
Abstract
Repetitive DNA is a significant component of eukaryotic genomes. We have developed a strategy to efficiently and accurately sequence repetitive DNA in the nematode Caenorhabditis elegans using integrated artificial transposons and automated fluorescent sequencing. Mapping and assembly tools represent important components of this strategy and facilitate sequence assembly in complex regions. We have applied the strategy to several cosmid assembly gaps resulting from repetitive DNA and have accurately recovered the sequences of these regions. Analysis of these regions revealed six novel transposon-like repetitive elements, IR-1, IR-2, IR-3, IR-4, IR-5, and TR-1. Each of these elements represents a middle-repetitive DNA family in C. elegans containing at least 3–140 copies per genome. Copies of IR-1, IR-2, IR-4, and IR-5 are located on all (or most) of the six nematode chromosomes, whereas IR-3 is predominantly located on chromosome X. These elements are almost exclusively interspersed between predicted genes or within the predicted introns of these genes, with the exception of a single IR-5 element, which is located within a predicted exon. IR-1, IR-2, and IR-3 are flanked by short sequence duplications resembling the target site duplications of transposons. We have established a website database (http://www.welch.jhu.edu/~devine/RepDNAdb.html) to track and cross-reference these transposon-like repetitive elements that contains detailed information on individual element copies and provides links to appropriate GenBank records. This set of tools may be used to sequence, track, and study repetitive DNA in model organisms and humans.
[The sequences reported in this paper have been deposited in GenBank under accession nos. U53139 and U86946–U86951.]
Repetitive DNA families have long been known to occupy the genomes of higher eukaryotes, but most of these families have remained uncharacterized in the years preceding genome sequencing projects (Britten and Kohn 1968). The Caenorhabditis elegans genome is estimated to harbor several hundred to several thousand repetitive DNA families, occupying ∼12%–17% of the worm genome (Sulston and Brenner 1974; Emmons et al. 1980). Both Cot hybridization and computer studies indicate that repetitive DNA is even more abundant in the genomes of flies, mice, and humans (Britten and Kohn 1968; Smit 1996). Transposable genetic elements comprise a significant fraction of this repetitive DNA, and represent a ubiquitous class of middle-repetitive DNA in these organisms. Such elements include the Ty elements in yeast (Boeke and Sandmeyer 1991), the Tc elements in the worm (Emmons et al. 1983; Collins et al. 1989; Leavitt and Emmons 1989; Dreyfus and Emmons 1991; Yuan et al. 1991; Collins and Anderson 1994), >20 transposons in the fly (Bingham and Zachur 1989;Blackman and Gelbart 1989; Engels 1989; Finnegan 1989a,b; Hartl 1989), and a variety of transposon-like repetitive elements in humans, including medium reiteration frequency sequences (MERs), short interspersed nuclear elements (SINEs), and long interspersed nuclear elements (LINEs) (Moran et al. 1996; Smit 1996; Smit and Riggs 1996). Other classes of repetitive DNA shared by many of these organisms include (1) multigene families encoding ribosomal and transfer RNAs, (2) multigene families encoding proteins such as histones, (3) satellite DNA, and (4) telomeric and subtelomeric repeats.
Inverted repeats are fairly abundant in C. elegans and are estimated to occupy ∼3–6 Mb of the ∼100-Mb genome (Sulston and Brenner 1974). Hybridization studies in C. elegans using the foldback fraction of Cot-purified genomic DNA previously have led to estimates of ∼2400 inverted repeats per haploid genome (Emmons et al. 1980). These inverted repeats had stems (repeat units) ranging from 50 to 9000 bp in length (with an average length of 670 bp) and intervening loops ranging from 230 to 9260 bp in length (with an average length of 1340) as judged by electron microscopy.
Repetitive DNA can be technically challenging to sequence. Therefore, some types of repetitive DNA, such as large segments of satellite DNA, might be best studied with a combination of mapping and limited sequence analysis. This approach was taken with the 1-Mb rDNA region of yeast chromosome XII, which consists solely of 100–200 tandemly repeated copies of the 9-kb rDNA unit. The terminal rDNA units were sequenced for the Saccharomyces cerevisiae genome project, but the intervening ∼1 Mb of DNA was characterized by mapping only (Link and Olson 1991). Other types of repetitive DNA may be too dispersed or too heterogeneous to analyze in this manner. Such sequences include inverted and tandem repeats located throughout the nematode genome and a variety of localized or dispersed repetitive sequences in the human genome.
We have developed a transposon-based strategy to sequence repetitive DNA efficiently using artificial transposons and automated fluorescent sequencing that, in principle, may be applied to any type of repetitive or otherwise challenging DNA. Using this strategy, we have accurately recovered the sequences of both inverted and tandemly arranged repetitive DNA. In the process, we have identified six novel middle-repetitive DNA families in C. elegans, including five inverted repeat families and a single tandem repeat family. We have established a website database (http://www.welch.jhu.edu/~devine/RepDNAdb.html) to track and cross-reference these transposon-like repetitive elements that contains detailed information on individual element copies and provides links to appropriate GenBank records. This set of tools may be used to sequence, track, and study repetitive DNA in model organisms and humans.
RESULTS
Artificial Transposons for Mapping and Automated Sequencing
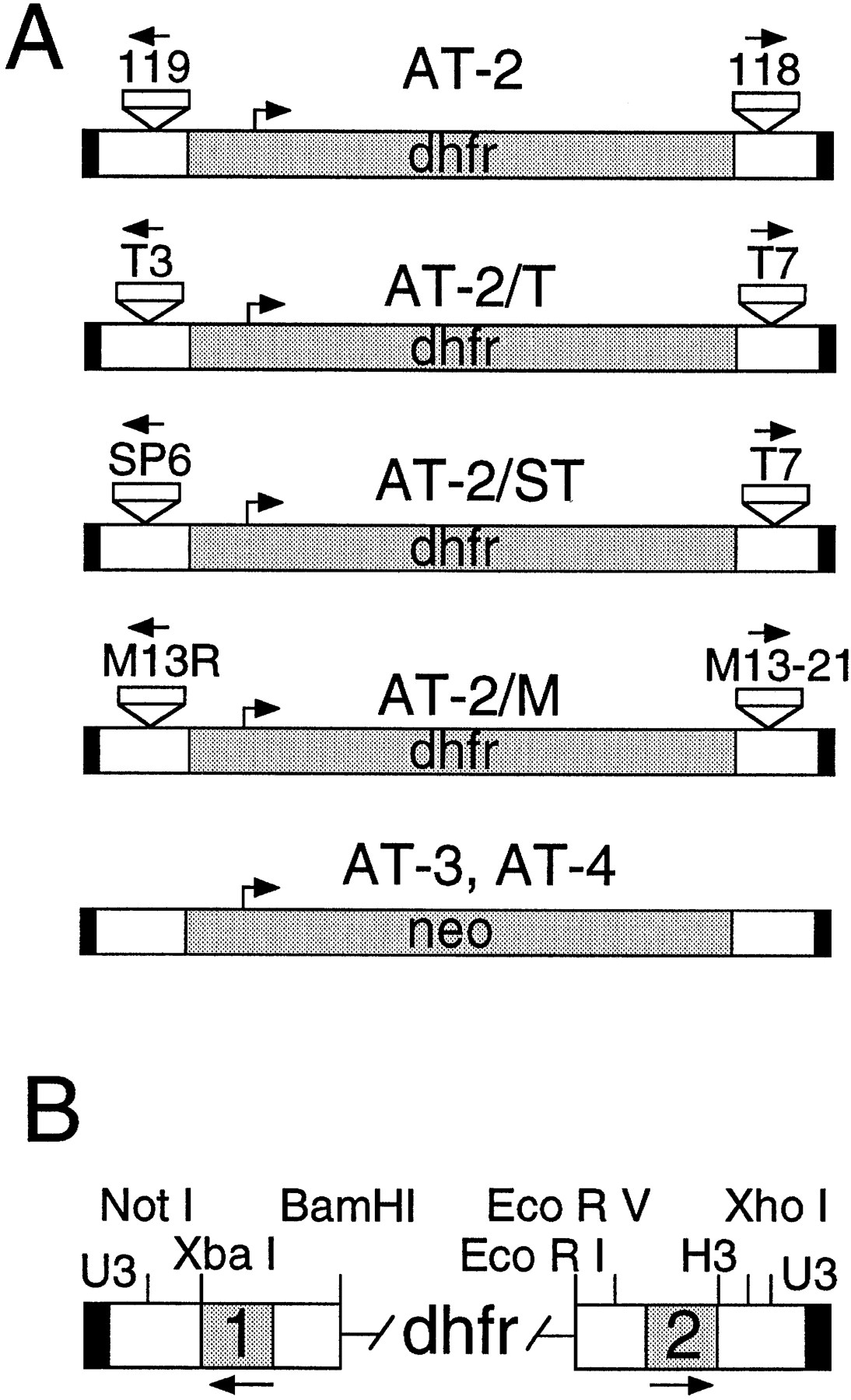
We have described previously methods to generate artificial transposons and to integrate them into plasmid targets in vitro using Ty1 integrase (Devine and Boeke 1994). We have now adapted this system for high throughput mapping and automated sequencing and have developed a number of new transposons, primers, and mapping tools for this purpose. New artificial transposons such as AT-3 and AT-4 that carry the neo gene were developed for use with kanamycin selection, and new transposons carrying subterminal T3, T7, SP6, M13–20, and R primer sites were developed for use with either dye primer or dye terminator fluorescent sequencing (Fig.1). The dye primer-versions of these primers (including ET primers) are available from commercial sources along with their associated mobility files, making them attractive for large-scale sequencing applications. Fluorescent dye primers for the SD118 and SD119 priming sites located near the termini of the artificial transposon AT-2 were also synthesized and developed for sequencing (Fig. 1). Finally, several transposon mapping tools were developed and adapted to a 96-well format (see Methods).
Artificial transposons for automated sequencing. (A) The artificial transposon AT-2 and several AT-2 derivatives are shown. The new priming sites are indicated near the termini as small boxes with arrows facing outward. AT-2 has sites for the 118 and 119 primers; AT-2/T has sites for the T3 and T7 primers; AT-2/ST has sites for the SP6 and T7 primers; AT-2/M has sites for the M13 -20 and R primers. AT-3 and AT-4, which carry the neo gene for use with kanamycin selection, are also shown. (B) An expanded view of the transposon termini. The dhfr gene that is used as the selectable marker and separates the two termini is indicated. The black box indicates the 4-bp U3 terminal Ty1 sequence; the white boxes indicate the regions carrying multiple restriction sites for mapping; and the shaded boxes labeled 1 and 2 indicate the locations of the inserted primer sites. Integration reactions with AT-2, AT-2/T, AT-2/M, and AT-2/ST generated ∼1 × 103 to 1 × 104 recombinants per reaction, whereas AT-3 and AT-4 generated 1 × 102 to 1 × 103 recombinants per reaction.
A Strategy for Sequencing Repetitive DNA
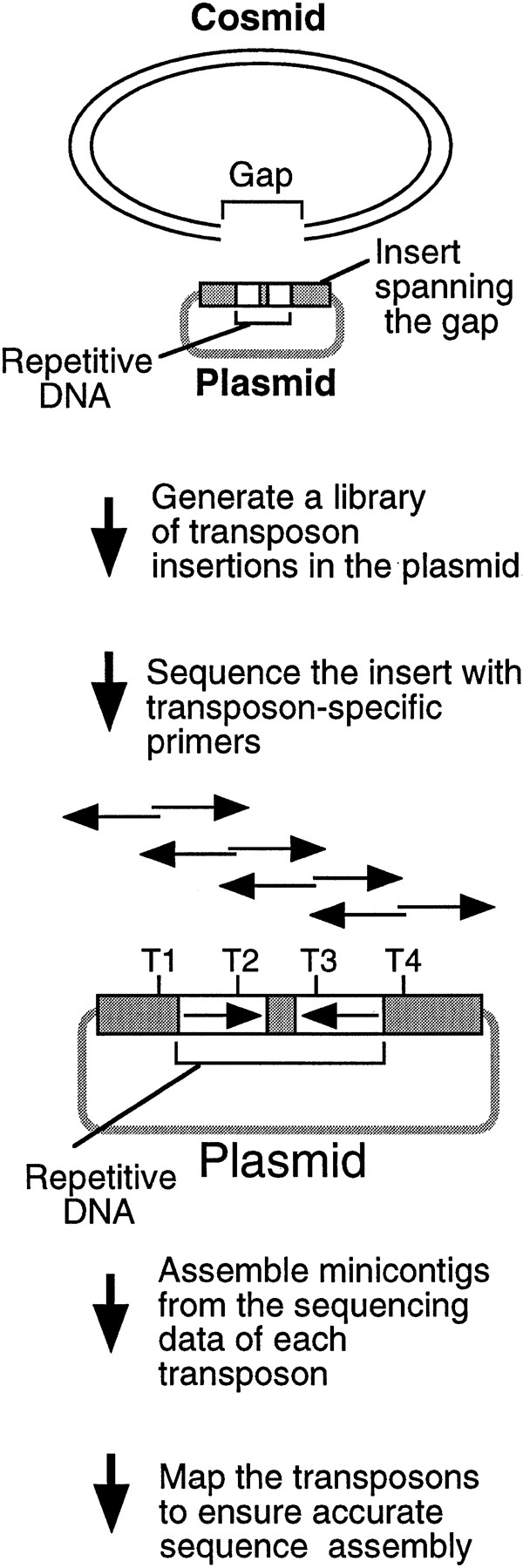
Although cosmids, BACs, and P1 clones may be sequenced efficiently using M13 libraries and random shotgun sequencing strategies (Wilson and Mardis 1997), assembly gaps are often present after the initial rounds of sequencing that must be closed with directed efforts. Such gaps are caused by a variety of factors including repetitive DNA, and are caused frequently by inverted repeats in C. elegans(Wilson et al. 1994; Chissoe et al. 1997). Many gaps can be closed with relatively simple efforts such as extending reads into the gap region; however, a number of gaps are refractory to such approaches (particularly those containing repetitive sequences). To tackle these regions systematically, we developed a transposon-based sequencing strategy to close gaps that could be applied to any type of repetitive or troublesome DNA without prior knowledge of the sequence structure (Fig. 2). The process consists of the following steps: (1) identifying a plasmid clone carrying an insert that spans the gap, (2) integrating artificial transposons into the target clone in vitro, (3) recovering raw sequencing data using the integrated transposons, (4) building minicontigs from the two sequences initiated from each transposon, and (5) mapping the order of the transposon insertions when necessary to ensure accurate sequence assembly.
Strategy for sequencing repetitive DNA. A plasmid is identified with an insert spanning the cosmid gap, a transposon library is generated, and the insert is sequenced using the transposons. T1–T4 represent transposon insertions. The two sequencing reactions initiated from individual transposon insertions are shown as back-to-back arrows, which can be assembled using the 5-bp target site duplications. The transposon order can be determined by mapping to ensure accurate sequence assembly.
A Sequence Assembly Gap Caused by a 379-bp Inverted Repeat
To test our strategy, we initially focused on a sequence assembly gap that was present in the C. elegans cosmid F18E3 following M13 production sequencing. A pUC18 clone was identified that carried a 2.6-kb insert spanning the gap, and the insert was sequenced using integrated transposons and transposon-specific dye primers as outlined below (Fig. 3). We first mapped a collection of 96 independent transposon recombinants using the restriction enzymePvuII (see Methods) to search for transposons within the plasmid insert (data not shown). Thirty-nine transposons mapping to this region were selected to recover the complete insert sequence on both DNA strands (Fig. 3A). Minicontigs were then assembled from the sequences initiated from each transposon using the flanking 5-bp target site duplications, and these contigs were assembled to derive a final consensus sequence. To independently test whether the final assembly was correct, all 39 transposon insertions were then positionally mapped by restriction mapping, and the order of the transposons was found to be fully consistent with that predicted by the sequence assembly (Fig.3B). The 2.6-kb consensus sequence successfully closed the gap present in the F18E3 cosmid (GenBank accession no. U53139).
Sequence analysis of the F18E3 gap region. (A) The F18E3-10 plasmid insert is depicted along with the 39 transposon insertions used to recover the complete sequence. These 39 were chosen randomly from 62 that mapped to the insert region by PvuII mapping. The vertical arrows represent individual transposon insertions. Those facing downward are in the forward orientation (the same orientation as the selectable marker in the transposon), whereas those facing upward are in the reverse orientation. Several transposons are numbered for cross-referencing with the gel (in B). (B) Positional mapping of the transposon insertions. A subset of the transposons mapped is shown. A diagram is located under the figure to indicate the source of each fragment. The 0.9-kb fragment present in the recombinants represents the transposon, which is liberated from the plasmid upon digestion with EcoRI and NotI. The size of the fragment labeled “short” provides the distance of the transposon from the unique EcoRI site at the right end of the insert, whereas the fragment labeled “long” contains the vector and a variable amount of insert sequence. (M) λHindIII marker; (F) F18E3-10 potential plasmid. (C) Representative dye primer sequencing with a transposon recombinant template. Approximately 350–500 bases of high quality sequence data was obtained routinely with various primers.

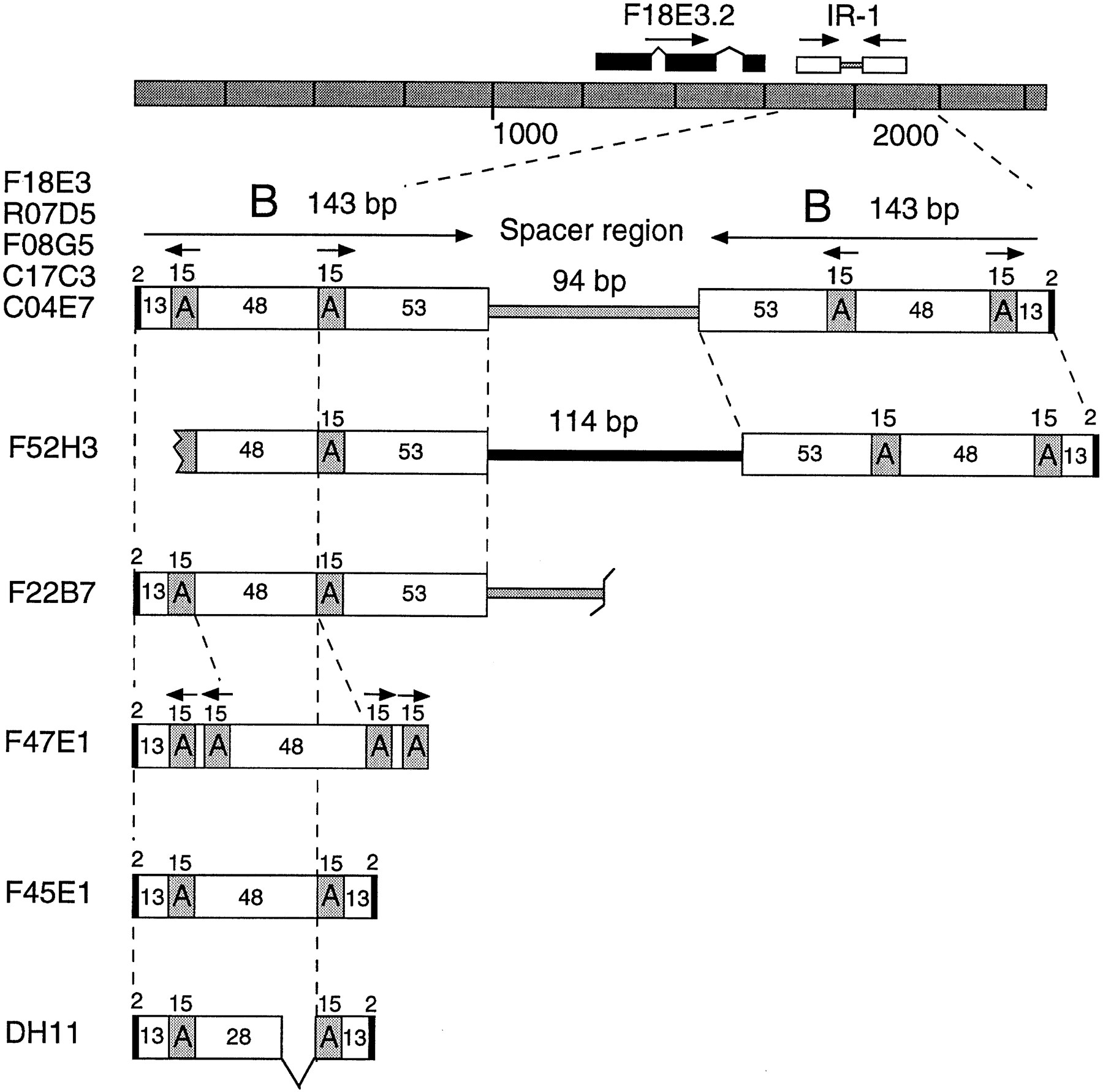
Further analysis of the 2.6-kb insert showed that it contained two families of repetitive DNA involved in a complex inverted repeat structure (Fig. 4). Because palindromic sequences are not propagated well in M13, the complex inverted repeat is likely to have caused the sequence assembly gap. The 379-bp repeat consists of two 143-bp inverted B modules separated by a 94-bp unique spacer. Each B module itself contains a central region of two inverted 15-bp A modules separated by 48 bp (Fig. 4). Interestingly, the entire 379-bp element was flanked by a 2-bp direct repeat of the sequence TA.
The IR-1 repetitive family. The F18E3 insert structure is depicted. Note that the IR-1 element is located adjacent to a potential gene, F18E3.2, whose predicted exons are shown as black boxes. Additional IR-1 element copies are diagrammed below. Five full-length copies 379 bp in length were identified (in cosmids F18E3, R07D5, F08G5, C17C3, and C04E7). Two truncated copies (F52H3 and F22B7) and three unusual copies (F47E1, F45E1, and DH11) were also identified (see also Table 1for additional copies). The 15-bp A modules and the 143-bp B modules are indicated along with the sizes of the various regions, including the 2-bp flanking duplications (small black boxes). Note that F52H3 contains an entirely different spacer sequence (black rectangle).
IR-1, a Family of Middle-Repetitive DNA in C. elegans
At least 16 copies of this 379-bp invertedrepeat-1 (IR-1) element or closely related sequences were identified in GenBank using the BLAST algorithm, indicating that it comprises a family of middle-repetitive DNA in C. elegans(Fig. 4; Table 1). The fact that additional copies were present in the database indicates that the inverted repeat structure could be sequenced with other methods. However, these methods were not systematic and were more labor-intensive than the current strategy.
IR-1 Elements inC. elegans
| Element no. | Chromosome | Location | Nearest Gene(s) | Size (bp) | Comment | Percent Identity | TSD (bp) | TSD |
| R07D5 | X | intergenic | unc-7, R07D5.2 | 379 | full length | 99.5 | 2 | AT |
| F18E3 | V | intergenic | F18E3.1 F18E3.2 | 379 | full length | 99.2 | 2 | TA |
| F08G5 | IV | intron | F08G5.5 | 379 | full length | 97.4 | 2 | TA |
| C17C3 | II | intron | C17C3.12 | 379 | full length | 96.6 | 2 | AA |
| F39B3 | X | intergenic | F39B3, Unknown | 400 | internal insertion | 96.0 | 2 | AA |
| C04E47 | X | intergenic | C04E7.2, C04E7.3 | 379 | full length | 94.5 | 2 | AC |
| F52H3 | II | intergenic | ZK892.5 F52H3.7 | 399 | truncated, unique spacer | 70.0 | 2 | AT* |
| F35E12 | V | — | — | 392 | internal insertion | 94.4 | 2 | AC |
| F22B7 | III | intergenic | F22B7.9, F22B7.10 | 194 | truncated | 97.5 | 2 | TT* |
| ZK849 | I | — | — | 1105 | internal insertion | — | 2 | TA |
| F47E1 | X | intron | F47E1.3 | 112 | truncated and rearranged | 82.1 | 2 | TA* |
| ZC8 | X | intergenic | ZC8.2, ZC8.3 | 1105 | internal insertion | — | 2 | TA |
| F54D1 | IV | intergenic | F54D1.5, F54D1.6 | 1083 | internal insertion | — | none | |
| F45E1 | X | intron | F45E1.7 | 103 | internal deletion | 99.0 | 2 | CA |
| F10D11 | I | intron | F10D11.d | 173 | internal deletion | — | 2 | TA |
| ZK354 | I | — | — | 173 | internal deletion | — | 2 | TA |
| F02E9 | I | — | — | 173 | internal deletion | — | 2 | TA |
| C44H4 | X | intron | C44H4.6 | 102 | internal deletion | — | 2 | TG |
| DH11 | II | intergenic | DH11.1, DH11.2 | 84 | internal deletion | 100 | 2 | TA |
[i] Copies of IR-1 found in the C. elegans genome. Of the 19 elements shown, 14 had BLAST P < 10−25, using the consensus element sequence as the query (Table 2). The remaining five elements (at the bottom) had BLAST P < 10−19. The element number is the same as the corresponding cosmid number for the clone harboring the element. Elements were classified as intergenic if they were between known or predicted genes, or as intron if located within the intron of a known or predicted gene. Percent identity is relative to the reference consensus sequence (Table2). An asterisk (*) next to the target site duplication (TSD) indicates that the element was truncated at one end, and the intact end was used to determine the expected duplication sequence. Elements within regions for which information is not yet available have dashes in the appropriate column. Similar tables are available at our website for this and the other element families (http://www.welch.jhu.edu/~devine/RepDNAdb.html). In the website tables, the element number is hot-linked to the appropriate GenBank record, facilitating rapid information retrieval.
Three classes of IR-1 elements were identified using the F18E3 query sequence, namely, (1) full-length elements 379 bp in length retaining the same structure as the original F18E3 copy, (2) truncated elements that lacked sequences at one end or the other, and (3) unique elements resembling the parent F18E3 but having altered structures possibly caused by rearrangements (Fig. 4; Table 1). Five full-length (379-bp) elements, retaining 94.5% to 99.5% sequence identity with F18E3, were identified. Remarkably, like the original F18E3 copy, each of these elements was flanked by a 2-bp sequence duplication. These 2-bp direct repeats are similar to the TA target site duplications created by theC. elegans transposons Tc1 and Tc3 (and marinerfamily elements found in host species ranging from fungi to insects to mammals) upon integration into new locations (van Luenen and Plasterk 1994). However, unlike these elements, the IR-1 element is not strictly limited to TA dinucleotides; some copies were flanked by AA, AT, AC, CA, or TG duplications (Table 1). IR-1 elements were found on all six of the C. elegans chromosomes, and were found exclusively within predicted introns or intergenic regions. For example, IR-1 element R07D5, which is located on chromosome X, is found within the intergenic region between the unc-7 gene and a second predicted gene, R07D5.2. Like the original F18E3 copy, this element is 379 bp in length, is flanked by a 2-bp direct repeat, and differs by only two nucleotides from the sequence of F18E3. For additional data on the IR-1 repetitive family (and the other families below), see our Repetitive DNA database (RepDNAdb) website (http://www.welch.jhu.edu/~devine/RepDNAdb.html) under “Caenorhabditis elegans.”
IR-2, a Palindromic Repetitive Element Flanked by a 4- to 9-bp Sequence Duplication
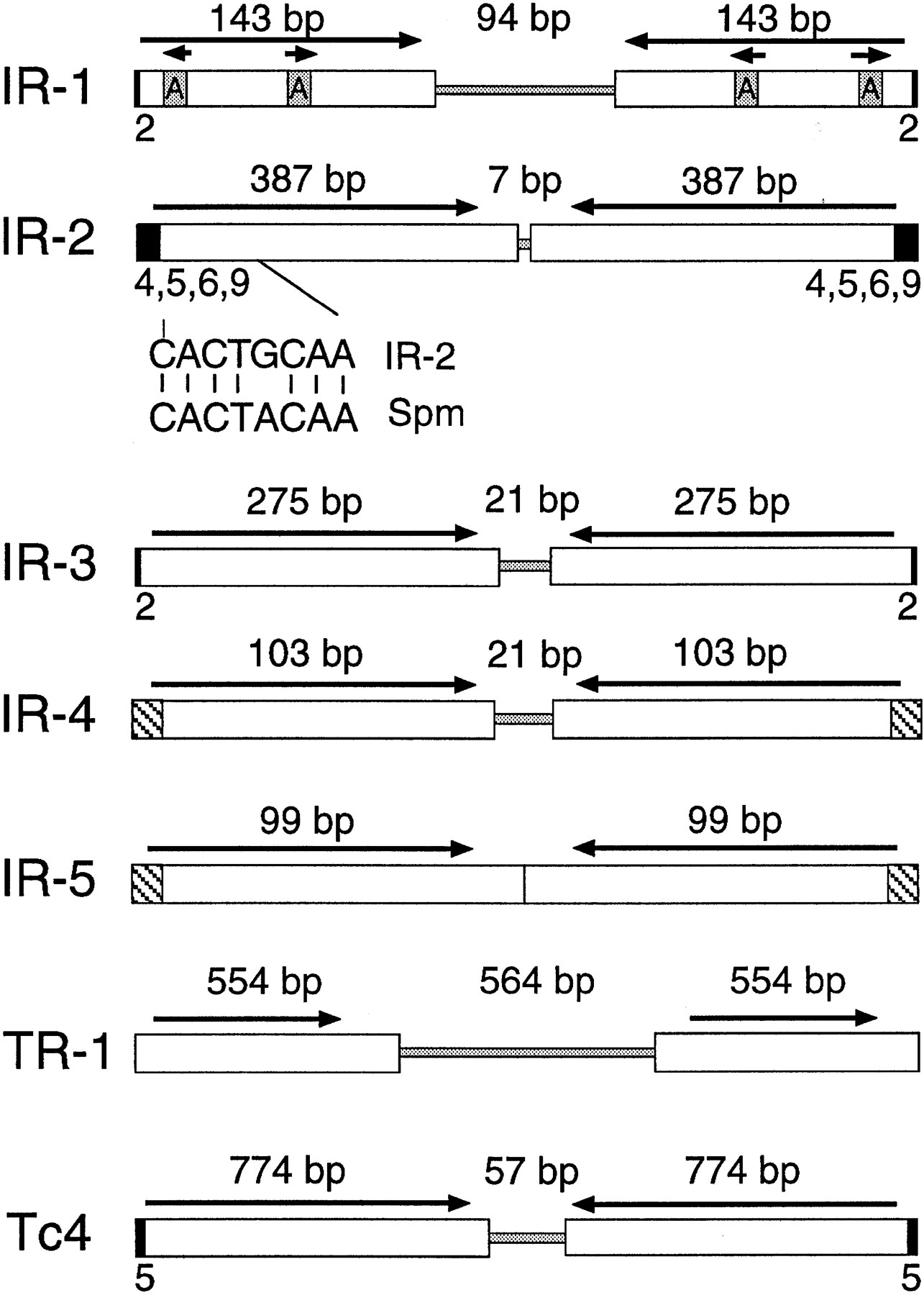
A second unrelated inverted repeat, IR-2, was identified in cosmid C35B1 by sequencing a 3.5-kb pUC18 insert spanning an assembly gap with the same strategy as that outlined for F18E3. In this case, transposon mapping was not used to confirm the finished sequence assembly. IR-2 is 781 bp in length and is a palindrome consisting of two inverted 378-bp modules separated by a 7-bp unique spacer (Fig. 5; Table 2). The C35B1 copy of IR-2 is flanked by a 4-bp duplication of the sequence 5′-TTGG-3′. Interestingly, the extreme termini of the IR-2 element closely resemble those of the Maize En/Spm transposon, with 7/8 bp identity (Federoff 1989; Fig. 5).
Structures of the IR and TR repetitive elements. The IR and TR elements are shown, indicating the presence of inverted or tandem repeats (white boxes), spacer regions (shaded regions in the center), and flanking sequence duplications (small black boxes). The terminal sequences of IR-2 and Spm are compared under the IR-2 map, with 7/8 base pair identity. The hatched areas of IR-4 and IR-5 indicate their similar terminal sequences. The Tc4 transposon of C. elegans, which has been shown to be a functionally active mobile element in C. elegans (Yuan et al. 1991), is shown for comparison. Note that only full-length representative copies are shown for each repetitive family and that each family was as structurally diverse as IR-1 (Fig. 4).
Features of Repetitive DNA Families
| Repetitive family | Initial copy | Accession no. for consensus | Element size (bp) | Minimum no. of copies in genome[i] | Potential TSD (bp) | Terminal 10-bp sequences(s) |
| IR-1 | F18E3 | U86946 | 379 | 16 | 2 | 5‘-CTCGGCATTC-3‘ |
| IR-2 | C35B1 | U86947 | 781 | 47 | 4,5,6,9 | 5‘-CACTGCAACT-3‘ |
| IR-3 | F32D1 | U86948 | 578 | 140 | 2 | 5‘-AAGGTGGTGT-3‘ |
| IR-4 | ZK6 | U86949 | 227 | 12 | None | 5‘-TATTACCGGT-3‘ |
| IR-5 | F23F1 | U86950 | 198 | 14 | None | 5‘-TATTACGGGA-3‘ |
| TR-1 | F49F1 | U86951 | 1672 | 3 | None | 5‘-ATTTACTTCT-3‘ |
| 5‘-AATACTACAC-3‘ |
[i] Copies found as of November 27, 1996 with BLASTP <10−25 using the consensus element to search GenBank and the C. elegans Consortium database, at which time ∼80% of the genome sequence was available. The following features are indicated: name of the family, the cosmid in which the initial element was identified, the GenBank accession number for the consensus element sequence derived from several element copies, the size of the full-length element, the minimal number of copies per genome, target site duplication (if any), and the sequence at the termini. In each case, the 5‘ end of the sequence represents the terminus of the element.
At least 12 copies of IR-2 or closely related sequences were identified in GenBank using the BLAST algorithm, indicating that it comprises a family of middle-repetitive DNA in C. elegans. More than 47 highly significant matches (with P < 10−25) were identified in the C. elegans database (C. elegansMapping and Sequencing Consortium, Sanger Centre, Hinxton, UK, and Washington University, St. Louis, MO), which also includes unfinished cosmids in progress and is estimated to include up to 80% of the complete genome sequence (as of November 1996). Several of these unfinished cosmid contigs terminated within the IR-2 palindrome, as would be expected of inverted repeats that lead to assembly gaps. Two of the matches represented previously reported inverted repeats on chromosome III (Wilson et al. 1994), whereas the other matches were found on the remaining chromosomes (I, II, IV, V, and X), and were located exclusively within predicted introns and intergenic regions. Remarkably, each of the full-length elements examined was flanked by a unique 4- to 9-bp sequence duplication. The C35B1 copy of IR-2 was flanked by a 4-bp duplication of the sequence 5′-TTGG-3′, whereas another IR-2 element was flanked by a 6-bp duplication of the sequence 5′-CGCATC-3′. A third element was flanked by a 9-bp repeat of the sequence 5′-CGCGCAAAG-3′. The remaining nine full-length elements examined were flanked by 5-bp duplications with the following sequences (5′ → 3′): CATAA; AACGT; CATGT; CAACG; AACAA; TTCCG; TGGTT; TTTTC; AGGTG.
IR-3, an Abundant Repetitive Element Found Primarily on the X Chromosome
A third repetitive element, IR-3, was identified in cosmid F32D1 by sequencing a 3-kb pUC18 insert spanning an assembly gap in this cosmid. IR-3 is a 578-bp palindrome comprised of two inverted 275-bp modules separated by a unique 21-bp spacer (Fig. 5; Table2). Like IR-1 and IR-2, database searches indicated that this element comprises a family of middle-repetitive DNA inC. elegans, present at a minimum of 140 copies per genome (as of November 1996, using a P-value cutoff of 10−25). Full-length elements were flanked by Tc1/mariner-like TA sequence duplications. Although copies of IR-3 mapped to all six chromosomes, this element was strikingly abundant on chromosome X, with 55 of the 108 elements examined (51%) mapping to this chromosome. Whereas IR-1 and IR-2 (and the IR-4 and IR-5 elements described below) were also found on chromosome X, these elements were much more evenly distributed among all (or most) of theC. elegans chromosomes, indicating that the remarkable distribution of IR-3 is not simply a result of an overabundance of chromosome X-specific sequences in the database.
IR-4 and IR-5, Two Similar Repetitive Families
Two additional palindromic repetitive families, IR-4 and IR-5, were discovered by closing assembly gaps in cosmids ZK6 and F23F1, respectively (Fig. 5; Table 2). A 0.6-kb pUC18 insert was sequenced to close the gap in ZK6, whereas a 1.0-kb pUC18 insert was sequenced to close the gap in F23F1. Although their full consensus sequences are very different, the IR-4 and IR-5 repetitive elements are similar in many respects. For example, they have almost identical terminal sequences, sharing the same sequence at 8 of the terminal 10 bp (Table2). These elements are also fairly similar with respect to their sizes, copy numbers, and genomic distributions: IR-4 is 227 bp in length, whereas IR-5 is 198 bp (Table 2); IR-4 is present at a minimum of 12 copies per genome, whereas IR-5 is present at a minimum of 14 copies per genome; and IR-4 is found on all six chromosomes, whereas IR-5 is found on five of the six chromosomes. All of the elements are interspersed between predicted genes or within the introns of predicted genes, with a single exception (IR-5 element M05B5), which is located within a predicted exon of the M05B5.3 gene. None of the element copies from either of the families have flanking sequence duplications.
TR-1, a Low-Copy Tandemly Repeated Element
A tandemly repeated sequence, TR-1, was identified in cosmid F49F1 by sequencing a 2.8-kb pUC18 insert spanning an assembly gap. This 1654-bp tandem repeat consists of two very similar direct repeats (554 and 546 bp in length, respectively) separated by a 564-bp unique spacer (Fig. 5; Table 1). Although the repeat units are slightly different in length, they are very similar in sequence and share 90% sequence identity. Two somewhat degenerate matches were identified in the Genome Sequencing Center’s database; one of these copies was partially inverted, whereas the other was truncated.
DISCUSSION
Artificial Transposons for Sequencing Repetitive DNA
Repetitive DNA is a significant component of eukaryotic genomes. The actual repeat units of repetitive DNA can be organized in a number of different ways: as localized inverted or tandem repeats, as localized mixed repetitive DNA containing both inverted and tandem repeats, or as dispersed repetitive DNA with distantly separated repeat units. Irrespective of the structure, most of the difficulties associated with recovering repetitive sequences are attributable to two basic problems: (1) poor propagation of intact DNA clones carrying repetitive sequences, or (2) misaligned sequence assemblies caused by the repeats. Clone propagation problems are caused either by clone under-representation in libraries resulting from the repeated sequences or by clone rearrangements promoted by the repeats. Misaligned sequence assemblies are caused by the inaccurate assignment of a given sequencing read to one of several similar but distinct repeat units.
Although a variety of sequencing strategies have been developed, most strategies have serious limitations in regions containing repetitive DNA. For example, primer walking is not possible in such regions because the primer anneals to multiple locations within the template and generates a mixed sequence. Transposon-based sequencing strategies, on the other hand, use unique priming sites located near the termini of integrated transposons to recover DNA sequences (Ahmed 1985; Adachi et al. 1987; Phadnis et al. 1989; Strathmann et al. 1991; Devine and Boeke 1994; Kimmel et al. 1997; Fig. 3) and thus avoid this problem. Second, because it is known that the two sequences obtained from a single transposon comprise a minicontig, they can be assembled into 600- to 1000-bp sequences that are twice as likely to span problem regions (Fig. 3). Finally, unlike random shearing, the linkage of the sequenced clone is maintained throughout the analysis. This provides a valuable option that is otherwise not available, namely, that the order of the transposons can be mapped and the resulting data may be used to independently determine the correct order of the sequence (Fig. 3).
Inverted repeats such as the IR elements (and other inverted repeats inC. elegans) cause sequencing problems primarily because they are not propagated well in single-stranded vectors. Clones carrying such regions are under-represented in M13 libraries presumably because they form hairpin structures (Chissoe et al. 1997). As a consequence, assembly gaps occur in cosmids at sites lacking clone coverage when M13 libraries are used in conjunction with shotgun sequencing. Because the same inverted repeats are propagated well in double-stranded plasmid vectors, the problem is partially solved by simply cloning the repetitive region into a double-stranded plasmid vector. Nevertheless, not all sequencing strategies are suitable for the subsequent sequencing of this repetitive DNA, and as mentioned above, the unique priming sites and additional assembly tools provided with a transposon approach are useful in such regions. Although assembly tools are not necessary in all cases, they are available when required, and can be applied to any type of repeat structure after problems are discovered.
Like inverted repeats, tandem repeats may also be difficult to sequence. First, tandem repeats can rearrange and delete intervening sequences, or can promote other types of gross rearrangements that yield clones that no longer resemble the corresponding parental clones. Short tandem repeats can also cause slippage during cycle sequencing whereby extension products anneal at different places within a tandem array and cause a mixed sequence. Longer tandems can also cause major assembly problems, particularly when the tandem repeats are highly similar and extend beyond the length of a single sequencing read length. Although transposons cannot solve clone propagation problems, transposon-based mapping and/or assembly tools can be used to accurately sequence and assemble clones such as TR-1 that can be propagated in plasmids (Fig. 5).
Nonrepetitive DNA
We have also used these transposon-based methods to sequence nonrepetitive DNA, and our results indicate that artificial transposon-mediated DNA sequencing is broadly useful with 1- to 15-kb plasmid inserts cloned from a variety of sources (Table3). We have also successfully integrated transposons into four different cosmid targets and have used the resulting transposon insertions to recover sequence information from these cosmids (Table 3). Several thousand artificial transposon insertions have been sequenced in our laboratories, and the data indicate that in vitro integration is highly random with Ty1 integrase irrespective of the base composition or sequence structure of the DNA target. This is in contrast to Ty1 transposition in vivo in yeast, where integration is specifically targeted to genes transcribed by RNA polymerase III in a host factor-dependent manner (Devine and Boeke 1996). Thus, in the absence of host factors, the DNA integration reaction mediated by Ty1 integrase is random and is therefore useful for DNA sequencing applications. As expected, target site duplications 5 bp in length are created routinely upon transposon integration in vitro, and these duplications are useful for assembling minicontigs. Moreover, because transposon libraries are fairly simple to construct compared with most alternatives, this approach is particularly useful for plasmid-based sequencing projects.
Plasmid and Cosmid Targets Analyzed
| Target name | DNA insert | Species[ii] | Vector |
| pSD523 | 8.0 kb genomic | S. cerevisiae | pRS200 |
| pCAR143 | 5.0 kb genomic | S. cerevisiae | pRS200 |
| pCAR206 | 2.1 kb genomic | S. cerevisiae | pMOB |
| pCAR158 | 2.0 kb genomic | S. cerevisiae | pBluescript |
| p3C-1 | 11 kb genomic | S. cerevisiae | YCp50-LEU2 |
| pSD553 | 4.5 kb genomic | S. cerevisiae | pRS316 |
| pSD546 | 3.0 kb genomic | S. cerevisiae | pRS323 |
| pEC89 | 3.0 kb genomic | S. cerevisiae | pBluescript |
| pKN116 | 5.0 kb genomic | S. pombe | pBluescript |
| p3.1 | 15 kb genomic | S. pombe | YCp50-LEU2 |
| cosmid JEDI C III | 45 kb genomic | L. donovani | cosmid |
| F18E3-10 | 2.6 kb genomic | C. elegans | pUC18 |
| C35B1 | 3.5 kb genomic | C. elegans | pUC18 |
| F32D1 | 3.0 kb genomic | C. elegans | pUC18 |
| ZK6 | 0.6 kb genomic | C. elegans | pUC18 |
| F23F1 | 1.0 kb genomic | C. elegans | pUC18 |
| F49F1 | 2.8 kb genomic | C. elegans | pUC18 |
| IMAGE clone 1 | 1–4 kb cDNA | H. sapiens | Lafmid |
| IMAGE clone 2 | 1–4 kb cDNA | H. sapiens | Lafmid |
| IMAGE clone 3 | 1–4 kb cDNA | H. sapiens | Lafmid |
| IMAGE clone 4 | 1–4 kb cDNA | H. sapiens | Lafmid |
| IMAGE clone 5 | 1–4 kb cDNA | H. sapiens | Lafmid |
| IMAGE clone 6 | 1–4 kb cDNA | H. sapiens | Lafmid |
| IMAGE clone 7 | 1–4 kb cDNA | H. sapiens | Lafmid |
| IMAGE clone 8 | 1–4 kb-cDNA | H. sapiens | Lafmid |
| IMAGE clone 9 | 1–4 kb cDNA | H. sapiens | Lafmid |
| pWAFp | 5.0 kb genomic | H. sapiens | pBluescript |
| pG2 | 7.0 kb cDNA | H. sapiens | pUC19 |
| pMG4 | 3.0 kb genomic | H. sapiens | pBluescript |
| pMG10 | 4.0 kb genomic | H. sapiens | pBluescript |
| pJEST | 2.5 kb cDNA | H. sapiens | pUC18 |
| pSD590 | 7.0 kb genomic | H. sapiens | pBluescript |
| p2A9 | 5.0 kb cDNA | H. sapiens | pUC8 |
| pBD28 | 6.0 kb cDNA | H. sapiens | pUC8 |
| cosmid F10080 | 40 kb genomic | H. sapiens | Lawrist 5 |
| cosmid F13544 | 35 kb genomic | H. sapiens | Lawrist 5 |
| cosmid F23932 | 45 kb genomic | H. sapiens | Lawrist 5 |
[i] Plasmid and cosmid targets for which artificial transposon insertion libraries have been constructed and used for mapping and/or sequencing.
[ii] Saccharomyces cerevisiae; Schizosaccaromyces pombe; Leishmania donovani; Caenorhabditis elegans; Homo sapiens.
Transposon-Like Repetitive Families
In addition to being a useful tool for gap closure, our sequencing strategy has also evolved into a useful tool for identifying novel transposon-like repetitive DNA families in genomes. In fact, we have identified six novel repetitive families in the C. elegansgenome using this strategy. The gaps themselves signal the possible presence of repetitive sequences (Chissoe et al. 1997), and the sequencing strategy allows accurate closure of the gap and discovery of the novel repetitive DNA (Figs. 2 and 3).
Several aspects of these repetitive families suggest that they represent transposable genetic elements in C. elegans. First, like transposons, multiple copies are present at dispersed sites throughout the genome. The sequences of these duplicated copies are often nearly or completely identical, suggesting that they originated from a single (or a small number of) master copies. Second, inverted or direct repeats are located at the termini of the IR and TR elements. Such highly characteristic repeats are observed frequently in transposable elements. For example, each of the Tc elements of C. elegans has inverted terminal repeats (Emmons et al. 1983; Collins et al. 1989; Leavitt and Emmons 1989; Dreyfus and Emmons 1991; Yuan et al. 1991; Collins and Anderson 1994), whereas the Ty retroelements of yeast have direct long terminal repeats (LTRs) (Boeke and Sandmeyer 1991). Finally, three of the six elements were flanked by short sequence duplications resembling the target site duplications of transposons. To date, transposition is the only known cellular mechanism for generating such duplications. For those transposons that have been studied carefully, target site duplications are known to be caused by staggered cleavage of the target DNA by an integrase or transposase, followed by strand joining with the transposon and subsequent gap repair.
Six transposon families have been identified previously in C. elegans, namely, Tc1–Tc6 (Emmons et al. 1983; Collins et al. 1989;Leavitt and Emmons 1989; Dreyfus and Emmons 1991; Yuan et al. 1991;Collins and Anderson 1994). Each of these transposons has terminal inverted repeats and target site duplications that are very similar to those identified for the IR elements. For example, IR-3 elements are flanked by 2-bp TA duplications that are identical to the target site duplications flanking Tc1 and Tc3 (andmariner). IR-1 is often flanked by these 2-bp Tc1/mariner-like TA duplications as well, but may also be flanked by a wide range of other dinucleotides, suggesting that it may represent a novel member of the Tc1/marinerfamily.
Tc4 is perhaps the most relevant C. eleganstransposon with respect to the IRs, because it is strikingly similar in structure and has been shown to be an active transposon (Yuan et al. 1991). Like the IR elements, Tc4 consists solely of two inverted 774-bp modules separated by a 57-bp spacer region, and is flanked by a 5-bp target site duplication (Fig. 5). Tc4 lacks significant open reading frames capable of encoding proteins that might carry out its transposition. However, larger (full-length) Tc4elements encoding significant open reading frames have been identified recently, and the proteins encoded are likely to be responsible for the transposition of all Tc4 elements (Li and Shaw 1993). It is attractive to speculate that the IR elements might also share such a relationship with currently unknown full-length IR elements.
The IR elements closely resemble a number of DNA transposons that are known to transpose by cut and paste mechanisms. These include the En/Spm elements of maize (Federoff 1989), the FB elements ofDrosophila (Bingham and Zachur 1989), and Tc1/mariner-like elements from a variety of organisms (Smit and Riggs 1996). Transposition is carried out with a transposase that binds to the terminal sequences of the element, excises the transposon at its termini, and then integrates it into a new target site; a target site duplication of characteristic length and/or sequence is typically generated. Transposon families of this type often contain both full-length and internally degenerate copies, and may include a large number of nonautonomous elements that retain only the terminal sequences required for transposition.
An unusual aspect of the IR and TR elements described here is that they lack substantial open reading frames. Transposons generally encode the proteins necessary for their own transposition, and these factors are often encoded within a central region of the transposon located between the terminal repeats. In the absence of such proteins, it is unclear how these elements could have become dispersed throughout the genome. One possibility is that, like Tc4, the IR elements might represent internally deleted copies of full-length functional elements located elsewhere in the genome. However, full-length elements such as those proposed have not been identified in the 80% of C. eleganssequenced to date, suggesting that these elements either reside in the unsequenced 20% of the genome or have been lost from C. elegans. Alternatively, the IR and TR elements represent nonautonomous transposons that rely entirely on unrelated elements for their mobility. This is unlikely because nonautonomous elements generally have the same terminal sequences as the transposon whose machinery they use, and these elements lack similarity to any of the known C. elegans transposons. Nevertheless, the fact that IR-4 and IR-5 share similar terminal sequences suggests that these elements share a common mechanism of transposition.
Higher eukaryotic genomes have long been known to contain repetitive DNA but the potential role of such DNA in genomes is largely unknown. It is now becoming possible to identify and study such repetitive DNA by examining the data emerging from genome sequencing projects. Data from the C. elegans project suggests that much of the middle-repetitive DNA of this organism represents potentially active transposable genetic elements as well as inactive relics of such elements. These transposon-like repetitive families represent a potential source of genomic mutation, and might also provide essentialcis- or trans-acting functions for the host organism (e.g., see Levis et al. 1993).
The C. elegans genome sequencing project began initially with the gene-rich regions of the six C. elegans chromosomes, and has now progressed to regions such as the telomeric arms of these chromosomes. The same genomic regions of organisms such asDrosophila consist largely of repetitive DNA in the form of heterochromatin. Although these regions will be particularly interesting from the viewpoint of identifying and studying transposons, and for gaining an understanding of heterochromatin, they might be especially difficult to sequence. Thus our strategy for sequencing repetitive DNA will be particularly useful in the finishing stages of genome projects where repetitive DNA might be abundant.
METHODS
Construction of Artificial Transposons and Plasmids
AT-2/M was constructed with custom PCR primers designed to introduce sequences homologous to the standard M13–20 and R primer sequences into the AT-2 transposon 25 bp from the termini (Fig. 1). The PCR product was cloned into the XbaI–HindIII sites of pAT-2 (Devine and Boeke 1994) using restriction sites incorporated near the 5′ termini of these primers. The resulting plasmid carrying AT-2/M is pSD610. AT-2/T and AT-2/ST were constructed in a similar manner, except that the standard T3, T7, and SP6 primer sequences were incorporated instead of M13–20/R (Fig. 1). The plasmids carrying AT-2/T and AT-2/ST are pSD611 and pSD612, respectively. AT-3 and AT-4 were constructed by inserting a 0.9-kb BamHI fragment from pGH54 (Joyce and Grindley 1984), carrying the neo gene from Tn903, into BamHI-digested pAT-1 (Devine and Boeke 1994). The plasmids carrying AT-3 and AT-4 are pSD570 and pSD571, respectively. Subcloning was accomplished with standard methods (Ausubel et al. 1987).
Integration of Artificial Transposons into DNA Targets in Vitro with Ty1 Integrase
In vitro integration reactions were carried out in 20–50 μl volumes containing 1–5 μg of target plasmid, 0.5 μg of transposon, and 2–5 μl of virus-like particles (providing the Ty1 integrase) in a buffer containing 10 mm Tris-HCl at pH 7.5, 10 mm MgCl2, 1 mm DTT, and 5% PEG 8000 (Devine and Boeke 1994). Recovered DNA was used to transform DH10B electrocompetent Escherichia coli by electroporation. Transformants carrying AT-2 recombinants were plated on M9 medium containing 100 μg/ml of trimethoprim and 50 μg/ml of ampicillin. Those carrying AT-3 and AT-4 recombinants were plated on LB medium containing 50 μg/ml of ampicillin plus 50 μg/ml of kanamycin.
Plasmid Preparation and Transposon Mapping
Recombinant plasmids carrying transposon insertions were prepared from DH10B E. coli cultures using a modified boiling method (Holmes and Quigley 1981; Kimmel et al. 1997) or a 96-well alkaline lysis method (Advanced Genetic Technologies Corp., Gaithersburg, MD). These plasmids were used for both mapping and sequencing. When necessary, transposon insertion sites were mapped by systematic restriction analysis using either regional mapping or positional mapping: Regional mapping was used to assign a particular transposon insertion to a known restriction fragment of a target plasmid such as the insert, whereas positional mapping was used to determine the precise location of a transposon insertion. Both types of mapping were performed by cutting transposon recombinants with restriction enzymes that cleave at both ends of the transposon to create a cleavage site at the site of transposon insertion (EcoRI and NotI, e.g., Figs. 1 and 3). For regional mapping, this cleavage site was generated with a pair of enzymes that also cut at the boundaries of the vector and the insert. Insertions were assigned to a restriction fragment according to the presence of a cleavage site. For positional mapping, the transposon cleavage site was mapped relative to a second restriction site in the plasmid (Fig. 3). Two additional types of interval mapping were also developed. One employed restriction mapping whereas the other involved PCR. The restriction mapping strategy was similar to the approaches above, except that restriction enzymes were chosen that did not cut within the transposon (such as PstI orPvuII). If a restriction fragment lacked a transposon, it remained the same size as in the parental plasmid. However, if it contained a new transposon insertion, it was increased in size by the size of the transposon (864 bp for AT-2, e.g.). The PCR mapping strategy was similar except that the PCR product itself was increased by the size of the transposon if an insertion occurred within the interval amplified by the PCR.
Sequencing
Dye terminator and dye primer DNA sequencing was performed with Taq FS polymerase (Perkin Elmer) or Thermosequenase (Amersham), following the manufacturers’ instructions. The SD118 and SD119 dye primers (also known as PI+ and PI−, respectively) were custom synthesized for use with the primer island transposon AT-2 (Perkin Elmer/ABD), whereas M13-21, R, T3, T7, and SP6 primers were purchased (Perkin Elmer/ABD, Amersham, Operon). ET primers (Amersham) were used according to the manufacturer’s instructions. Dye primer sequencing with the SD118 and SD119 primers was carried out using the following cycling parameters: 20 cycles of 96°C for 10 sec; 50°C for 5 sec; 70°C for 60 sec; followed by 20 cycles of 96°C for 10 sec; 70°C for 60 sec. Dye terminator sequencing with the SD118 and SD119 primers was performed with 25 cycles of 94°C for 15 sec; 50°C for 15 sec; 60°C for 4 min. Similar protocols were used for the other primers. Sequencing reactions were analyzed on ABI 373A or 377 automated sequencers. Sequences were assembled using the Sequencher DNA analysis software package by Genecodes Corporation or with XGAP (Dear and Staden 1991) and PHRAP (P. Green, unpubl.). The 5-bp target site duplications flanking the transposons were used as fusion sites to assemble minicontigs of 600–1000 bp from the two sequences initiated within each transposon. Mapping was also used to confirm assemblies when necessary. Finished consensus sequences were used to search GenBank (NCBI) or the Genome Sequencing Center database at Washington University at St. Louis (http://genome.wustl.edu:80/gsc/gschmpg.html) using the BLAST algorithm and the standard settings. Repetitive sequences and the accompanying cosmid records (indicating chromosome map location and gene information) were retrieved from GenBank or the Genome Sequencing Center database, and a web site was established to provide detailed information on these elements with links to their cosmid records (http://www.welchlink.welch.jhu.edu/~devine/RepDNAdb.html). Mobility files for the SD118 and 119 dye primers and additional protocols are also available at this site.
We thank Bob Waterston for helpful discussions and the Genome Sequencing Center for assistance. We acknowledge the C. elegansMapping and Sequencing Consortium for providing genome sequence data. We also thank the individuals who provided the plasmids shown in Table 3. This work was supported by American Cancer Society Postdoctoral Fellowship PF4040 (to S.E.D.), a grant from the Johns Hopkins University (to S.E.D. and J.D.B.), and a sponsored research agreement from Perkin Elmer, Applied Biosystems (to S.E.D. and J.D.B.). S.E.D. and J.D.B. have a financial interest in this work. This arrangement has been reviewed and approved by the committee on conflict of interest at Johns Hopkins University School of Medicine.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Notes
[5] Corresponding author.
Notes
[6] E-MAIL [email protected]; FAX (410) 614-2987.
REFERENCES
- ↵T. AdachiM. MizuuchiE.A. RobinsonE. AppellaM.H. O’DeaM. GellertK. Mizuuchi(1987) DNA sequence of the E. coli gyrB gene: Application of a new sequencing strategy. Nucleic Acids Res. 15:771–784.
- ↵A. Ahmed(1985) A rapid procedure for DNA sequencing using transposon-promoted deletions in Escherichia coli. Gene 39:305–310.
- ↵F. AusubelR. BrentR.E. KingstonD.D. MooreJ.G. SeidmanJ.A. SmithK. Struhl(1987) Current protocols in molecular biology. (Greene Publishing, New York, NY).
- ↵P.M. BinghamZ. Zachur(1989) Retrotransposons and the FB transposon from Drosophila melanogaster. in Mobile DNA, eds D.E. BergM.M. Howe(American Society for Microbiology, Washington, DC.) pp 485–502.
- ↵R.K. BlackmanW.M. Gelbart(1989) The transposable element hobo of Drosophila melanogaster. in Mobile DNA, eds D.E. BergM.M. Howe(American Society for Microbiology, Washington, DC.) pp 523–529.
- ↵R.J. BrittenD.E. Kohn(1968) Repeated sequences in DNA. Science 161:529–540.
- ↵J.D. BoekeS.B. Sandmeyer(1991) Yeast transposable elements. in The molecular and cellular biology of the yeast Saccharomyces : Genome dynamics, protein synthesis, and energetics, eds J. BroachJ. PringleE. Jones(Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY), pp 193–264.
- ↵J.J. CollinsP. Anderson(1994) The Tc5 family of transposable elements in Caenorhabditis elegans. Genetics 137:771–781.
- ↵J. CollinsE. ForbesP. Anderson(1989) The Tc3 family of transposable genetic elements in Caenorhabditis elegans. Genetics 112:47–55.
- ↵S. DearR. Staden(1991) A sequence assembly and editing program for efficient management of large projects. Nucleic Acids Res. 19:3907–3911.
- ↵S.E. DevineJ.D. Boeke(1994) Efficient integration of artificial transposons into plasmid targets in vitro: A useful tool for DNA mapping, sequencing, and functional genetic analysis. Nucleic Acids Res. 22:3765–3772.
- ↵(1996) Integration of the yeast retrotransposon Ty1 is targeted to regions upstream of genes transcribed by RNA polymerase III. Genes & Dev. 10:620–633, ibid.
- ↵D.H. DreyfusS.W. Emmons(1991) A transposon-related palindromic repetitive sequence from C. elegans. Nucleic Acids Res. 19:1871–1877.
- ↵S.W. EmmonsB. RosenzweigD. Hirsh(1980) Arrangement of repeated sequences in the DNA of the nematode Caenorhabditis elegans. J. Mol. Biol. 144:481–500.
- ↵S.W. EmmonsL. YesnerK.S. RuanD. Katzenberg(1983) Evidence for a transposon in Caenorhabditis elegans. Cell 32:55–65.
- ↵W.R. Engels(1989) P Elements in Drosophila melanogaster. in Mobile DNA, eds D.E. BergM.M. Howe(American Society for Microbiology, Washington, DC.) pp 437–484.
- ↵N.V. Federoff(1989) Maize transposable elements. in Mobile DNA, eds D.E. BergM.M. Howe(American Society for Microbiology, Washington, DC.) pp 375–411.
- ↵D.J. Finnegan(1989a) The I factor and I-R hybrid dysgenesis in Drosophila melanogaster. in Mobile DNA, eds D.E. BergM.M. Howe(American Society for Microbiology, Washington, DC.) pp 503–517.
- ↵D.E. BergM.M. Howe, eds(1989b) F and related elements in Drosophila melanogaster. Mobile DNA (American Society for Microbiology, Washington, DC.) pp 519–521, ibid.
- ↵D.L. Hartl(1989) Transposable element mariner in Drosophila species. in Mobile DNA, eds D.E. BergM.M. Howe(American Society for Microbiology, Washington, DC.) pp 5531–5536.
- ↵D.S. HolmesM. Quigley(1981) A rapid method for the preparation of bacterial plasmids. Anal. Biochem. 114:193–197.
- ↵C.M. JoyceN.D. Grindley(1984) Method for determining whether a gene of Eschericia coli is essential: Application to the polA gene. J. Bacteriol. 158:636–643.
- ↵B. KimmelM.J. PalazzolaC. MartinJ.D. BoekeS.E. Devine(1997) Transposon-mediated DNA sequencing. in Genome analysis: A laboratory manual, eds E. GreenB. BirrenR. MyersP. Hieter(Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY) . (In press)..
- ↵A. LeavittS.W. Emmons(1989) The Tc2 transposon in Caenorhabditis elegans. Proc. Natl. Acad. Sci. 86:3232–3236.
- ↵R.W. LevisR. GanesanK. HoutchensL.A. TolarF.M. Sheen(1993) Transposons in place of telomeric repeats at the Drosophila telomere. Cell 75:1083–1093.
- ↵W. LiJ.E. Shaw(1993) A variant Tc4 transposable element in the nematode C. elegans could encode a novel protein. Nucleic Acids Res. 21:59–67.
- ↵A.J. LinkM.V. Olson(1991) Physical map of the Saccharomyces cerevisiae genome at 100-kb resolution. Genetics 127:681–698.
- ↵J.V. MoranS.E. HolmesT.P. NaasR.J. DeBerardinisJ.D. BoekeH.J. Kazazian(1996) High frequency retrotransposition in cultured mammalian cells. Cell 87:917–927.
- ↵S.H. PhadnisH.V. HuangD.E. Berg(1989) Tn5supF, a 264-base-pair transposon derived from Tn5 for insertional mutagenesis and sequencing DNAs cloned in phage lambda. Proc. Natl. Acad. Sci. 86:5908–5912.
- ↵A.F.A. Smit(1996) The origin of interspersed repeats in the human genome. Curr. Opin. Genet. Dev. 6:743–748.
- ↵A.F.A. SmitA.D. Riggs(1996) Tiggers and DNA transposon fossils in the human genome. Proc. Natl. Acad. Sci. 93:1443–1448.
- ↵M. StrathmannB.A. HamiltonC.A. MayedaM.I. SimonW.M. MeyerowitzM.J. Palazzolo(1991) Transposon-facilitated DNA sequencing. Proc. Natl. Acad. Sci. 88:1247–1250.
- ↵J.E. SulstonS. Brenner(1974) The DNA of C. elegans. Genetics 77:95–104.
- ↵H.G. van LuenenR.H. Plasterk(1994) Target site choice of the related transposable elements Tc1 and Tc3 of Caenorhabditis elegans. Nucleic Acids Res. 22:262–269.
- ↵R. WilsonR. AinscoughK. AndersonC. BaynesM. BerksJ. BonfieldJ. BurtonM. ConnellT. CopseyJ. Cooper(1994) 2.2 Mb of contiguous nucleotide sequence from chromosome III of C. elegans. Nature 368:32–38.
- ↵R.K. WilsonE.R. Mardis(1997) Shotgun sequencing. in Genome analysis: A laboratory manual, eds E. GreenB. BirrenR. MyersP. Hieter(Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY) . (In press)..
- ↵J.Y. YuanM. FinneyN. TsungH.R. Horvitz(1991) Tc4, a Caenorhabditis elegans transposable element with an unusual fold-back structure. Proc. Natl. Acad. Sci. 88:3334–3338.