple functions in the same pathway would have to be assumed; no biochemical evidence. |
| Missing function | Suggested candidates | Criteria for selection | Comments |
| Geranylgeranyl pyrophosphate synthase beta-subunit 2688215_Borbu 5105454_Aerpe 4633648_Arcfu IDSA_METJA IDSA_METTH 3257488_Pyrho | Phyletic pattern (archaea+ Bacteria except for Mycoplasmae), note that the DXPS-pathway bacteria may need such activity, too (see text). | Multiple functions in the same pathway would have to be assumed; no biochemical evidence. | |
| TPR-repeat proteins including geranylgeranyl pyrophosphate synthase alpha-subunit (large NCBI COG 0457) | Phyletic pattern (archaea+ Bacteria except for Mycoplasmae), note that the DXPS-pathway bacteria may need such activity, too (see text). | Multiple functions in the same pathway would have to be assumed; no biochemical evidence. |
Could an alternative PMK in archaea have been recruited from the NMP kinase superfamily? Members of this superfamily are present in each of the completely sequenced archaeal genomes, but they typically are more similar to NMP kinases than to the taxonomically heterogeneous group of PMK-like proteins (data not shown). No conservation between the substrate-binding domains of the latter group of proteins and the middle regions of the archaeal paralogs could be detected. Thus, an orphan NMP kinase-like protein seems a less likely candidate for phosphomevalonate kinase function in archaea. Representative proteins, however, are provisionally included in the short list of alternatives in Table 1.
Suggesting a candidate for the diphosphomevalonate decarboxylase function in archaea based on sequence similarity to the yeast prototype is no less challenging. Orphan paralogs remain in the galactokinase superfamily even after reserving one homolog per archaeal genome for the phosphomevalonate kinase activity. Arguing against these candidates, however, is the fact that the C-terminal halves of eukaryotic diphosphomevalonate decarboxylase sequences are unique for this family of enzymes, and they do not retrieve any other classes of sequences in database searches. Thus, it is not clear whether any orphan member of the galactokinase superfamily in archaea is endowed with all sequence elements required for the diphosphomevalonate decarboxylase activity.
Yet another missing step of the mevalonate pathway in archaea is isopentenyl delta-pyrophosphate isomerase. Although MutT-like domains in IPPI enzymes were detected in this study, and MutT-domain proteins are found in archaea (Fig. 2C), all of the archaeal proteins are shorter than the known IPPI proteins, which contain apparently unique domains in addition to the MutT-like pyrophosphate-binding sites. Thus, the IPPI function is still unaccounted for in archaea. IPPI orthologs also are missing from the completely sequenced genome of B. burgdorferi and from the extensive sequence databases of gram-positive cocci.
Alternative Approaches Towards Detection of the “Missing Link”—Combining the Evidence
Detection of homologs and partitioning them into orthologs and paralogs are the first steps in the reconstruction of metabolic pathways in a biochemically uncharacterized species. Whenever clear candidates cannot be identified by such approach, alternative strategies are needed. It has been suggested that, as secondary and tertiary structures of proteins may be better conserved than their sequences, these higher-order structures, known or predicted, should be compared directly (Aurora and Rose 1998; Pennec and Ayach 1998;Lehtonen et al. 1999). For example, proteins with an experimentally determined MutT-like fold, unrecognizable at the sequence similarity level, would be attractive candidates for the IPPI function. Unfortunately, very few three-dimensional structures of archaeal proteins have been resolved, while use of inferred secondary structures of proteins for the same purpose produced ambiguous results (e.g.,Aurora and Rose 1998; A.R. Mushegian, unpubl.).
Recently, additional techniques of inferring functional links between proteins were suggested. Enright et al. (1999) and Marcotte et al. (1999) compiled databases of orthologous domains that exist as stand-alone open reading frames (ORFs) in some species, yet are fused into multidomain proteins in others. These authors reasoned that a function for an uncharacterized domain may be predicted based on its fusion to a better-studied domain (the Rosetta stone approach). Analysis of domain organization was part of our ortholog definition procedure (see Methods). Notwithstanding the two domain fusions noted previously, one in the mevalonate pathway, where mammalian HMG-CoA reductases contain additional conserved membrane domain implicated in cholesterol sensing (Tseng et al. 1999), and another in DXP pathway, where some bacteria have YgbP and YgbB fused into a polyprotein (Herz et al. 2000), we did not find any “Rosetta stones” that could help to find missing candidates in archaea or Borrelia.
Analysis of gene colinearity in completely sequenced microbial genomes has shown that the long-range conservation of the order of orthologous genes on a chromosome is observed only between closely related species (Tatusov et al. 1996; Overbeek et al. 1999). At the other extreme, several small sets of orthologous genes are arrayed in the same order in most bacteria and archaea, and analysis of the products encoded by these universally conserved operons suggests that they code for the subunits of stoichiometric protein complexes (Mushegian and Koonin 1996; Dandekar et al. 1998). On the medium evolutionary scale, relative stability of gene linkages may indicate functional coupling (Overbeek et al. 1999).
We analyzed ORFs located close to the known genes of mevalonate pathway in bacterial and archaeal genomes. Interestingly, in Borrelia, an operon-like string (gi 2688613–gi 2688618) contains five ORFs belonging to the mevalonate pathway, and also the sixth product (BB0684; gi 2688617), annotated in the database as a carotenoid biosynthesis protein. The latter annotation comes from an uncharacterized homolog in Erwinia , which was found in an operon with the known genes of carotenoid biosynthesis, including geranylgeranyl pyrophosphate synthase.
This observation prompted us to look for similar proteins at the vicinity of the genes involved in the mevalonate pathway in bacteria and archaea. The results of this analysis are shown in Figure4. Strikingly, in most cases the orthologs of BB0684 gene were found adjacent to one or more genes involved in the mevalonate pathway or in the downstream step of isoprenoid biosynthesis (annotated as octaprenyl-diphosphate synthase, but exact specificity of this geranyl-diphosphate synthase homolog is not known; it is not unlikely to be the geranyl-diphosphate synthase itself — see footnote to Table 1 and, in more detail, Wang and Ohnuma 1999). The BB0684 were conspicuously close to these relevant genes in four of the five archaeal genomes (euryarchaeotes M. jannaschii,Methanobacterium thermoautotrophicum, and Archaeoglobus fulgidus, as well as the phylogenetically more distant crenarchaeote Aeropyrum pernix), and also in theStreptococcus/Enterococcus lineage of the gram-positive bacteria. Orthologs of BB0684 were therefore added to the short list of the candidates for the IPPI function.
Conserved strings include genes of the mevalonate pathway in archaea and bacteria. Blocks connected by an arrow indicate neighboring genes with a common transcriptional orientation, possibly representing operons. Blocks containing numbers represent known mevalonate pathway genes, as in Figure 1. Blocks designated as 2 indicate hydroxy-3-methylglutaryl-CoA synthase; 3 indicates 3-hydroxy-3-methylglutaryl-CoA reductase; 4 indicates mevalonate kinase; 5 indicates yeast-like phosphomevalonate kinase; 6 indicates diphosphomevalonate decarboxylase; and 8 indicates octaprenyl-diphosphate synthase, a member of the geranyl pyrophosphate synthase family. Other designations: A indicates ancient conserved protein (COG #1355), K indicates putative kinase related to uridylate- and acetylglutamate kinases, Cindicatescarotenoid biosynthesis protein (flavin-dependent oxidoreductase), and H indicates putative metal-dependent hydrolase. Pyrococcus abyssi has the same structure as Pyrococcus horikoshii, with one gene insertion between ancient conserved protein and mevalonate kinase. InStreptococcus pyogenes, genes 2 and 3 are flanking the mevalonate kinase operon, but are transcribed in opposite orientation. The mevalonate kinase gene has not been sequenced yet inSulfolobus solfataricus. The putative metal-dependent hydrolase has no orthologs in Aeropyrum andArchaeoglobus. GenBank identification nos. are given below the boxes, where available. Apparently-missing GI numbers in strings correspond to overlapping genes, typically short open reading frames (ORFs) on the opposite strand.
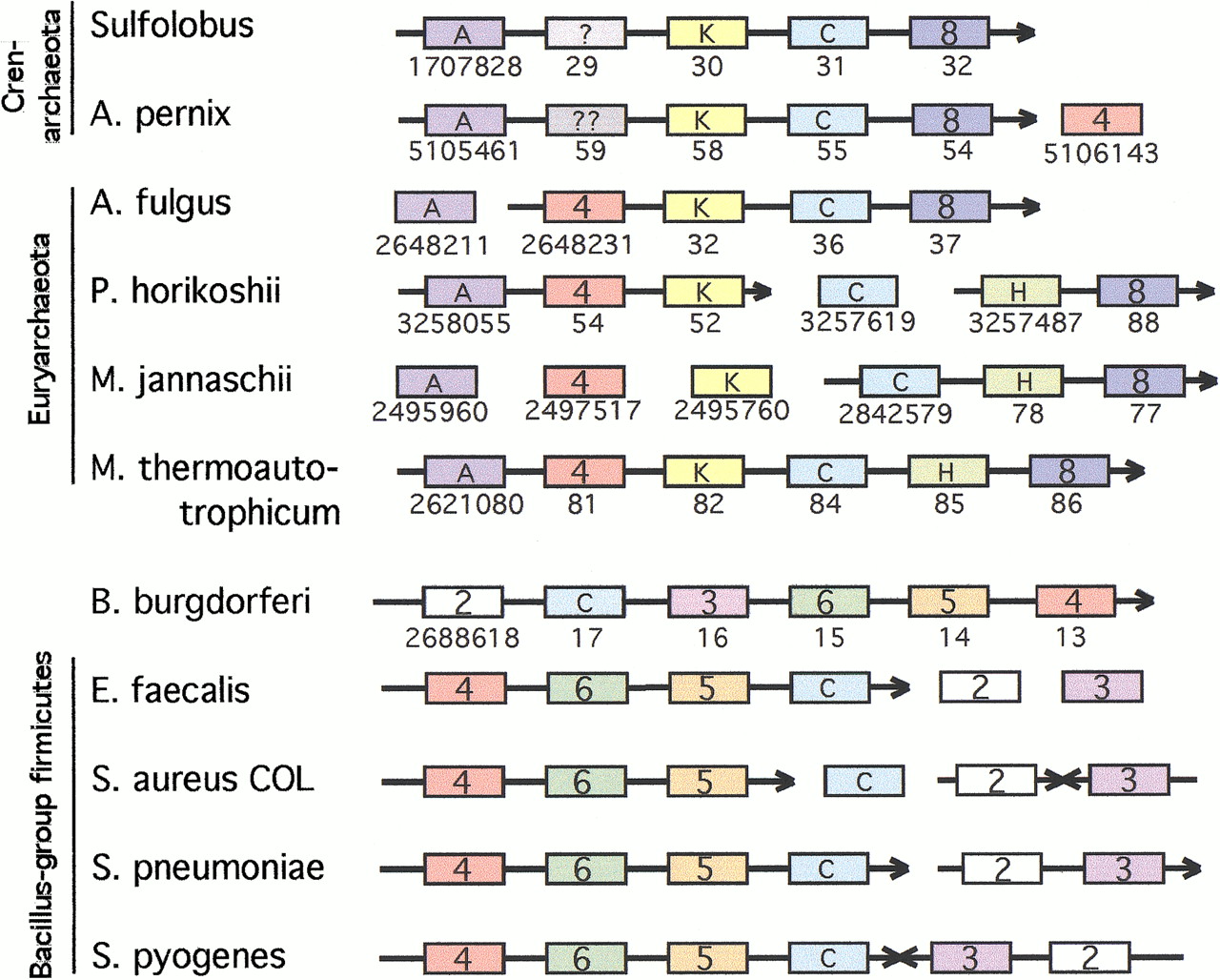
In archaea, two other sets of orthologs frequently are found close to the mevalonate pathway genes (Fig. 4). Both these sets of archaeal orthologs have only paralogs, but no orthologs, in bacteria. First, a predicted metal-dependent hydrolase with beta-lactamase-like fold (Aravind 1998) was found adjacent to the BB0684 ortholog and/or to octaprenyl diphosphate synthase in three genomes. The importance of this linkage is not clear, as there are no orthologs for this gene inA. pernix and A. fulgidus, and the predicted hydrolase activity seems to be incompatible with the required chemistry. Second, a putative kinase, related distantly to acetylglutamate kinases and uridylate kinases, was found adjacent to the mevalonate kinase and/or BB0684-like gene in four of five cases (Fig. 4). Some of these linkages also have been reported by the WIT database. Considering that PMK and mevalonate kinase always appear in the same operon in the five bacterial species (Fig. 3), and that the precedent of borrowing PMK from another family is set by Metazoa (Fig.2B), this putative kinase is a plausible candidate for the PMK or IPMK function in archaea (Table 1).
Yet another approach for predicting gene function relies on delineating groups of orthologous proteins in completely sequenced, phylogenetically diverse genomes. Using the sets of defined orthologs, one can derive phyletic patterns, i.e., regular expressions that encode the presence or absence of orthologous proteins in different genomes (Tatusov et al. 1997; Pellegrini et al. 1999). It has been predicted that these patterns, especially the uncommon ones, will be of use for metabolic reconstructions (Tatusov et al. 1997). A catalog of co-occurrences has been produced showing that, predictably, if a set of proteins occurs together in some but not all species, at least subsets of such proteins are known to act together (Pellegrini et al. 1999).
A convenient way to search for all orthologs shared by archaea andBorrelia is provided by the Clusters of Orthologous Groups (COGs) resource (http://www.ncbi.nlm.nih.gov/COG). We detected 165 COGs defined as having members in Borrelia and all four archaea represented in the COG database. Many of these groups contain omnipresent proteins with known functions irrelevant for our purposes (e.g., ribosomal proteins, chaperones, ion channels, or some well-studied metabolic enzymes) and were excluded from the further consideration. We also removed large families of closely related proteins, on the premise that putative IPPI will have only distant, if any, homologs within a given genome, similarly to what is observed with the known IPPIs and some other genes in the pathway. Only four COGs remained on the list, including the COG1304 to which BB0684 and its orthologs belong (Table 1).
Although the BB0684 family was pinpointed by two complementary approaches, i.e., by gene neighborhood analysis and phyletic pattern analysis, the results of sequence analysis of this family are less straightforward. Indeed, a flavin-dependent oxidoreductase activity is confidently predicted for these proteins based on similarity with well-studied glycolate oxidases (http://www.ncbi.nlm.nih.gov/COG/aln/COG1304.aln). An oxidoreductase activity would be appropriate in the context of the DXP pathway, for transitioning from 2-C-methyl-erythritol 2,4-cyclodiphosphate to IMP (see below), but it is difficult to accommodate a redox reaction within a path from phosphomevalonate to IPP or DMAPP. Interestingly, however, several plant desaturases have been shown to possess oxidoreductase activity (e.g., Hugueney et al. 1992; Rahier et al. 1997). Desaturases convert single C—C bonds in the hydrocarbons into the double bonds, and one can speculate that this type of reaction might be utilized for the rearrangement of the double bonds upon IPP and DMAPP interconversion.
Recent discovery of isopentenyl monophosphate kinase in plants and bacteria suggests that the mevalonate pathway and the DXP pathway may converge at the formation of IPP (Lange and Croteau 1999; Fig. 1). Accordingly, IPPI activity should be expected in all DXP-pathway bacteria, and yet only Escherichia and Mycobacteriumhave the orthologs of yeast IPPI. Is it possible that other completely sequenced bacteria and archaea have an IPPI enzyme nonorthologous to the yeast one? We constructed a phyletic pattern, using the “no” operator for two mycoplasmas, the “yes” operator for most of the other completely sequenced bacteria and archaea, and a relaxed requirement, i.e., either presence or absence, for yeast,Escherichia, Mycobacterium, and Rickettsia(the latter species lacks many of the DXP pathway enzymes [Fig. 1]). Interestingly, there are only eight COGs with such slightly degenerate phyletic pattern. While members of five COGs (0177, 0608, 0750, 0681, and 0504) have established functions unrelated to isoprenoid biosynthesis, three remaining COGs, present in all species but mycoplasmas, are geranylgeranyl pyrophosphate synthase beta subunit (COG0142; #8 in Fig. 1), undecaprenyl pyrophosphate synthase (COG0020), and TPR-repeat-containing proteins including alpha subunit of geranylgeranyl pyrophosphate synthase (COG0457). Is it possible that, in addition to transferring IPP, some of these enzymes moonlight as IPP isomerases? Members of these COGs are included in Table 1 as additional candidates for the IPPI function.
Yet another possibility is that archaea lack IPPI altogether. Notably, this may not hold true in bacteria, as DMAPP is required for modification of bacterial tRNA, performed by isopentenyl-tRNA synthase, the product of Mod5 gene in yeast (Benko et al. 2000). Whereas the archaea lack the orthologs of Mod5 and also do not seem to have the corresponding tRNA modification (http://medstat.med.utah.edu/RNAmods/), the orthologs or Mod5 are found in all completely sequenced bacteria except for the mycoplasmas, and in gram-positive cocci (A.A. Smit and A. Mushegian, unpubl.). This suggests that DMAPP is in fact formed in most bacteria, even those that do not have a conventional IPPI. Appropriate phyletic pattern searches produce lists of more than 40 candidate COGs, some comprising proteins with uncharacterized functions. A definitive solution for the case of the missing IPPI in archaea and some bacteria will only be possible after determination of the structure of C5 and C10isoprenoids in the species differing in the presence of the yeast-type IPPI enzyme.
We also tried to predict the diphosphomevalonate decarboxylase replacement in archaea. Examination of gene positions in archaeal genomes produced no results, and a sufficiently specific phyletic pattern could not be derived. Therefore, we inspected archaeal proteins with similarity to various decarboxylases. Two protein families, annotated as decarboxylases, in fact never have been shown to possess this activity (gi 2129193 and gi 2129181 in M. jannaschii; these proteins appear to belong to the phosphopentomutase and FAD-dependent oxidoreductase families, respectively). A number of real decarboxylases found in archaea have well-defined roles in different metabolic pathways. Of interest, however, was a family homologous to S- adenosyl methionine (SAM) decarboxylase from E. coli (Table1), with members found in all sequenced archaea except M. thermoautotrophicum, but only in a few bacteria (e.g.,Escherichia, Xanthomonas, Bacillus,Thermotoga, and Aquifex). It is likely that this family of decarboxylases has a distinct specificity in archaea (Table1).
Altogether, combination of computational approaches produced a list of 13 protein families, which are the candidates for the three missing functions (Table 1). This list is short enough as to allow a direct biochemical evaluation of the candidates.
Evolution of Isoprenoid Biosynthesis
Evolution of lipid side chain biosynthesis in various kingdoms of life has not been reconstructed in detail yet. Archaea seem to be unique in using the mevalonate pathway to build isoprenoid lipid side chains, and several archaeal proteins in this pathway can be identified by close similarity to the eukaryotic prototypes. On the other hand, displacements of individual orthologs as well as of whole cascades of orthologs coding for isoprenoid biosynthesis enzymes are detected in many species with extensively sequenced genomes. Under such circumstances, the evolution of a pathway cannot be straightforwardly reconstructed, as different genes in a pathway have different phylogenetic histories.
As a first approach in reconstructing the complex evolutionary history of isoprenoid biosynthesis and lipid side chain formation, one may consider what it would take to build the DXP and mevalonate routes. As with any function, the challenge is to propose a mechanism plausibly explaining the persistence of the intermediate steps until they are linked into a coherent pathway.
Could the switch from auxotrophy to the IPP biosynthesis have been achieved via the DXP pathway? The first enzyme in this pathway, DXP synthase, is a transketolase-like enzyme (Lois et al. 1998; Lange et al. 1998) that uses widely available glyceraldehyde phosphate and pyruvate as substrates. DXP is converted into 2-C-methylerythritol 4-phosphate by DXP reductoisomerase, an enzyme that could have been recruited by merging a unique C-terminal half with a glycine-rich N-terminal domain related to the dinucleotide-binding site of many oxidoreductases (Takahashi et al. 1998). 2-C-methylerythritol 4-phosphate in E. coli is converted into 4-diphosphocytidyl-2-C-methyl-erythritol by the YgbP protein (Rohdich et al. 1999), a nucleotidyltransferase of the large and widespread GlmU family (A.R. Mushegian, unpubl. obs.). The enzyme implicated in the next step of IPP biosynthesis (phosphorylation of 4-diphosphocytidyl-2-C-methyl-erythritol) as well as the last step of IPP synthesis, IMP kinase YchB, was apparently recruited from the diverse galactokinase superfamily. The 4-diphosphocytidyl-2-C-methylerythritol 2-phosphate cyclase YgbB appears to be unrelated to other proteins. To convert 2-C- methylerythritol 2,4-cyclodiphosphate into IMP, yet uncharacterized cyclohydrolase and oxidoreductase activities may be required.
Thus, at least two domain inventions of YdbP and DXP reductoisomerase C-terminal domain are required for the DXP pathway, but the functional relevance of these and other intermediate steps in the absence of the complete path remains unclear.
The mevalonate pathway could have emerged in several steps. First, HMG-CoA synthase and HMG-CoA reductase-like enzymes may have been selected for catabolic utilization of exogenous mevalonate-like carbon sources, analogous to what is observed in the present-day bacteriumPseudomonas mevalonii (Jordan-Starck and Rodwell 1989). The former enzyme belongs to a ubiquitous family of acyltransferases, while the latter enzyme family appears to be a unique group of oxidoreductases, unrelated to other enzymes. Next, the trio of enzymes concerned with anabolic utilization of mevalonate may have emerged by recruitment of galactokinase superfamily members to perform mevalonate kinase, phosphomevalonate kinase and, following domain acquisition from yet another unknown source, diphosphomevalonate decarboxylase activities, completing the path.
The evolution of the mevalonate pathway thus also had to involve several “leaps”. These include acquisition of the unique domains in HMG-CoA reductase and diphosphomevalonate decarboxylase. Moreover, four successive reactions in the mevalonate pathway either had to emerge simultaneously in a “super-leap” in anabolic direction, or needed a chance to evolve gradually, i.e., using some mechanism for selection-free tryouts of the incomplete versions of the pathway. Whichever of the two alternative pathways of isoprenoid biosynthesis became complete first, its advent would allow such tryouts to the other pathway. Combination of a MutT-like enzyme with another unique domain would have enabled isomerization of exogenous IPP and DMAPP-like precursors, the process that could precede the emergence of both pathways.
To proceed from these general considerations towards the reconstruction of events that may have happened in different evolutionary lineages, we note that mevalonate and DXP pathways, or their telltale representatives HMG-CoA reductase and DXP reductoisomerase, occur in most species to the exclusion of one another, except in higher plants, where both pathways are present but compartmentalized in cytoplasm and chloroplasts, respectively (recently sequenced genome of Vibrio cholerae encodes both enzymes, but lacks the other components of the mevalonate pathway while having the complete DXP pathway). Therefore, admittedly without any insight into the possible physiological or mechanistic reasons for such steady exclusion, we assume that the two pathways cannot coexist in one cell, and emergence of one complete pathway in the background of the other has to result in a (random) loss of one of the two. Thus, once both paths are completed in a cell, one of them, i.e., the DXP path in the lineage leading to eukarya and archaea, or the mevalonate path in bacteria, is discarded. This and later events in the evolution of isoprenoid biosynthesis and lipid side chain formation are summarized in Figure5.
Frequent horizontal transfers in the evolution of isoprenoid biosynthesis. Alternative pathways and displacements are color coded, mostly according to Fig. 1. Evolution of fatty acid biosynthesis has not been dissected in detail but is also shown for comparative purposes. Green lines, mevalonate pathway; blue lines, DXPS pathway; yellow, displacement of phosphomevalonate kinase in metazoans; red arrow and red-and-green line, displacement of three enzymes of mevalonate pathway, resulting in the chimeric pathway in archaea. Gray arrows, transfer of fatty acid biosynthesis genes to eukaryotes (solid line) and to selected archaea (double line). Green double line represents the transfer of HMG-CoA reductase to Vibrio cholerae, apparently from archaea (Heidelberg et al. 2000). The donor of the mevalonate pathway to Borrelia and cocci is unknown; a yet undiscovered isopentenyl diphosphate isomerase enzyme in these species may be shared with other bacteria (blue checkers) or with archaea (red checkers).
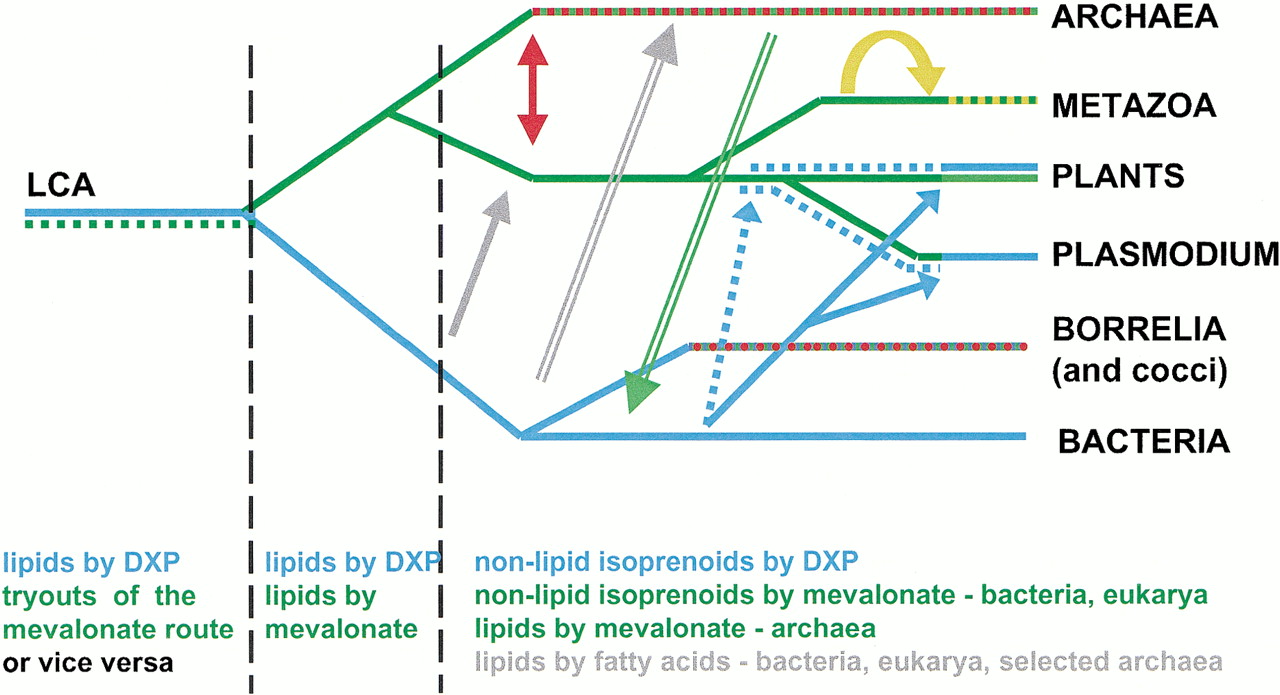
Within the archaea/eukaryotes, a displacement of three genes occurred via one of two scenarios. It is not clear whether the eukaryote-type pathway experienced displacements in the archaeal lineage before the divergence of Crenarchaeota and Euryarchaeota, or the archaeal-type pathway was the ancient one, and displacements occurred in the eukaryote lineage.
In the subsequent evolution of isoprenoid biosynthesis at least three horizontal transfers of whole pathways seem to have occurred (Fig. 5). The known eukaryotic recipients of the DXP pathway arePlasmodium lineage, green algae and higher plants. Apicomplexan protozoa, including Plasmodium, share a common ancestor with algae and plants and indeed have a complement of the plant-like genes encoded by their nuclear genomes (Roos et al. 1999), so it is possible that the transfer occurred in the common ancestor of these taxa. The fate of the two pathways, however, is different in plants and Plasmodium. In the former, both DXP and mevalonate pathway were retained and compartmentalized, while in the latter, the mevalonate pathway was probably displaced (completion of thePlasmodium genome project is awaited to confirm this).
At least two transfers of the mevalonate pathway into bacteria, followed by displacement of the DXP pathway, are also discernible. Bacterial recipients of the mevalonate pathway include the spirochaete lineage (B. burgdorferi) and a polyphyletic subset of gram-positive bacteria, Streptococcus, Enterococcus, and Staphylococcus (but not Bacillus to which the latter genus is related more closely than to the former two). Interestingly, sequences of mevalonate pathway genes from these bacteria are deep branches on the phylogenetic trees (Fig. 3 and data not shown), and they do not cluster with the orthologs from animal genomes, which would seem an obvious source of horizontally transferred genes. This tree topology could, in principle, be explained by accelerated sequence evolution in the respective bacterial lineages, but the observation that PMK in these bacteria is of the yeast type, thus far not detected in animals, seems to support the alternative hypothesis, i.e., that the acquisition of the mevalonate pathway byBorrelia and cocci was from species other than their present-day animal hosts or vectors.
In addition to these displacements of groups of genes, lineage-specific displacements of individual orthologs within the mevalonate pathway continued to happen, in particular displacement of galactokinase-like phosphomevalonate kinase by an NDK-related enzyme in metazoans, and additional replacements of IPPI in a subset of bacteria.
When were isoprenoid derivatives recruited to serve as lipid side chains? Any scenario of this event has to explain several puzzling observations. First, whereas two pathways of isoprenoid biosynthesis seem to be mutually exclusive, each of them can coexist with fatty acid biosynthesis, as illustrated by the gene repertoires of most bacteria and even some archaea, such as Aeropyrum andArchaeoglobus (the special case of the enzymes for fatty acid metabolism in these two species will be discussed elsewhere); why, then, do many archaea, including Methanococcus,Methanobacterius, and Pyrococcus, have only isoprenoids, but not fatty acids? Second, most biosynthetic enzymes in archaea are closer to their bacterial counterparts than to eukaryotic ones, suggesting a large-scale horizontal exchange of metabolic genes between bacterial and archaeal lineages (Koonin et al. 1997; Wolf et al. 1999). In contrast, archaeal enzymes of isoprenoid biosynthesis (reactions 1–4 in Fig. 1) typically exhibit the Woesean affinity between archaeal and eukaryal orthologs, similar to the informational proteins involved in such processes as DNA replication or protein biosynthesis (Koonin et al. 1997; Rivera et al. 1998). Why is the archaeal way of lipid side chain biosynthesis such an exception, i.e., why has it not been displaced by a bacterial-type set of enzymes?
Analysis of hyperthermophilic bacteria Thermotoga andAquifex suggests that an extreme environment might not require isoprenoid lipids (Fig. 1). Therefore, it is possible that the absence of fatty acids in many archaea is historical, rather than functional. At the time of massive gene exchange, both archaeal and bacterial lineages could have had isoprenoid lipid side chains, using mevalonate and DXP pathways, respectively, as the default biosynthetic routes. Because of the incompatibility of the DXP and mevalonate pathways, the former failed to establish itself in archaea. Fatty acids could have emerged later in the bacterial lineage, and may have been adopted by eukaryotes and by some but not all archaea as a result of symbiogenetic or lateral transfer of bacterial genes.
Conclusions
With dozens of genomes already sequenced completely, computational reconstruction of the metabolic pathways becomes a central step of genome analysis. Prediction of a pathway and analysis of its evolution are medium-scale tasks, which link functional and evolutionary interpretation of individual protein sequences with the whole-genome modeling, and were referred to recently as modular biology (Hartwell et al. 1999). We attempted to define one such biological module by predicting candidates for three consecutive reactions of the mevalonate pathway in archaea, and to gain insight into the evolution of isoprenoid biosynthesis and the lipid side chain formation in various kingdoms of life. Despite taking several complementary methods to their limits, we could not close on self-evident candidates for any of these reactions. However, such computational methods were capable of producing prioritized short lists of targets amenable to subsequent biochemical and genetic investigation.
The widespread phenomenon of displacement of orthologous genes (Koonin et al. 1996; Galperin et al. 1998) is the main reason why computational reconstruction of metabolic pathways remains difficult, despite the formidable progress in experimental biochemistry, development of metabolic databases, and computational analysis of genomes. The evolution of the mevalonate pathway is an example of how even a relatively short route can be diversified by repeated displacements. In most cases, it is unclear if a displacement is a “frozen accident”, or whether it confers a selective advantage as a result of biochemical differences between the old and the new enzymes. Even in the absence of such information, displacements may shed light on various aspects of biology, as may be the case with Borrelia where comparative analysis of mevalonate pathway enzymes raises the possibility of ancient association with nonanimal hosts. On a general note, our analysis of the mevalonate route illustrates the interplay of functional convergence (displacement of orthologs) and divergence (recruitment of related sequences to perform different functions) in the evolution of biochemical pathways.
Displaced genes may be good targets for species-specific intervention. Indeed, fosmidomycin and its derivative FR-900098 inhibit DXP reductoisomerase, and has been recently shown to possess anti-Plasmodium activity in vitro (Jomaa et al. 1999). Along the same lines, the yeast-type phosphomevalonate kinase inBorrelia, which thus far appears to have only distant paralogs in humans, or the replacement for IPPI, when it is identified, might be promising targets for anti-Lyme Disease therapy.
METHODS
Databases and Sequence Similarity Searches
Sequence databases at National Center for Biotechnology Information (NCBI) were used for all searches, including the nonredundant sequence database (NR), the EST database (dbest), and the database of unfinished microbial genomes. For database searches, we used the BLAST family of programs, including PSI-BLAST (Altschul et al. 1997). The WiseTools (Birney et al. 1996) were used to search dbest using Hidden Markov Models obtained from the aligned sequences.
The following databases were used as sources of biochemical information: KEGG (http://www.genome.ad.jp/kegg), Biochemical Pathways index of Boehringer-Mannheim (http://www.expasy.ch/cgi-bin/search-biochem-index), and WIT (http://igweb.integratedgenomics.com/IGwit/).
Ortholog Definition
Criteria for distinguishing orthologs from paralogs have been proposed (Tatusov et al. 1996, 1997; Yuan et al. 1998). Essentially, in two completely sequenced genomes, orthologs are each other's best matches when a protein encoded by one genome is used to scan the database of protein sequences encoded by the other genome. Orthologs from two evolutionary lineages have to identify the same gene/protein in the third lineage. Orthologs typically align along their whole lengths; insertions-deletions of globular domains longer than 60 amino acids were not allowed in our work, except for the full-length fusions of two or more functional enzymes in some species. This approach is similar to the one used in the COG database of orthologous groups from complete genomes (Tatusov et al. 2000), but some sets of orthologs defined in this study do not have corresponding COG (for example, phosphomevalonate kinase or diphosphomevalonate decarboxylase). Moreover, some COGs include both orthologs and paralogs within a large superfamily, while in this study the clustered partitions of the superfamily tree could have been treated separately (e.g., a subset of genes within COG 1304. See Table 1 and text for discussion).
Phylogenetic Methods
Multiple sequence alignments were produced using the programs ClustalX (Thompson et al. 1997) and MACAW (Schuler et al. 1991). Regions without noticeable sequence conservation were removed from the alignments, and the remaining ungapped blocks were concatenated. Programs from the Phylip package (Felsenstein 1999) were used to produce 1000 bootstrap replica and to build phylogenetic trees using either maximum likelihood or neighbor-joining methods.
Other Methods
Genes were considered to be adjacent in the genome if they fulfilled the criteria outlined by Overbeek et al. (1999), i.e., if they were coded in the same DNA strand and were parts of gene arrays separated by no more than 300 nucleotides (in this definition, two close proteins do not have to be each other's immediate neighbor). Gene fusion and fission analysis was carried out manually by aligning the homologs; defining the N- and C-termini of the longest region with recognizable similarity; collecting all protein domains outside these borders, which were longer than 60 amino acids; and repeating the ortholog definition procedure for these subsequences. Fold recognition was based on modified PSI-BLAST protocol (Wolf et al. 1999). Secondary structures were predicted with the programs PHD (Rost 1996) and PSIPRED (Jones 1999).
We thank A. Eroshkin, A. Teplyakov, and Y. Wolf for help with the data analysis, and C. Taylor and D. DellaPenna for critical reading of the manuscript. A.M. is supported in part by grant GM58831 from the National Institutes of Health (NIH). Analysis of the unfinished genomes of the gram-positive cocci was made possible by generous submission to the public databases of the preliminary sequence data from The Institute of Genome Research and the University of Oklahoma.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Notes
[1] Corresponding author.
[2] These and some other gi numbers relate to the resubmission of the proteins to the NR database as the cured SWISS-PROT entries; sequential numbering of the entries does not imply their linkage in the genome unless specifically indicated.
Notes
[3] E-MAIL [email protected]; FAX (858) 625–0158.
[4] Article and publication are at www.genome.org/cgi/doi/10.1101/gr.145600.
REFERENCES
- ↵S.F. AltschulT.L. MaddenA.A. SchafferJ. ZhangZ. ZhangW. MillerD.J. Lipman(1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402.
- ↵Aravind, L. 1998. An evolutionary classification of the metallo-β-lactamase fold proteins. In Silico Biologyarticle 980108. http://www.bioinfo.de/isb/1998/01/0008/.
- ↵R. AuroraG.D. Rose(1998) Seeking an ancient enzyme in Methanococcus jannaschii using ORF, a program based on predicted secondary structure comparisons. Proc. Natl. Acad. Sci. 95:2818–2823.
- ↵A.L. BenkoV. VaduvaN.C. MartinA.K. Hopper(2000) Competition between a sterol biosynthetic enzyme and tRNA modification in addition to changes in the protein synthesis machinery causes altered nonsense suppression. Proc. Natl. Acad. Sci. 97:61–66.
- ↵E. BirneyJ.D. ThompsonT.J. Gibson(1996) PairWise and SearchWise: Finding the optimal alignment in a simultaneous comparison of a protein profile against all DNA translation frames. Nucleic Acids Res. 24:2730–2739.
- ↵P. BorkT. DandekarY. Diaz-LazcozF. EisenhaberM. HuynenY. Yuan(1998) Predicting function: From genes to genomes and back. J. Mol. Biol. 283:707–725.
- ↵P. BorkE.V. Koonin(1994) A P-loop-like motif in a widespread ATP pyrophosphatase domain: Implications for the evolution of sequence motifs and enzyme activity. Proteins Struct. Funct. Genet. 20:347–355.
- ↵P. BorkC. SanderA. Valencia(1993) Convergent evolution of similar enzymatic function on different protein folds: The hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 2:31–40.
- ↵T. DandekarB. SnelM. HuynenP. Bork(1998) Conservation of gene order: A fingerprint of proteins that physically interact. Trends Biochem. Sci. 23:324–328.
- ↵S. Dhe-PaganonJ. MagrathR.H. Abeles(1994) Mechanism of mevalonate pyrophosphate decarboxylase: Evidence for a carbocationic transition state. Biochemistry 33:13355–13362.
- ↵A. DischJ. SchwenderC. MullerH.K. LichtenthalerM. Rohmer(1998) Distribution of the mevalonate and glyceraldehyde phosphate/pyruvate pathways for isoprenoid biosynthesis in unicellular algae and the cyanobacterium Synechocystis PCC 6714. Biochem. J. 333:381–388.
- ↵J.A. Eisen(1998) Phylogenomics: Improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 8:163–167.
- ↵W. EisenreichM. SchwarzA. CartayradeD. ArigoniM.H. ZenkA. Bacher(1998) The deoxyxylulose phosphate pathway of isoprenoid biosynthesis in plants and microorganisms. Chem. Biol. 5:R221–R233.
- ↵A.J. EnrightI. IlipoulosN.C. KyrpidesC.A. Ouzounis(1999) Protein interaction maps for complete genomes based on gene fusion events. Nature 402:86–90.
- ↵J. Felsenstein(1993) PHYLIP (Phylogeny Inference Package) version 3.5c. Distributed by the author. (Department of Genetics, University of Washington, Seattle, WA).
- ↵M.Y. GalperinD.R. WalkerE.V. Koonin(1998) Analogous enzymes: Independent inventions in enzyme evolution. Genome Res. 8:779–790.
- ↵L.H. HartwellJ.J. HopfieldS. LeiblerA.W. Murray(1999) From molecular to modular cell biology. Nature 402:C47–C52.
- ↵J.F. HeidelbergJ.A. EisenW.C. NelsonR.A. ClaytonM.L. GwinnR.J. DodsonD.H. HaftE.K. HickeyJ.D. PetersonL. Umayam(2000) DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483.
- ↵S. HerzJ. WungsintaweekulC.A. SchuhrS. HechtH. LuttgenS. SagnerM. FellermeierW. EisenreichM.H. ZenkA. Bacher(2000) Biosynthesis of isoprenoids: YgbB protein converts 4-diphosphocytidyl-2C-methyl-D-erythritol 2-phosphate to 2C-methyl-D-erythritol 2,4-cyclodiphosphate. Proc. Natl. Acad. Sci. 97:2486–2490.
- S.M. HoutenW. KuisM. DuranT.J. de KoningA. van Royen-KerkhofG.J. RomeijnJ. FrenkelL. DorlandM.M. deBarseW.A. Huijbers(1999) Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat. Genet. 22:175–177.
- ↵S.M. HoutenG.J. RomeijnJ. KosterR.G. GrayP. DarbyshireG.P. SmitJ.B. de KlerkM. DuranK.M. GibsonR.J. Wanders(1999) Identification and characterization of three novel missense mutations in mevalonate kinase cDNA causing mevalonic aciduria, a disorder of isoprene biosynthesis. Hum. Mol. Genet. 8:1523–1528.
- ↵P. HugueneyS. RomerM. KuntzB. Camara(1992) Characterization and molecular cloning of a flavoprotein catalyzing the synthesis of phytofluene and zeta-carotene in Capsicum chromoplasts. Eur. J. Biochem. 209:399–407.
- ↵D.T. Jones(1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195–202.
- ↵H. JomaaJ. WiesnerS. SanderbrandB. AltincicekC. WeidemeyerM. HintzI TurbachovaM. EberlJ. ZeidlerH.K. Lichtenthaler(1999) Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 285:1573–1576.
- ↵T.C. Jordan-StarckV.W. Rodwell(1989) Pseudomonas mevalonii 3-hydroxy-3-methylglutaryl-CoA reductase. Characterization and chemical modification. J. Biol. Chem. 264:17913–17918.
- ↵M. Kates(1993) Membrane lipids of archaea. in The biochemistry of archaea (archaebacteria) ed M. Kates(Elsevier, Amsterdam), pp 261–295.
- ↵E.V. Koonin(1993) A superfamily of ATPases with diverse functions containing either classical or deviant ATP-binding motif. J. Mol. Biol. 229:1165–1174.
- ↵E.V. KooninL. Aravind(1998) Genomics: Re-evaluation of translation machinery evolution. Curr. Biol. 8:R266–R269.
- ↵E.V. KooninA.R. MushegianP. Bork(1996) Non-orthologous gene displacement. Trends Genet. 12:334–336.
- ↵E.V. KooninA.R. MushegianM.Y. GalperinD.R. Walker(1997) Comparison of archaeal and bacterial genomes: Computer analysis of protein sequences predicts novel functions and suggests a chimeric origin for the archaea. Mol. Microbiol. 25:619–637.
- ↵B.M. LangeR. Croteau(1999) Isopentenyl diphosphate biosynthesis via a mevalonate-independent pathway: Isopentenyl monophosphate kinase catalyzes the terminal enzymatic step. Proc. Natl. Acad. Sci. 96:13714–13719.
- ↵B.M. LangeM.R. WildungD. McCaskillR. Croteau(1998) A family of transketolases that directs isoprenoid biosynthesis via a mevalonate-independent pathway. Proc. Natl. Acad. Sci. 95:2100–2104.
- ↵J.V. LehtonenK. DenessioukA.C. MayM.S. Johnson(1999) Finding local structural similarities among families of unrelated protein structures: A generic non-linear alignment algorithm. Proteins Struct. Funct. Genet. 34:341–355.
- ↵L.M. LoisN. CamposS.R. PutraK. DanielsenM. RohmerA. Boronat(1998) Cloning and characterization of a gene from Escherichia coli encoding a transketolase-like enzyme that catalyzes the synthesis of D-1-deoxyxylulose 5-phosphate, a common precursor for isoprenoid, thiamin, and pyridoxol biosynthesis. Proc. Natl. Acad. Sci. 95:2105–2110.
- ↵H. LüttgenF. RohdichS. HerzJ. WungsintaweekulS. HechtC.A. SchuhrM. FellermeierS. SagnerM.H. ZenkA. Bacher(2000) Biosynthesis of isoprenoids: YchB protein of Escherichia coli phosphorylates the 2-hydroxy group of 4-diphosphocytidyl-2C-methyl-D-erythritol. Proc. Natl. Acad. Sci. 97:1062–1067.
- ↵E.M. MarcotteM. PellegriniH.L. NgD.W. RiceT.O. YeatesD. Eisenberg(1999) Detecting protein function and protein-protein interactions from genome sequences. Science 285:751–753.
- ↵A.S. MildvanD.J. WeberC. Abeygunawardana(1999) Solution structure and mechanism of the MutT pyrophosphohydrolase. Adv. Enzymol. Relat. Areas Mol. Biol. 73:183–207.
- ↵M.H. Moghadasian(1999) Clinical pharmacology of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Life Sci. 65:1329–1337.
- ↵A.R. MushegianE.V. Koonin(1996) Gene order is not conserved in bacterial evolution. Trends Genet. 12:289–290.
- OMIM Entry 251170. . February 7, 2000. http://www.ncbi.nlm.nih.gov/htbin-post/Omim/dispmim?251170.
- OMIM Entry 260920. . February 7, 2000. http://www.ncbi.nlm.nih.gov/htbin-post/Omim/dispmim?260920.
- ↵R. OverbeekM. FonsteinM. D'SouzaG.D. PuschN. Maltsev(1999) The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. 96:2896–2901.
- ↵M. PellegriniE.M. MarcotteM.J. ThompsonD. EisenbergT.O. Yeates(1999) Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. 96:4285–4288.
- ↵X. PennecN. Ayache(1998) A geometric algorithm to find small but highly similar 3D substructures in proteins. Bioinformatics 14:516–522.
- D. PotterH.M. Miziorko(1997) Identification of catalytic residues in human mevalonate kinase. J. Biol. Chem. 272:25449–25454.
- ↵D. PotterJ.M. WojnarC. NarasimhanH.M. Miziorkom(1997) Identification and functional characterization of an active-site lysine in mevalonate kinase. J. Biol. Chem. 272:5741–5746.
- ↵A. RahierM. SmithM.. Taton(1997) The role of cytochrome b5 in 4alpha-methyl-oxidation and C5(6) desaturation of plant sterol precursors. Biochem. Biophys. Res. Commun. 236:434–437.
- ↵M.C. RiveraR. JainJ.E. MooreJ.A. Lake(1998) Genomic evidence for two functionally distinct gene classes. Proc. Natl. Acad. Sci. 95:6239–6244.
- ↵F. RohdichJ. WungsintaweekulM. FellermeierS. SagnerS. HerzK. KisW. EisenreichA. BacherM.H. Zenk(1999) Cytidine 5′-triphosphate-dependent biosynthesis of isoprenoids: YgbP protein of Escherichia coli catalyzes the formation of 4-diphosphocytidyl-2-C-methylerythritol. Proc. Natl. Acad. Sci. 96:11758–11763.
- ↵D.S. RoosM.J. CrawfordR.G. DonaldJ.C. KissingerL.J. KlimczakB. Striepen(1999) Origin, targeting, and function of the apicomplexan plastid. Curr. Opin. Microbiol. 2:426–432.
- ↵B. Rost(1996) PHD: Predicting one-dimensional protein structure by profile-based neural networks. Methods Enzymol. 266:525–539.
- ↵G.D. SchulerS.F. AltschulD.J. Lipman(1991) A workbench for multiple alignment construction and analysis. Proteins Struct. Funct. Genet. 9:180–190.
- ↵A. TachibanaT. TanakaM. TaniguchiS. Oi(1996) Evidence for farnesol-mediated isoprenoid synthesis regulation in a halophilic archaeon, Haloferax volcanii. FEBS Lett. 379:43–46.
- ↵S. TakahashiT. KuzuyamaH. WatanabeH. Seto(1998) A 1-deoxy-D-xylulose 5-phosphate reductoisomerase catalyzing the formation of 2-C-methyl-D-erythritol 4-phosphate in an alternative nonmevalonate pathway for isoprenoid biosynthesis. Proc. Natl. Acad. Sci. 95:9879–9884.
- ↵R.L. TatusovM.Y. GalperinD.A. NataleE.V. Koonin(2000) The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28:33–36.
- ↵R.L. TatusovE.V. KooninD.J. Lipman(1997) A genomic perspective on protein families. Science 278:631–637.
- ↵R.L. TatusovA.R. MushegianP BorkN.P. BrownW.S. HayesM. BorodovskyK.E. RuddE.V. Koonin(1996) Metabolism and evolution of Haemophilus influenzae deduced from a whole-genome comparison with Escherichia coli. Curr. Biol. 6:279–291.
- ↵A. TeplyakovP. SebastiaoG. ObmolovaA. PerrakisG.S. BrushM.J. BessmanK.S. Wilson(1996) Crystal structure of bacteriophage T4 deoxynucleotide kinase with its substrates dGMP and ATP. EMBO J. 15:3487–3497.
- ↵J.D. ThompsonT.J. GibsonF. PlewniakF. JeanmouginD.G. Higgins(1997) The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882.
- ↵T.T. TsengK.S. GratwickJ. KollmanD. ParkD.H. NiesA. GoffeauM.H. Saier Jr.(1999) The RND permease superfamily: An ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1:107–125.
- ↵K. WangS. Ohnuma(1999) Chain-length determination mechanism of isoprenyl diphosphate synthases and implications for molecular evolution. Trends Biochem. Sci. 24:445–451.
- ↵Y.I. WolfL. AravindN.V. GrishinE.V. Koonin(1999) Evolution of aminoacyl-tRNA synthetases-analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 9:689–710.
- Y.I. WolfL. AravindE.V. Koonin(1999) Rickettsiae and Chlamydiae: Evidence of horizontal gene transfer and gene exchange. Trends Genet. 15:173–175.
- Y.I. WolfS.E. BrennerP.A. BashE.V. Koonin(1999) Distribution of protein folds in the three superkingdoms of life. Genome Res. 9:17–26.
- ↵Y.P. YuanO. EulensteinM. VingronP. Bork(1998) Towards detection of orthologues in sequence databases. Bioinformatics 14:285–289.
When this manuscript was under revision, a study of lateral gene transfer in the evolution of mevalonate and DXPS pathways was published (Boucher, Y. and Doolittle, W.F. 2000. Mol. Microbiol. 37: 703–716). In addition to the gene displacements also documented here, the analysis of phylogenetic tree of the HMG-CoA reductase family allowed the authors to detect a paralogous displacement between two related but distinct classes of this enzyme. Indeed, in Archaeoglobus fulgidus the bacterial-type (class 2) enzyme replaces the more expected anabolic (class 1) HMG-CoA reductase typical of other archaea.