
In its most simple terms, the human genome consists of two distinct fractions of DNA: repetitive and unique sequence. Traditionally, a portion of the unique fraction is thought to comprise the obvious functional constituents of our genome, including exons, introns, and regulatory DNA elements. With the exception of telomeric and centromeric repeat sequences, the functional significance of the vast majority of the repetitive fraction is less clear. Since the early experiments of reassociation kinetics of single-stranded human DNA (Britten and Kohne 1968), various gradations of repetitiveness have always been recognized on the basis of both the copy number and the degree of sequence similarity. The number of repeats range from the prolific (LINES, SINES, α-satellite, etc., in the 100,000’s) to the relatively few. By virtue of the fact that multigene families exist, genes themselves may be repetitive in nature. Many of the most well-studied members of gene families (hemoglobins and HOX genes), however, appear to be sufficiently divergent (Ohno 1970) or localize to discrete clusters of tandem arrays (rRNA genes, HLA genes, immunoglobulin gene segments). These are often distinguished based on the sequence divergence of individual members or their clustered position within the human genome. The term “unique” DNA, therefore, is relative, determined largely by what we already know about any given genome. The more our genome becomes sequenced, the more the total amount of “apparent” unique sequence will dwindle, with a concomitant burgeoning of the repeat classes. The basic paradigm regarding the repetitive and unique nature of DNA sequence underlies any effort to sequence a genome. In fact, the reason that any genome can be sequenced and assembled is that there is sufficiently enough unique sequence interdigitated among the repetitive fraction, the repetitive fraction is sufficiently divergent, and/or the repetitive fraction can be distinguished as such. A simple corollary exists among the sequencing community: The fewer and less complicated the repeats, the easier a genome is to sequence.
At a recent National Institutes of Health (NIH) meeting entitled, “Genomic Alterations in Genetic Disease: Mechanisms of Structural Rearrangement,” a much more complex picture of the organization of repeat sequences in the human genome emerged. Regions of the genome, conspicuously located within the subtelomeric and pericentromeric portions of chromosomes, which harbor large tracts (50–200 kb) of duplicated genomic segments that exhibit a remarkable degree of sequence similarity (95%–9%) are being identified. Unlike “traditional” repeat elements, these segments appear to carry complete or partial genomic structure of known genes, suggesting that they have recently been transposed from elsewhere in the genome. Therefore, they have the appearance of normal gene-encoding unique DNA, and are not, at first glance, easily distinguished as repetitive sequences. Interestingly, many of these large genomic segments of paralogous (sequence similarity due to duplication) sequence were discovered on either side of the breakpoint clusters of well-known microdeletion/microduplication syndromes, such as Prader–Willi syndrome (PWS) in 15q11–13, Williams syndrome, Smith–Magenis syndrome (SMS) in 17p11.2, and Velocardiofacial (VCFS) syndrome in 22q11.2, which suggests that they may have a role in mediating aberrant recombination associated with instability in these regions. Our own recent estimate from available genomic sequence in GenBank (130.1 Mb) seems to give further credibility to this complexity in our genome. A total of 1.1 Mb of genomic sequence, encompassing 21 different genes, was identified that showed remarkable sequence identity (95%–98%) to other large genomic segments or other sequenced cDNAs mapping to different locations in the genome. Most of these segments were identified among sequences mapping to the pericentromeric regions of chromosomes (2p11, 10p11, 15q11, 16p11, and 22q11), which suggests a hitherto unrecognized property of our genome to duplicate and transpose genomic segments to these regions. At the end of the NIH meeting, two general conclusions were reached regarding these complex repeat regions: (1) These repeat sequences are particularly difficult to resolve both from the perspective of mapping and sequencing; and (2) the sequence and organization of these repeat regions will be critical in understanding the process of genomic instability and disease in these regions.
Human Pericentromeric Regions Are Hot Spots for Recent Duplication Events
Recent comparative analyses of discrete genetic loci among primates (Eichler et al. 1996, 1997; Regnier et al. 1997; Zimonjic et al. 1997) further support the existence of a rather recent mechanism for genome duplication. The data indicate an ability of the genomes of higher primates to duplicate or transpose gene-rich genomic segments ranging from 5 to 50 kb in length to the pericentromeric regions of various chromosomes (Borden et al. 1990; Wong et al. 1990; Buiting et al. 1992; Bernardi et al. 1993; Tomlinson et al. 1994; Eichler et al. 1996, 1997; Frippiat et al. 1997; Kehrer-Sawatzki et al. 1997; Regnier et al. 1997; Zimonjic et al. 1997) (Table 1). The term pericentromeric refers to a large transition zone that begins immediately distal to the α-satellite repeat and extends into the first distinguishable cytogenetic Giemsa-stained band on either side of the centromere.
Summary of Pericentromeric-Directed Duplications in Man
| Segment | Ancestral locus | Duplicated loci | Est. length (kb) | Est. age (mya) |
| Adrenoleukodystrophy | Xq28 | 2p11, 10p11, 16p11, 22q11 | 9.7 | 5–7 |
| Creatine transporter | Xq28 | 16p11* | 26.5 | 5–7 |
| ERY-1/MN7 | 15q13 | 15q11*, 16p11* | ∼26 | 5–10 |
| KGFlocus | 15q15/21q21.1 | 2q21, 9p11, 9q12, 18p11, 18q11, 21q11 | ? | 5–10 |
| Glutamyl transpeptidase | 22q11.2 | 22q11*, 18, 19 and 20 | ? | ∼5 |
| MS29segment | 6p25 | 16p11* | ∼15 | 1–3 |
| Neurofibromatosis | 17q11.2 | 12q12, 14q11, 15q11, 18p11, 21q11, 22q11 | 1–4.6 | 1–25 |
| Variable IG heavy | 14q32.3 | 15q11 and 16p11 | ? | <5 |
| Variable IG κ | 2p11 | 1q12, 9p11, 9q11, 22q11 | 10–30 | 5–15 |
| Variable IG λ | 22q11 | 8q11 | ? | ? |
| von Willebrand factor | 12p12 | 22q11 | ∼30 | ∼5 |
[i] (*) Multiple copies of the segment are present within this region. (KGF) Keratinocyte growth factor gene; (ERY-1) END repeat family of transcripts; (IG) immunoglobulin gene segment. Estimated age (in mya) is based on divergence from ancestral sequence (this value may change as more copies of each duplicated segment are identified). Sequence divergences range from 10% to <1%. In many cases, the precise size of the duplication is undetermined.
Phylogenetic analyses suggest that most of these events have occurred relatively recently during primate evolution [1–15 mya (million years ago)] such that quantitative and qualitative differences in the distribution of these segments are observed among representative members of the higher primates (Eichler et al. 1996, 1997; Regnier et al. 1997; Zimonjic et al. 1997). The duplications often involve the movement of material between nonhomologous chromosomes and appear to be followed by subsequent intrachromosomal events that distribute copies in a nontandem fashion (Buiting et al. 1992; Kehrer-Sawatzki et al. 1997). In the few examples in which intrachromosomal events have been analyzed, the duplicated segments have been estimated to be separated by 1–3 Mb. Analysis of the sequences flanking the duplication junctions (Borden et al. 1990; Eichler et al. 1996, 1997) revealed a specific class of interspersed repeat sequences (CAAAAG or CAGGG) located near the integration sites within the pericentromeric regions. The existence of such sequences at the junctions of the duplications may indicate that they play a functional role in mediating the process of interchromosomal transfer of genetic material (Eichler et al. 1996,1997). It is interesting that a very similar duplication bias and mechanism have recently been demonstrated for the subtelomeric regions of chromosomes (Rouquier et al. 1998; Trask et al. 1998). This suggests that both pericentromeric and subtelomeric regions share an unusual function to duplicate genomic material from nonhomologous chromosomes.
Pericentromeric Regions Are Prone to Genetic Instability
Based on the existing data (Table 1), three pericentromeric regions demonstrate a particularly strong bias to acquire paralogous segments. More than half of the duplicated segments (6 of 11 loci, or 55%) have integrated within cytogenetic band interval 16p11.2 (Borden et al. 1990; Wong et al. 1990; Zimmer et al. 1990; Buiting et al. 1992;Zachau 1993; Tomlinson et al. 1994; Arnold et al. 1995; Ermert et al. 1995; Eichler et al. 1996, 1997). Likewise, 22q11.2 and 15q11.2 emerge as additional hot spots for integration with 5/11 and 3/11 of the duplicated loci having been directed to these regions of the human genome, respectively. In some cases, such as the creatine transporter,ERY-1 (END repeat family of transcripts), and neurofibromatosis loci, multiple copies of gene segments have been duplicated to 15q11 and 16p11 (Buiting et al. 1992; Amos-Landgraf et al. 1997; Kehrer-Sawatzki et al. 1997). This suggests that such regions are not only prone to interchromosomal duplications but undergo subsequent intrachromosomal duplication events distributing additional copies in the region.
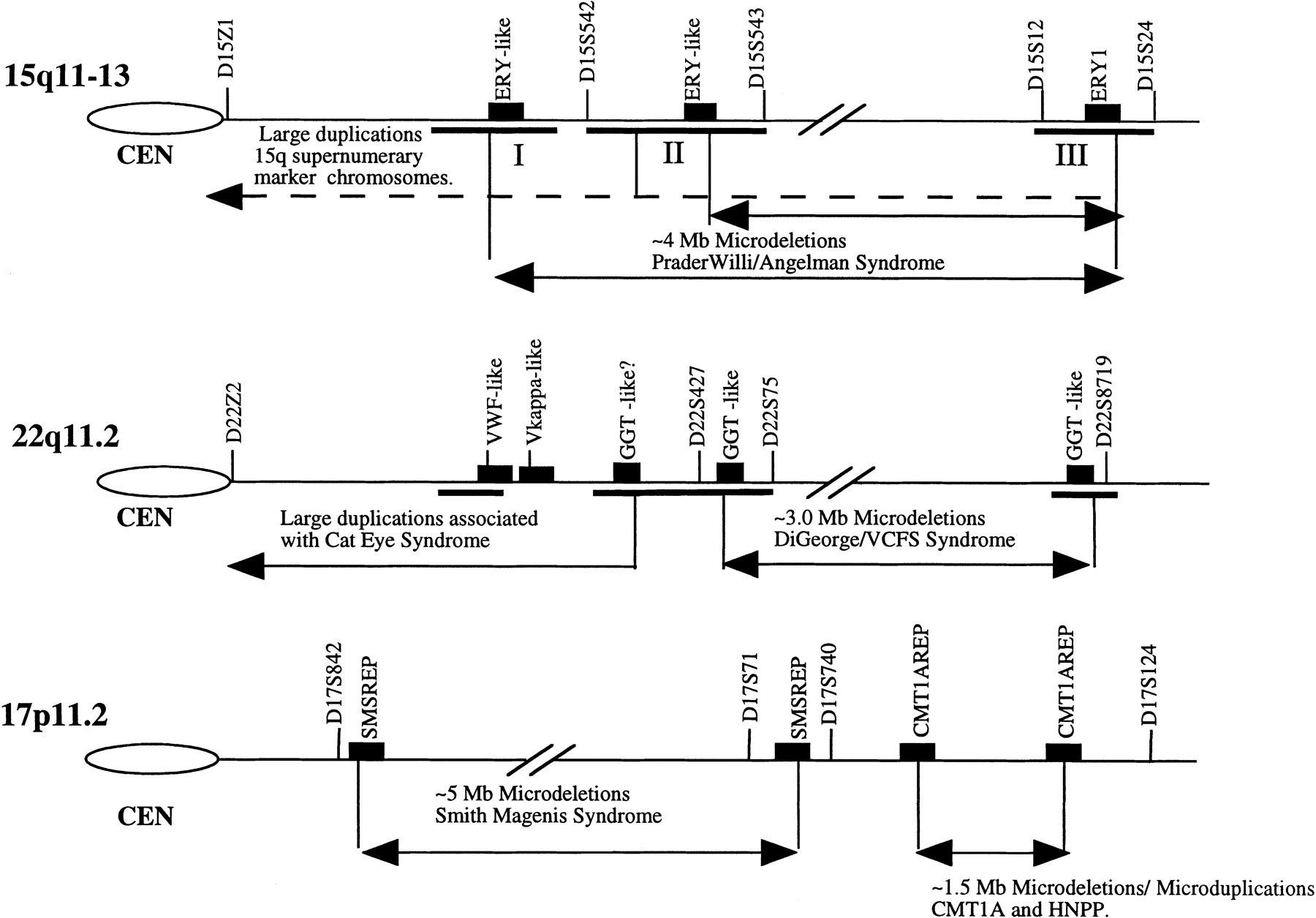
Perhaps it is not surprising that two of these intervals, 15q11.2 and 22q11, represent some of the most unstable regions of the human genome. These regions are frequently associated with sporadic duplication and deletions (Table 2). For example, duplication of the 15q11–q14 interval is responsible for the most common form of marker chromosome formation among humans, accounting for >50% of all observed bisatellited supernumerary chromosomes (Huang et al. 1997). A 4-Mb microdeletion of the 15q11–13 region accounts for ∼70%–75% of all patients with Prader–Willi/Angelman syndrome (Mutirangura et al. 1993; Carrozzo et al. 1997). In addition to supernumerary marker chromosomes (SMCs) and microdeletions of the Prader–Willi/Angelman critical region (PWACR) proximal 15q, several sporadic and inherited cases of interstitial duplications and triplications of the 15q11–13 region have been reported (Browne et al. 1997). Similar to 15q11.2, microdeletion and microduplication of 22q11.2 are commonly observed cytogenetic anomalies. The molecular defect in nearly 80% of all VCFS and DiGeorge syndrome (DGS) is the result of a large interstitial deletion encompassing 3 Mb of 22q11.2 sequence (Morrow et al. 1995). In contrast, Cat-eye syndrome (CES) patients have been shown to arise from duplications of 22q11.2 that result from either supernumerary marker chromosome formation or less commonly observed interstitial duplications (McDermid et al. 1996). Taken together, the data suggest that the regions 15q11 and 22q11 are hot spots for deletion and duplication. Recent physical mapping in these regions of 15q11 and 22q11 has, without exception, identified the presence of paralogous low-copy repeat sequences that map very near the duplication and deletion breakpoints (Buiting et al. 1992; Halford et al. 1993; Morris and Thacker 1993; Mears et al. 1994; Amos-Landgraf et al. 1997; Huang and Miao 1997). It has been suggested that these large blocks of paralogous sequence are responsible for the genomic instability of these regions (Fig. 1).
Examples of Pericentromeric Instability in the Human Genome
| Instability | Cytogenetic interval | Event (Mb) |
| DiGeorge syndrome | 22q11.2 | microdeletion (3) |
| Velocardiofacial syndrome | 22q11.2 | microdeletion (3) |
| Cat-eye syndrome | proximal 22q11 | SMC duplication |
| Williams syndrome | 7q11.23 | microdeletion (1.5) |
| Prader–Willi syndrome | 15q11-15q13 | microdeletion (4) |
| Angelman syndrome | 15q11-15q13 | microdeletion (4) |
| Inverted 15 (dup) | 15q11-15q14 | SMC duplication |
| 15q duplication | 15q11 proximal | interstitial duplication |
| Charcot–Marie Tooth | 17p11.2 | microdeletion (1.5) |
| HNPP | 17p11.2 | interstitial duplication (1.5) |
| Smith–Magenis syndrome | 17p11.2 | microdeletion (5) |
| 16p duplication | 16p11-13 | interstitial duplication |
[i] The most common cytogenetic event associated with each instability is indicated.
Pericentromeric gene duplication and genetic instability. The propensity of chromosomal regions 22q11, 15q11, and 17p11 to undergo microduplication/microdeletion is shown. Most common breakpoint regions are indicated by horizontal bars. The map locations of known intrachromosomally duplicated gene segments are shown as solid boxes in relation to genetic markers of these regions. More intrachromosomal duplicated copies of the GGT-like repeat block [which consists of variable smaller repeat units of the γ-glutamyl transferase (GGT) and breakpoint-cluster-like (BCR) genes] exist than are shown. Similarly, multiple copies of the ERY-1 gene have been identified within each cluster as well as copies located within 16p11.2 SMS–REP represents a duplicated gene cluster (>200 kb) composed of multiple genes [signal recognition particle, a keratin gene (KER-I), a coactisin gene (CLP), and the TRE oncogene]. The CMT1A–REP duplication segment consists of a portion of the COX10 gene as well as a novel cDNA, C170RF1. Many other pericentromerically duplicated segments identified within these regions have not been assigned to this map.
Genetic Disease Implications
The fact that duplicated segments of nonprocessed genes (which we term “duplicon” to distinguish from other repeat sequences) have been targeted to the same regions (Table 1) of the genome that are characterized by frequent sporadic genomic rearrangement (Table 2) leads to the question, Is the presence of these duplicated segments in these regions a cause or consequence of the documented instability? In the case of the PWS, at least, recent data argue that the mechanism of pericentromeric duplication provides the molecular basis for predisposition to microdeletion (Buiting et al. 1992; Amos-Landgraf et al. 1997; Christian et al. 1997). A precedence for this type of genomic architecture and deletion mechanism has already been established for CMT1A/HNPP (Charcot-MarieTooth disease type 1A andhereditary neuropathy with liability topressure palsies) and SMS (Reiter et al. 1996; Chen et al. 1997). In these latter two cases, the microdeletion and microduplication breakpoints have been unequivocally mapped within the duplicated segments, indicating a causal relationship between the duplication of genomic segments and recurrent chromosomal structural rearrangements. There are three striking similarities of the molecular basis of these disorders that are pertinent to the “process” of pericentromeric duplication: (1) The duplications are recent (<10 mya based on the level of sequence divergence); (2) the paralogy domains are interspersed (usually separated by a few megabases of intervening sequence and did not arise by tandem duplication); and (3) the duplications that have created the liability to genomic instability have been biased to the pericentromeric regions of chromosomes. The finding of a recent pericentromeric-directed mechanism within the human genome involving duplications of gene clusters is consistent with the model for microdeletion established by these three “genomic” diseases. Thus, an understanding of the molecular mechanism responsible for pericentromeric duplications (both interchromosomal and intrachromosomal) would appear to be critical to our understanding of the molecular etiology of instability in these regions.
To Sequence or Not to Sequence?
The duplicative nature of these regions poses a particular dilemma in the assembly of the human genomic reference sequence. The considerable paralogy of these regions (95%–99%) over large distances (5–150 kb) indicates that high-throughput procedures such as PCR-based STS mapping (Green and Olson 1990) and BAC-end sequencing (Venter et al. 1996) by themselves may be inadequate for developing sequence-ready physical maps. Mapping of YACs and radiation hybrids using such STSs, particularly in large duplicated regions, often fails to resolve a unique coordinate (McDermid et al. 1996; Bouffard et al. 1997). The end result of such efforts has been the development of incomplete physical and genetic maps (http://www.ncbi.nlm.nih.gov/HUGO) or multiple maps, none of which can accommodate all of the physical attributes of the region. There is, of course, the daunting prospect that duplicated regions themselves may be polymorphic in the human population. As a result, any two individuals may possess a different genomic architecture in these regions. Because most resources (YAC, BAC, and chromosome-specific cosmid libraries) are derived from different individuals, combining such resources to build a single human physical map may be confounding at best. These problems are further compounded by the fact that YAC clones mapped to these areas are notoriously unstable. Parenthetical reports of YAC clones exhibiting an extraordinary degree of interstitial deletion or chimerisim abound within the pericentromeric regions of chromosomes (Jackson et al. 1996;McDermid et al. 1996; Christian et al. 1997). Considering the propensity of these regions to undergo interchromosomal duplication events, the observation of chimeric clones may have a biological basis and not simply represent an artifactual rearrangement. The general consensus of the participants of the NIH meeting, also aptly described as the “Masochist Mapping and Sequencing Club,” was that the unusual paralogous nature of these regions demands intense (and therefore more costly) scrutiny both from the perspectives of mapping and sequencing.
The announcement of a new initiative (the Venter/Perkin-Elmer venture) to complete the sequencing of the human genome using a whole-shotgun approach may complicate our understanding of these regions even further (Venter et al. 1998). The bulk of the (10× coverage) sequence will be generated from ∼2-kb insert plasmid clones for which there is absolutely no information on their map position within the human genome. Assembly of sequence reads from paralogous copies of genomic segments that may exhibit 95%–99% sequence identity will be difficult, at best, if not impossible, at worst, to resolve. Even with the use of end sequences from a 10-kb insert clone or BAC clone scaffold map, it will likely be very difficult to disentangle the sequence reads that have been misassembled among the paralogous copies or to properly map, in retrospect, the origin of such sequence. Such pitfalls are largely avoided with the traditional map and then shotgun approach, as all sequence reads are contained within a single BAC and are not being sampled from an entire human genome. To make matters even worse, the Venter/Perkin-Elmer venture promises to apply its whole shotgun approach to multiple individuals with the objective of identifying and precisely locating single-nucleotide polymorphisms (SNPs). Where the degree of paralogy can closely approach the level of human polymorphism, it is difficult to imagine how SNPs may be distinguished accurately from paralogous sequence variants (Fig.2). To be fair, however, the majority of human genes clearly do not have paralogous counterparts within the pericentromeric regions (our current estimates suggest ∼1%. Nevertheless, many of the genes that are duplicated [adrenoleukodystrophy (ALD), neurofibromatosis, ERY-1, von Willebrand factor, etc.] are not without clinical consequence. The understanding of mutational variants that impact human phenotype will proceed much more smoothly once the paralogous nature of these genes has been deduced.
SNP or paralogous sequence variant? A CONSED sequence alignment of 50 bp from an intronic segment is duplicated to six different sites in the human genome. Both forward and reverse (r) sequence reads were generated from dye-terminator sequencing reactions of PCR products amplified from monochromosomal somatic cell hybrids. Only one variant nucleotide was identified among the copies. The presence of duplicated copies on each chromosome has been verified by FISH and genomic hybridization.

The question that would appear to remain, then, is should such regions be targeted by the Human Genome Project (HGP) for sequencing? The critics would argue that these regions represent junk DNA (a term borne out of ignorance and not necessarily a biological property of our genome) and therefore should not be a priority for sequencing. The countermand to this argument is that these regions appear to be involved in recurrent chromosomal structural rearrangements associated with human genetic disease. By dint of this fact, these segments do confer a biological function—genomic rearrangement—a property that may be deleterious to the individual but advantageous from the perspective of an evolving species. Others would argue that these regions represent intractable portions of the genome that should be left for sequencing at a later date. The problem with this proverbial ostrich—bury-your-head-in-the-sand—approach is that such regions cannot be easily distinguished. Therein lies the potential dilemma for the HGP. Unlike other repetitive elements such as classical centromeric and telomeric repeat sequences, these duplicated segments do not have, at the sequence level of a BAC, properties that identify them as repeats. Prima facie, these sequences appear to carry genes with defined intron–exon boundaries and, therefore, for all intents and purposes, represent repeat sequences incognito. Furthermore, the ancestral loci often originate in nonpericentromeric localized regions (ERY-1 in 15q13, ALD in Xq28, MS29 in 6p25, etc.)—regions that have already been or will be targeted for sequencing.
Intentionally or unintentionally, the human genome sequencing community will be forced to tackle these difficult regions. A sequencing deferral is not an option. The only question then that really remains is not if but how the HGP will choose to analyze these hot spots of gene duplication? We will either target these regions in a systematic fashion from the standpoint of recognizing their paralogous nature and thereby understanding their role in biology and disease or we will sequence these regions by default and hope to reconstruct their genomic complexity and impact on genomic instability after the fact.
The latter approach has the potential to generate gaps in the human sequence that will ultimately translate into lacunae in our understanding the organization, evolution, and associated pathology of the human genome. Sequencing divorce of mapping offers quantity over quality, forfeiting a true understanding of the organization and the paralogous nature of the human genome. The HGP is just beginning to provide useful insight into the complex organization and evolution of our genome. Let us not short-change ourselves and future generations of scientists by opting for the “cheapest and quickest” route in the generation of a “complete” human genomic reference sequence.
Notes
[3] Corresponding author.
Notes
[4] E-MAIL [email protected]; FAX (510) 422-2282.
REFERENCES
- ↵J. Amos-LandgrafY. JiA. WandstratD. DriscollS. SchartzR. Nicholls(1997) Am. J. Hum. Genet. (Suppl.) 61: A3..
- ↵N. ArnoldJ. WienbergK. EmertH. Zachau(1995) Genomics 26:147–156.
- ↵F. BernardiP. PatracchiniD. GammatiM. PinottiC. SchwienbacherG. BalleriniG. Marchetti(1993) Hum. Mol. Genet. 2:545–548.
- ↵P. BordenR. JaenichenH. Zachau(1990) Nucleic Acids Res. 18:2101–2107.
- ↵G.G. BouffardJ.R. IdolV.V. BradenL.M. IyerA.F. CunninghamL.A. WeintraubJ.W. TouchmanR.M. Mohr-TidwellD.C. PelusoR.S. Fulton(1997) Genome Res. 7:673–692.
- ↵R.J. BrittenD.E. Kohne(1968) Repeated sequences in DNA. Science 161:529–540.
- ↵C. BrowneN. DennisE. MaherF. LongJ. NicholsonJ. SillibourneC. Barber(1997) Am. J. Hum. Genet. 61:1342–1352.
- ↵K. BuitingV. GregerB. BrownsteinR. MohrI. VoiculescuA. WinterpachtB. ZabelB. Horsthemke(1992) Proc. Natl. Acad. Sci. 89:5457–5461.
- ↵R. CarrozzoE. RossiS. ChristianK. KittikamronC. LivieriA. CorriasL. PucciA. FoisP. SimiL. Bosio(1997) Am. J. Hum. Genet. 61:228–231.
- ↵K. ChenP. ManianT. KoeuthL. PotockiQ. ZhaoA. ChinaultC. LeeJ. Lupski(1997) Nature Genet. 17:154–163.
- ↵S. ChristianS. MartinJ. FantesN. BhattB. HuangD. Ledbetter(1997) Am. J. Hum. Genet. (Suppl.) 61: A7..
- ↵E. EichlerF. LuY. ShenR. AntonacciV. JurecicN. DoggettR. MoyzisA. BaldiniR. GibbsD. Nelson(1996) Hum. Mol. Genet. 5:899–912.
- ↵E. EichlerM. BudarfM. RocchiL. DeavenN. DoggettA. BaldiniD. NelsonH. Mohrenweiser(1997) Hum. Mol. Genet. 6:991–1002.
- ↵K. ErmertH. MitlohnerW. SchemppH. Zachau(1995) Genomics 25:623–629.
- ↵J. FrippiatP. DardS. MarshG. WinterM. Lefranc(1997) Eur. J. Immunol. 27:1260–1265.
- ↵E. GreenM. Olson(1990) Science 250:94–98.
- ↵S. HalfordE. LindsayM. NayuduA. CareyA. BaldiniP. Scambler(1993) Hum. Mol. Genet. 2:191–196.
- ↵B. HuangJ. CrollaS. ChristianM. Wolf-LedbetterM. MachaP. PapenhausenD. Ledbetter(1997) Hum. Genet. 99:11–17.
- ↵G. HuangG. Miao(1997) Trends Biotechnol. 15:200–202.
- ↵M. JacksonC. SeeL. MulliganB. Lauffart(1996) Genomics 33:258–270.
- ↵H. Kehrer-SawatzkiT. SchwickardtG. AssumG. RocchiW. Krone(1997) Hum. Genet. 100:595–600.
- ↵H. McDermidK. McTaggartM. RiaziT. HudsonM. BudarfB. EmanuelC. Bell(1996) Genome Res. 6:1149–1159.
- ↵A. MearsA. DuncanM. BudarfB. EmanuelB. SellingerJ. Siegel-BarteltC. GreenbergH. McDermid(1994) Am. J. Hum. Genet. 55:134–142.
- ↵T. MorrisJ. Thacker(1993) Proc. Natl. Acad. Sci. 90:1392–1396.
- ↵B. MorrowR. GoldbergC. CarlsonR. Das GuptaH. SirotkinJ. CollinsI. DunhamH. O’DonnellP. ScamblerR. Shprintzen(1995) Am. J. Hum. Genet. 56:1391–1403.
- ↵A. MutiranguraA. JayakumarJ. SutcliffeM. NakaoM. McKinneyK. BuitingB. HorsthemkeA. BeaudetA. ChinaultD. Ledbetter(1993) Genomics 18:546–552.
- ↵S. Ohno(1970) Evolution by gene duplication. (Springer Verlag, Berlin, Germany).
- ↵V. RegnierM. MeddebG. LecointreF. RichardA. DuvergerV. NguyenB. DutrillauxA. BernheimG. Danglot(1997) Hum. Mol. Genet. 6:9–16.
- ↵L. ReiterT. MurakamiT. KoeuthL. PentaoD. MuznyR. GibbsJ. Lupski(1996) Nat. Genet. 12:288–297.
- ↵S. RouquierS. TaviauxB.J. TraskV. Brand-ArponG. van den EnghJ. DemailleD. Giorgi(1998) Nat. Genet. 18:243–250.
- ↵I. TomlinsonG. CookN. CarterR. ElaswarapuS. SmithG. WalterL. BuluwelaT. RabbittsG. Winter(1994) Hum. Mol. Genet. 3:853–860.
- ↵B. TraskC. FriedmanA. Martin-GallardoL. RowenC. AkinbamiJ. BlankenshipC. CollinsD. GiorgiS. IadonatoF. Johnson(1998) Hum. Mol. Genet. 7:13–26.
- ↵J. VenterH. SmithL. Hood(1996) Nature 381:364–366.
- ↵J.C. VenterM.D. AdamsG.G. SuttonA.R. KerlavageH.O. SmithM. Hunkapiller(1998) Science 280:1540–1542.
- ↵Z. WongN. RoyleA. Jeffreys(1990) Genomics 7:222–234.
- ↵H. Zachau(1993) Gene 135:167–173.
- ↵F. ZimmerH. HameisterH. SchekH. Zachau(1990) EMBO J. 9:1535–1542.
- ↵D. ZimonjicM. KelleyJ. RubinS. AaronsonN. Popescu(1997) Proc. Natl. Acad. Sci. 94:11461–11465.