
Only a small portion of the genome of higher organisms encodes information for amino acid sequences of proteins, and of the noncoding sequence an unknown fraction plays a vital role in regulating gene expression. It is widely appreciated that comparisons among genome sequences will provide a key opportunity to identify functional regions of noncoding DNA by virtue of the conservation of their primary sequences. Underlying this assumption (that sequence conservation implies functional constraint) is an old idea in the theory of molecular evolution: that substitution rates vary among sites depending on constraint. In this issue, Bergman and Kreitman (2001)study the properties of sequence divergence in noncoding regions by comparing 100 kb of sequence in promoter regions and introns of 40 genes of Drosophila melanogaster and Drosophila virilis. Using a heuristic filtered dotplot, they identify blocks >8 bp in length with >70% sequence conservation between the two species. Altogether, ∼22%–26% of the noncoding sequence fell into such conserved blocks. On average, there were 10.7 blocks per kilobase pair of D. melanogaster DNA, and the blocks varied widely in length with an average of 19 bp. Distributions of block lengths, distributions of lengths of insertion/deletion events, and patterns of nucleotide substitutions were all statistically indistinguishable in contrasts of intergenic and intronic sequences. This is a surprising result, as one would expect that transcriptional, splicing, and secondary structure requirements would all place different constraints on intronic vs. intergenic noncoding sequences. But, if there are different constraints, they do not reveal themselves through differences in patterns of sequence divergence between distantDrosophila species.
One gets an uneasy feeling when aligning such divergent sequences (∼80% of the nucleotides were unalignable) because the statistical properties of the aligned blocks are likely to depend heavily on the method chosen to find the alignments in the first place. The authors are well aware of this, and so they contrast the blocks found by their heuristic to several other algorithms, including DiAlign,DBA, VISTA and Lamark. These other methods identify conserved blocks by weighting different properties of the sequence, and thus the correspondence among methods is not perfect. However, 98% of the blocks found by Bergman and Kreitman were also identified by at least one of the other methods. Furthermore, the conclusion that the substitution process is homogeneous across introns and intergenic sequences appears to be robust across methods of identifying blocks. Although the distribution of block sizes does depend on the algorithm for finding conserved blocks, it is not radically different across these methods.
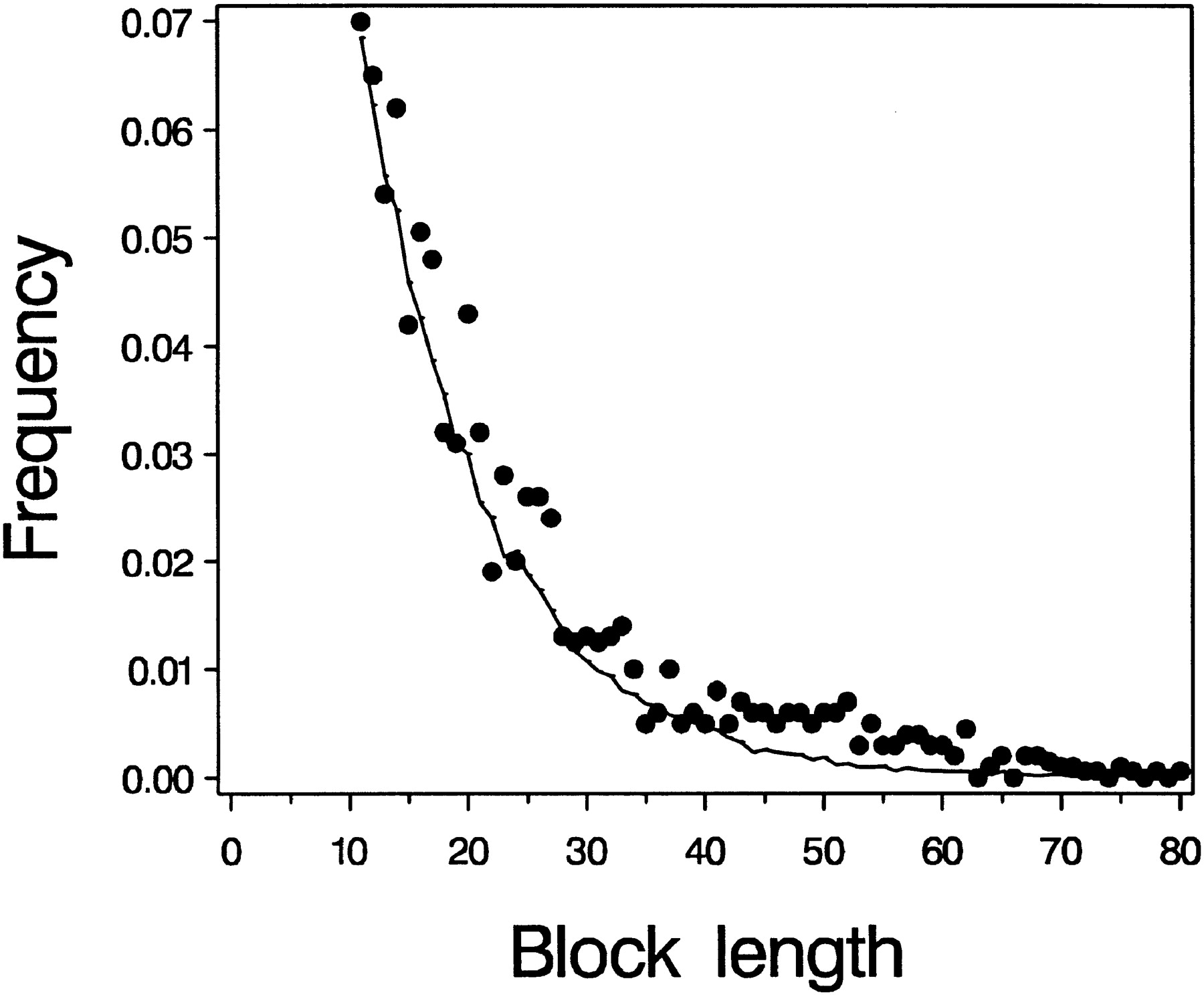
The distribution of conserved block lengths fits well to a lognormal distribution, begging the question of what sort of processes might produce this distribution. Considering the simpler case of block identities, or runs of sequence with perfect matches, Karlin et al. (1985) derived an expression for the expected length of the longest perfect match between random sequences. The mathematics behind this allowed some of the early inferences of “significance” of matches in noncoding DNA. Because on average every unconstrained site in the genomes of D. melanogaster and D. virilis is expected to have suffered a substitution, the analogy to finding conserved blocks in otherwise random sequence is reasonable. Simulations of base substitution between a pair of species are easily done by starting with one string and distributing random changes with equal probability per site. The data of Bergman and Kreitman seem to fit such a distribution reasonably well for the case of 95 substitutions per kilobase pair (Fig. 1). What does it mean when a pure mutation-drift model fits these data? First of all, 95 substitutions per kilobase is well below the average divergence betweenD. melanogaster and D. virilis, so by this criterion these conserved blocks clearly are low in divergence. It might be unexpected that the fit in Figure 1 is this good, because some of the conservation in noncoding regions ought to be caused by binding sites of regulatory proteins or RNAs, and there is no reason for the lengths of binding sites to fit this distribution. The good fit to a mutation-driftmodel suggests that regions of apparent sequence conservation might have a reduced rate of mutation (with the caveat that many models can fit a distribution such as this). Typically, models of molecular evolution consider homogeneous mutation rates, and differences in sequence divergence are attributed to differences in rates of substitution, because most mutations in functionally conserved regions are deleterious and are rapidly eliminated. Generally it is considered less parsimonious to invoke altered mutation rates to explain conserved regions, but evidence for variation in mutation rate is strong, especially for simple sequence repeats (e.g., Bachtrog et al. 2000), and our understanding of the precise base-to-base variability in mutation rates is quite fragmentary. Furthermore, DNA polymerase or DNA repair mechanisms may produce runs of high fidelity sequence interspersed with regions that are more error-prone. In short, it is not easy to discount the idea that some of the apparently conserved blocks of sequence seen in studies such as Bergman and Kreitman's might simply be mutational coldspots.
Observed and simulated distribution of lengths of conserved blocks in noncoding DNA. The dots are the data taken from Bergman and Kreitman (2001) and reflect the distribution of conserved block lengths (∼70% identity and length >10 nucleotides) found between Drosophila melanogaster and Drosophila virilis in intergenic and intronic sequences. The curve was obtained by introducing 95 random substitutions per kilobase, and scoring run lengths of blocks of absolute identity from these simulated data. The plot shows only blocks of 11 bp or longer because of the potential bias against identifying shorter blocks in the observed data.
So how can we ever distinguish between mutational coldspots and functional constraint? At several points Bergman and Kreitman illustrate the utility of analyzing three or more species together. With multiple species, especially with known phylogenetic relations, it is possible to infer the direction in which substitutions and indels have occurred. Many studies have made inferences of molecular evolutionary processes from multiple species sequences, including a demonstration that secondary structure serves as a constraint in intron sequences (Kirby et al. 1995; Leicht et al. 1995). With the genome ofD. pseudoobscura on its way, and the continuing development of computational tools for identifying local alignments in noncoding DNA (e.g,. Schwartz et al. 2000), the prospects for extending the work of Bergman and Kreitman appear excellent. In particular, a key conclusion of this paper is that understanding the process of how substitutions occur in noncoding DNA will be of great assistance in finding regions that are conserved or otherwise deviate from a pattern of purely neutral processes.
Notes
[1] E-MAIL [email protected]; FAX (814) 865-9131.
[2] Article and publication are athttp://www.genome.org/cgi/doi/10.1101/gr.201601.
REFERENCES
- ↵D. BachtrogM. AgisM. ImhofC. Schlötterer(2000) Mol. Biol. Evol. 17:1277–1285.
- ↵C.M. BergmanM. Kreitman(2001) Genome Res. 11:1335–1345.
- ↵S. KarlinG. GhandourD.E. Foulser(1985) Mol. Biol. Evol. 2:35–52.
- ↵D.A. KirbyS.V. MuseW. Stephan(1995) Proc. Natl. Acad. Sci. 92:9047–9051.
- ↵B.G. LeichtS.V. MuseM. HanczycA.G. Clark(1995) Genetics 139:299–308.
- ↵S. SchwartzZ. ZhangK.A. FrazerA. SmitC. RiemerJ. BouckR. GibbsR. HardisonW. Miller(2000) Genome Res. 10:577–586.