
Abstract
Circular RNA (circRNA) is a class of RNA molecules that forms a closed loop with their 5′ and 3′ ends covalently bonded. CircRNAs are known to be more stable than linear RNAs, have distinct properties and functions, and are promising biomarkers. Existing methods for assembling circRNAs heavily rely on the annotated transcriptomes, hence exhibiting unsatisfactory accuracy without a high-quality transcriptome. We present TERRACE, a new algorithm for full-length assembly of circRNAs from paired-end total RNA-seq data. TERRACE uses the splice graph as the underlying data structure that organizes the splicing and coverage information. We transform the problem of assembling circRNAs into finding paths that “bridge” the three fragments in the splice graph induced by back-spliced reads. We adopt a definition for optimal bridging paths and a dynamic programming algorithm to calculate such optimal paths. TERRACE features an efficient algorithm to detect back-spliced reads missed by RNA-seq aligners, contributing to its much-improved sensitivity. It also incorporates a new machine-learning approach trained to assign a confidence score to each assembled circRNA, which is shown to be superior to using abundance for scoring. On both simulations and biological data sets, TERRACE consistently outperforms existing methods by a large margin in sensitivity while achieving better or comparable precision. In particular, when the annotations are not provided, TERRACE assembles 123%–413% more correct circRNAs than state-of-the-art methods. TERRACE presents a significant advance in assembling full-length circRNAs from RNA-seq data, and we expect it to be widely used in future research on circRNAs.
Splicing is a ubiquitous and essential post-transcriptional modification of precursor mRNAs. In this process, introns are excised and the two flanking exons are stitched together. Canonical splicing covalently connects the 3′ end of an upstream exon to the 5′ end of the immediate downstream exon. A class of noncanonical splicing, known as back-splicing, stitches the 3′ end of a downstream exon to the 5′ end of an upstream exon via a back-splicing junction (BSJ), forming a closed circular structure called circular RNA (circRNA). Both canonical and back-splicing junctions may experience alternative splicing; consequently, a gene can express multiple circRNAs (Li et al. 2018b; Ji et al. 2019). CircRNAs are more prevalent and conserved than previously thought. More than 60% of human genes express at least one circRNA (Ji et al. 2019). Notably, BIRC6, a gene that has only 12 linear transcripts, was found to express 243 circRNAs (Ji et al. 2019). CircRNAs usually have a lower expression level than their linear forms, but certain circRNAs are significantly more abundant and constitute the major isoform of their genes (Szabo and Salzman 2016; Ji et al. 2019; Kristensen et al. 2022; Vromman et al. 2023). Expression of circRNAs is also often tissue-specific and developmental stage–specific in a spatiotemporal pattern (Memczak et al. 2013; Rybak-Wolf et al. 2015; Venø et al. 2015).
An increasing volume of research has been evidencing the regulatory functionality of circRNAs and their use in disease diagnosis (Memczak et al. 2013; Kristensen et al. 2022). To name a few, circBIRC6 has been found to play a role in cell pluripotency (Yu et al. 2017); circHIPK3, in cell growth (Zheng et al. 2016); and circular Foxo3, in suppression of cancer cell proliferation and survival (Yang et al. 2016). Because of its circular structure, circRNAs are resistant to most RNA degradation mechanisms (Li et al. 2018b). In particular, its lack of free 5′ and 3′ ends protects the molecule itself from exonucleases (Suzuki and Tsukahara 2014), making them more stable and causing them to have a longer half-life than their linear counterparts (Jeck et al. 2013; Jeck and Sharpless 2014). CircRNAs have been reported to serve as novel biomarkers in carcinogenesis and pathogenesis (Rybak-Wolf et al. 2015; Wang et al. 2016; Kristensen et al. 2022) and have found their potential use in noninvasive diagnosis (Vo et al. 2019).
It is therefore of great interest to detect expressed circRNAs in cells. The RNA sequencing (RNA-seq) protocols that target on linear RNAs or mRNAs with poly(A) tails will neglect circRNAs (Kristensen et al. 2022). Tailored RNA-seq experiments have been designed for circRNAs, for example, using RNase R to digest linear RNAs and therefore to enrich circRNAs followed by sequencing (Ji et al. 2019; Vo et al. 2019). These approaches are efficient in raising the sensitivity of circRNA detection but are often low-throughput and costly. The total RNA-seq technology, on the other hand, can capture and sequence the entire population of RNA molecules, including circRNAs, in a biological sample. Total RNA-seq is high-throughput and has been widely used in many studies on RNAs. Large-scale total RNA-seq data sets are available in public repositories such as the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) (Clough and Barrett 2016), the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) (Leinonen et al. 2011), and The ENCODE Project (The ENCODE Project Consortium 2012), providing a rich resource to further study circRNAs.
Numerous computational methods to detect circRNAs from total RNA-seq were published lately (for a recent review, see Vromman et al. 2023). However, many of them require a fully annotated transcriptome, including CYCLeR (Stefanov and Meyer 2023), psirc (Yu et al. 2021), CircAST (Wu et al. 2019), CIRCexplorer2 (Zhang et al. 2016), and CIRCexplorer3 (Ma et al. 2019). Those methods’ dependency on an existing annotation significantly limits their capability to detect novel circRNAs and their applicability to nonmodel species without a well-annotated transcriptome. Other tools, including CIRI-full (Zheng et al. 2019), Circall (Nguyen et al. 2021), CircMiner (Asghari et al. 2020), CIRI2 (Gao et al. 2018), CIRI-AS (Gao et al. 2016), and CircMarker (Li et al. 2018a), can be operated annotation-free, are constrained to identifying BSJ only, in a deficiency of assembling full-length circRNAs. Additionally, some methods have to be combined with experimental enrichment of circRNA (Stefanov and Meyer 2023). Because of the complexity of alternative splicing and low circRNA abundance, current computational methods unfortunately fail to accurately detect BSJs while also producing exceedingly unsatisfying full-length assemblies. Therefore, the problem of in silico assembly of circRNAs, from highly sensitive BSJ identification to full-length circRNA reconstruction, remains largely unresolved.
Here we present accuraTe assEmbly of circRNAs using bRidging and mAChine lEarning (TERRACE), a new tool for assembling full-length circRNAs from paired-end total RNA-seq data. TERRACE stands out by assembling circRNAs accurately without relying on annotations, a feature absent in most existing tools. TERRACE starts with a fast, light-weight algorithm to identify back-spliced reads (i.e., reads containing BSJs). We realize that the key to assembling circRNAs is to correctly “bridge” the three fragments in a back-spliced read. We formulate this task as seeking paths in a weighted splice graph that can connect the three fragments and that has its “bottleneck” weight maximized, and solve this formulation using an efficient dynamic programming algorithm. TERRACE also features a new machine-learning model that is trained to assign confidence scores to assembled circRNAs, which we show outperforms abundance-based ranking.
Results
Experimental setup
TERRACE
We implemented the algorithm explained in Methods as a new circRNA assembler named TERRACE. TERRACE takes the alignment (in BAM format; can be produced by any RNA-seq aligner such as STAR [Dobin et al. 2013] or HISAT [Kim et al. 2015]) of total RNA-seq reads as input and produces a list of full-length, annotated circRNAs in the GTF format. TERRACE is designed to assemble reliable circRNAs without the need for an annotation but comes with the flexibility of an optional reference annotation file.
Methods for comparison
Although there is an abundance of methods that predict BSJs, very few of them produce the full-length annotation (for a classification, please refer to the CYCLeR paper by Stefanov and Meyer 2023). Full-length circRNA assemblers that do not require a reference annotation are even rarer. We only identify CIRI-full as such a tool, and it serves as the state of the art in the field. We include CIRCexplorer2, CIRI-full, and CircAST for comparison when the reference annotation is provided. CircAST needs to be provided with a list of BSJs from CIRI2 or CIRCexplorer2; we choose to use CIRI2, as the results are worse when CIRCexplorer2 is used. CYCLeR is not suitable for comparison because it necessitates both control total RNA-seq samples and circRNA-enriched samples, whereas our study exclusively focuses on total RNA libraries. Sailfish-cir and CIRCexplorer3 are tools that primarily target circRNA quantification and, hence, are not included.
Data sets
We use both real data set and simulated data set for evaluation. The real data set is chosen from the isoCirc (Xin et al. 2021) paper (obtained from the NGDC BioProject database [https://ngdc.cncb.ac.cn/bioproject/] under accession number PRJCA000751), which consists of eight human tissue samples (lung, brain, skeletal muscle, heart, testis, liver, kidney, and prostate). Full-length circRNAs for these samples are cataloged by isoCirc using a combination of a reference gene annotation and long reads, which we use as the ground truth. The simulated data set is generated using CIRI-simulator (Gao et al. 2015), previously used by several methods (Gao et al. 2016; Jia et al. 2019; Zhang et al. 2020; Nguyen et al. 2021). The expressed circRNAs are available in the simulation that we use as ground truth. We simulate 10 samples and report the average performance.
Evaluation
We define an assembled circRNA to be correct if the coordinates of its BSJ and its intron-chain all exactly match a circRNA in the ground truth. We then calculate recall, defined as the proportion of circRNAs in the ground truth that are correctly assembled, and precision, which is the percentage of assembled circRNAs that are correct. It is common that a method exhibits high precision but low sensitivity or vice versa. Thus, we report F-score, calculated as (2 × recall × precision) / (recall + precision). We also draw the precision-recall curve for TERRACE. With the curve, we can calculate an adjusted precision with respect to another method, defined as the precision of TERRACE at a point on the curve when its recall matches the recall of the compared method. We draw and compare two precision-recall curves for TERRACE, by using either the “abundance” or the “score” inferred by the random forest.
Comparison of assembly accuracy
Figure 1, A–H, shows the accuracy of TERRACE and CIRI-full on the real data set when the reference annotation is not provided. TERRACE yields a significantly higher number of correct circRNAs compared with CIRI-full across all samples (228% more on average) at a comparable precision (43% vs. 40% on average). The F-score of TERRACE is consistently higher than that of CIRI-full (22% vs. 10% on average). Both the precision-recall curves of TERRACE are above those of CIRI-full, proving TERRACE's much enhanced accuracy. Specifically, the average adjusted precision of TERRACE is 75% (using the random-forest curve; it is 72% using the coverage curve) compared with CIRI-full at 40%.
Comparison of assembly accuracy without annotation. F-scores (%) are indicated on top of data points. (A) Brain, (B) lung, (C) skeletal muscle, (D) heart, (E) testis, (F) liver, (G) kidney, (H) prostate.
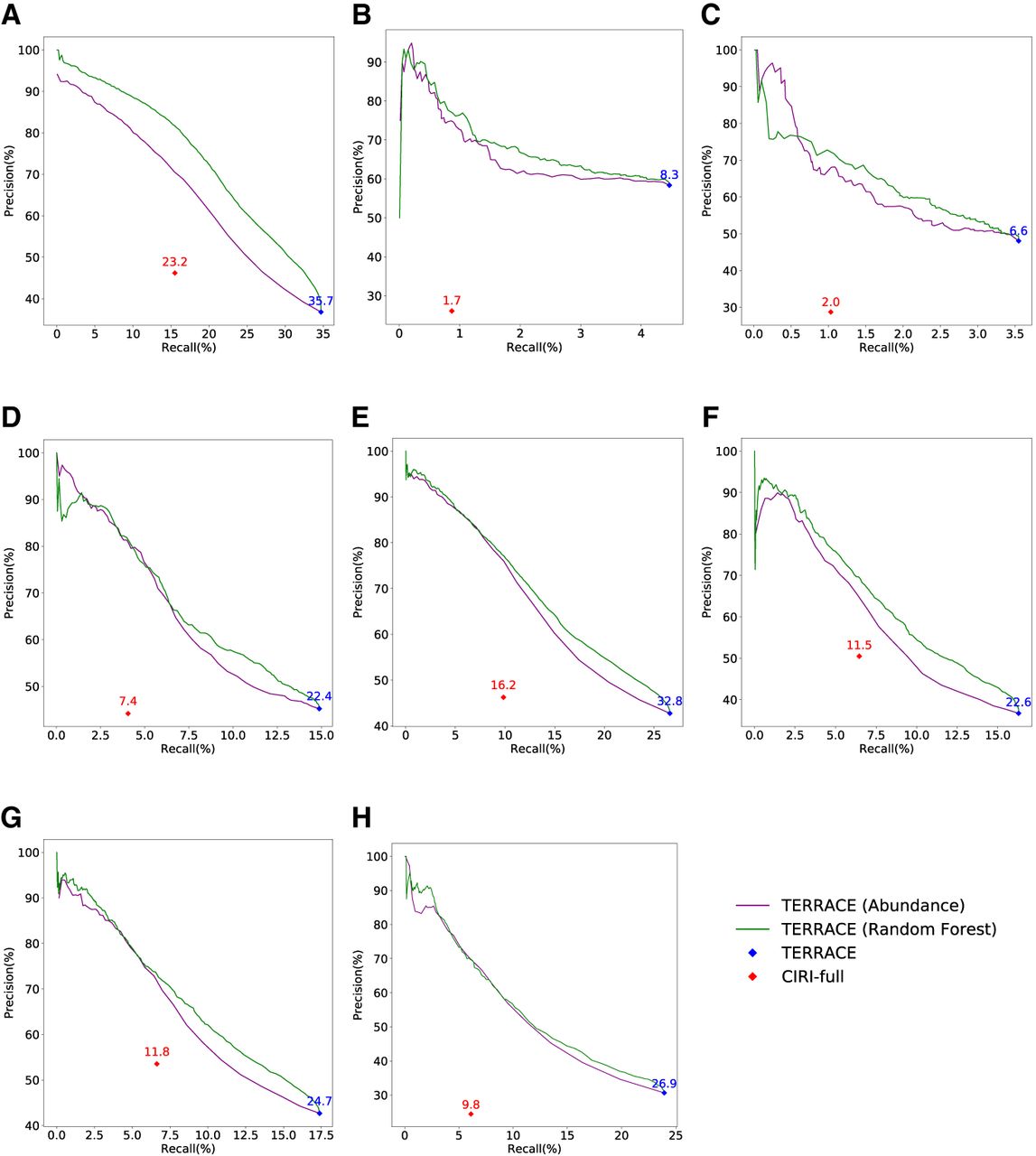
The confidence scores produced by random forest result in a superior curve than that using abundance as the varying parameter. This improvement can be attributed to the machine-learning model's ability to learn a more effective and accurate scoring function from a broad class of features (including abundance). Of note, when the abundance is high, simply using it can identify correct circRNAs accurately in a performance similar to random forest (the low-recall regions in the figures). However, when the abundance becomes low, correct and incorrect circRNAs become indistinguishable by just using abundance. The trained scoring function effectively tackles this problem, ensuring a bigger improvement in precision in the high-recall regions.
Figure 2, A–H, compares the accuracy of different methods running with an annotation. TERRACE reconstructs 17% more correct circRNAs than CIRCexplorer2 while obtaining higher precision on all samples. TERRACE assembles 271% more correct ones than CIRI-full at a comparable precision (42% vs. 40% on average). CircAST exhibits higher precision but at a cost of much reduced recall. Again, TERRACE obtains a higher F-score than all other methods on all samples. Also, the precision-recall curves of TERRACE consistently lie well above all the other data points, demonstrating its improved accuracy. In particular, the average adjusted precision of TERRACE (using the random-forest curve) is 76% compared with CIRI-full at 40%, 50% compared with CIRCexplorer2 at 40%, and 86% compared with CircAST at 59%.
Comparison of assembly accuracy with annotation. F-scores (%) are indicated on top of data points. (A) Brain, (B) lung, (C) skeletal muscle, (D) heart, (E) testis, (F) liver, (G) kidney, (H) prostate.
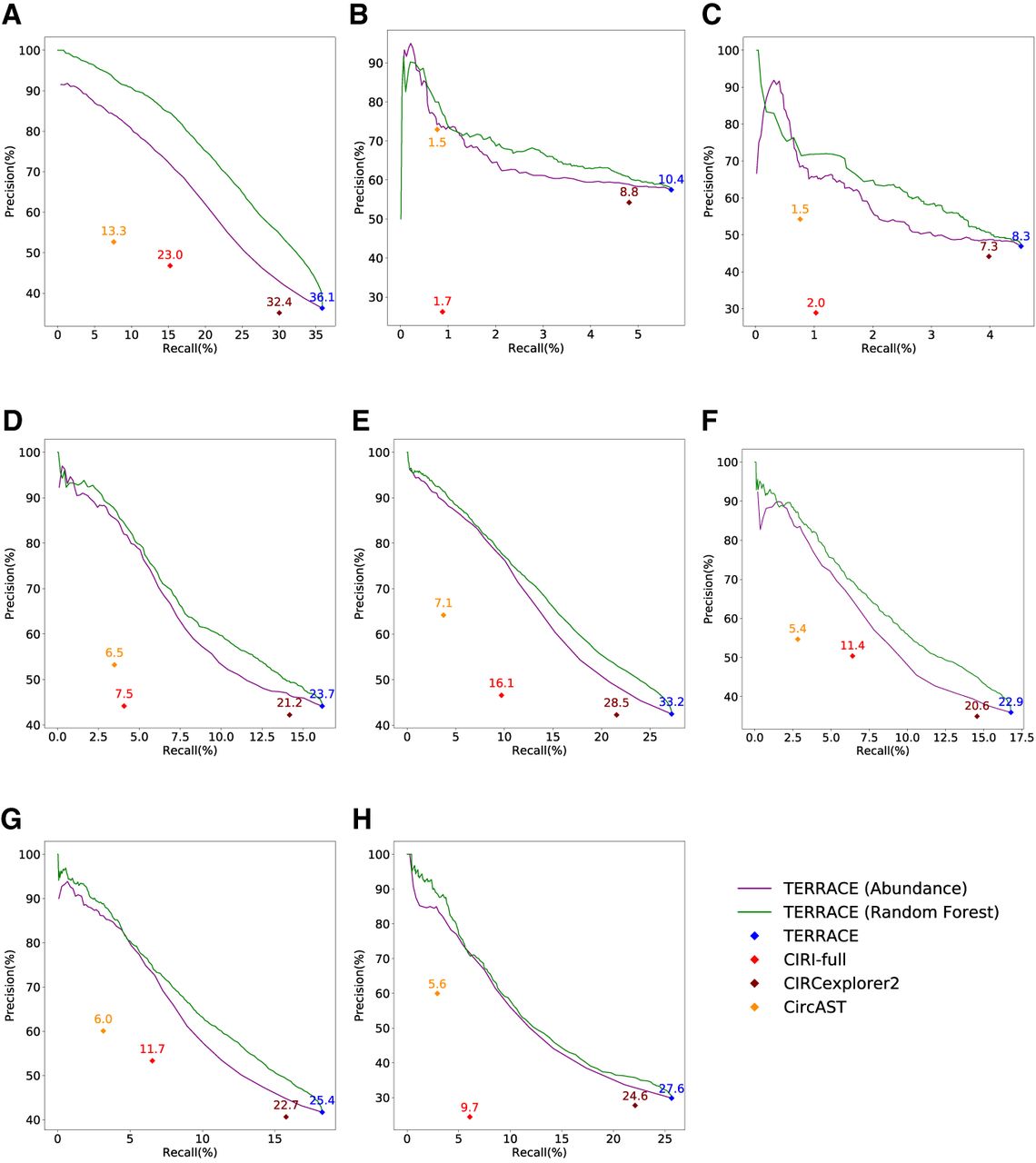
We also compare the precision-recall curves of TERRACE with the curves generated by varying the abundance thresholds of other methods. Supplemental Figures S4 and S5 confirm the superiority of TERRACE over the curves of other methods. To better quantify the improvement, we measure the area under the precision-recall curve. Because the precision or recall ranges of different methods may vary substantially, we compare TERRACE with each alternative method by locating a shared range (constrained by recall or by precision), and calculate the partial area under the precision-recall curve (pAUC). For details, see Supplemental Tables S3–S5.
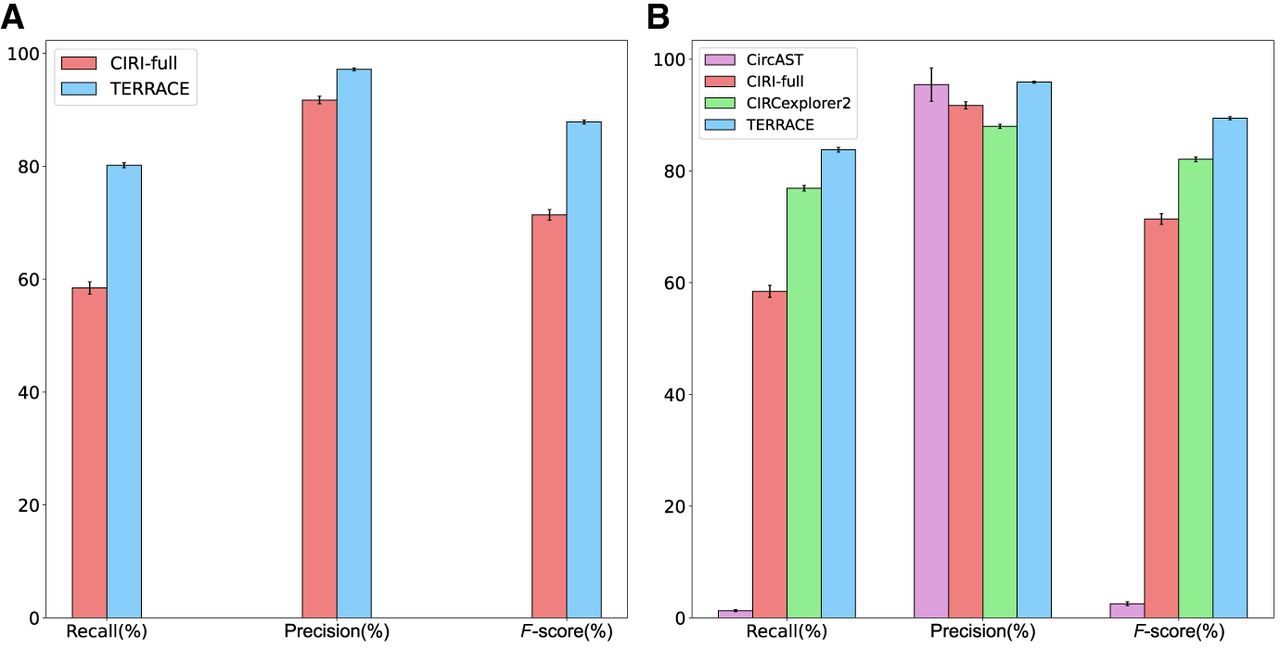
Figure 3, A and B, shows the accuracy of TERRACE compared with other tools on simulated data. When annotation is not provided, TERRACE identifies an average of 37% more correct circular transcripts and achieves better precision than CIRI-full. In the presence of annotations, TERRACE consistently outperforms CIRI-full and CIRCexplorer2 on all measures. TERRACE exhibits an average precision comparable to CircAST while maintaining much better recall, resulting in a much higher overall F-score. We conducted additional experiments to evaluate TERRACE's performance across a range of simulation parameters, including read length, circular transcript coverage, and linear transcript coverage. Across all variations, TERRACE demonstrated improved overall performance compared with other methods (see Supplemental Tables S6, S7). We observe that recall rates for TERRACE is <80% when the circular coverage is reduced to 5×. This is because we require the BSJ of a back-spliced read to match a splice junction inferred from read alignments. When the sequencing coverage is low, there exist sparse regions where the aligner is unable to infer a splice junction. Because of the absence of these junctions, many correct back-spliced reads get discarded, resulting in low recall. One possible approach to address this would be to adjust the splice junction requirement based on coverage depths.
Average accuracy of different tools on simulated data, (A) without annotation and (B) with annotation. The error bars show the SD over 10 simulated samples.
We realize that the recall rates of various methods including TERRACE are quite low on real data (Figs. 1, 2), particularly in lung and muscle tissues. This trend is also evident in the precision values. One plausible explanation for this discrepancy could be linked to the use of annotated circRNAs from long-read data as the ground truth for evaluation. Upon analyzing the count of annotated circRNAs across the samples and comparing them to the limited number detected by various methods (refer to Supplemental Table S8), we hypothesize that long-read and short-read total RNA-seq data (which are used here) capture divergent sets of expressed circRNAs. This may result in accurate predictions from short reads being misclassified as incorrect, leading to an underestimation of both precision and sensitivity. As evidence, we notice that the read coverage across the gene loci in muscle and lung samples is highly nonuniform: Many reads cluster densely within a few gene loci, leaving other genes with sparse coverage. This causes all methods, including TERRACE, to construct much fewer numbers of correct circRNAs compared with the ground truth. The number of circRNAs annotated using long reads is in the similar range with other samples, so the coverage nonuniformity seems not to exist in the long-read data set. Nevertheless, we emphasize that although the ground truth used for this study may not fully capture the absolute accuracy, it still serves as a fair benchmark for comparing the relative accuracy of different methods. Given the underestimated recall rates of TERRACE on the biological samples, we resort to simulations to illustrate that achieving high recall is possible in an unbiased setting. The performance of TERRACE on simulated samples strongly reinforces this assertion.
Comparison with long-read assemblers
We also conducted experiments to compare the results of TERRACE with a long-read assembler, CIRI-long (Zhang et al. 2021). CIRI-long analyzes various sequencing protocols to measure their effect on circRNA detection and concludes that the optimal procedure for circRNA detection using nanopore technology should include RNase R, A-tailing, reverse transcription with SMARTer RT under RNase H conditions, and 1 kb (long) fragment size selection. We use nanopore sequencing reads from the optimal protocol to run CIRI-long, and Illumina short reads (RNase R and Total) from the same study to run TERRACE. The data sets from two biological replicates of mouse brains are available from the NGDC Genome Sequence Archive (GSA; https://ngdc.cncb.ac.cn/gsa) under accession number CRA003317. Because an established ground truth is not available, we resort to finding the number of overlapping circRNAs between CIRI-long and TERRACE as illustrated in Supplemental Figures S7 and S8. The low number of overlapping circRNAs compared with the number detected uniquely by each tool reconfirms the hypothesis that long reads and short reads express divergent sets of circRNAs. Additionally, the large number of unique detections by TERRACE is an indication of many de novo circRNAs that are likely to be true given the relatively higher precision of TERRACE on the benchmarking data sets.
Discussion
The substantial growth in research dedicated to circRNAs in recent years underscores their significance in biology and medicine. Despite the abundance of experimental and computational techniques designed for circRNA detection, inherent limitations persist within these methods. Current experimental protocols often require special enrichment of libraries for accurate detection, whereas computational methods are hindered by their dependence on annotation and inability to reconstruct full-length molecules. TERRACE made a significant advancement toward closing this critical gap. TERRACE utilized a fast algorithm that effectively detects previously overlooked BSJs. A bridging system, which we originally proposed for improving (linear) transcript assembly, was redesigned to reconstruct the full-length circRNAs. Instead of using abundance for ranking, TERRACE learned a better score function with a broader class of informative features. TERRACE outperforms existing tools drastically, especially in scenarios in which annotations are unavailable. We anticipate widespread adoption of TERRACE, particularly in studies involving species lacking well-annotated transcriptomes.
Further improvements can be made for TERRACE. The precision-recall curves (see Figs. 1, 2) are not satisfactory. We would investigate the extraction of more features and advanced learning approaches. For instance, we can incorporate the count of splicing positions or partial exons supporting a BSJ as extra features. Additionally, we could explore training a model with complete sequences of bona fide circRNAs, possibly sourced from established circRNA databases, to better differentiate between accurate and erroneous ones. An LSTM model may be used for such sequence-based training. We also could consider extending TERRACE to incorporate additional types of data. Leveraging long reads and/or circRNA-enriched libraries for detection may reveal less obvious circRNAs, enhancing the assembly accuracy.
Methods
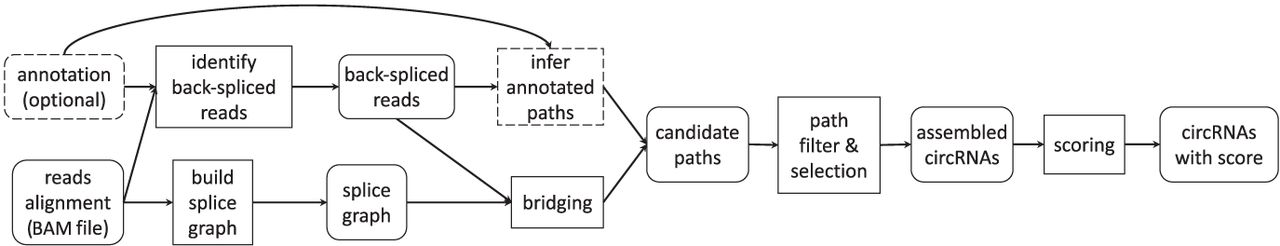
TERRACE takes the alignment of paired-end total RNA-seq reads and, optionally, a reference annotation as input. TERRACE first identifies back-spliced reads, each of which will be assembled into a set of full-length circular candidate paths in the underlying splice graph. The candidate paths, optionally augmented by the annotated transcripts, are subjected to a selection process followed by a merging procedure to produce the resultant circRNAs (for an illustration, see Supplemental Figs. S1–S3, S6). A score function is learned that can assign a confidence score to each assembled circRNA (for a list of features utilized to learn the score function, see Supplemental Note). TERRACE is outlined in Figure 4. An extended version of the Methods with full descriptions of all the steps is provided as a Supplemental Methods section. Analysis and comparison of runtime and memory usage are available in Supplemental Note and Supplemental Tables S1 and S2, respectively.
Outline of TERRACE. Rounded boxes represent data and data structures. Rectangles represent procedures. Dashed boxes indicate optional.
Software availability
The source code of TERRACE is freely available at GitHub (https://github.com/Shao-Group/TERRACE) and as Supplemental Code. TERRACE is also available as a conda package at https://anaconda.org/bioconda/terrace. The scripts, evaluation pipelines, and instructions that can be followed to reproduce the experimental results of this work are available at GitHub (https://github.com/Shao-Group/TERRACE-test). The alignment files (by STAR) of these samples and the raw sequences and alignment files of the simulated data are hosted at Penn State Data Commons (doi:https://doi.org/10.26208/AZ99-RQ38). For additional information, please refer to the Supplemental Material.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work is supported by the US National Science Foundation (2019797 and 2145171 to M.S.) and by the US National Institutes of Health (R01HG011065 to M.S.). We thank Qimin Zhang for helpful input. We thank Fangqing Zhao and Jinyang Zhang for their assistance with CIRI-full.
Author contributions: T.Z. and M.S. designed the project. All authors designed and implemented the methods. T.Z. conducted the experiments. All authors discussed the results and approved the final manuscript.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279106.124.
References
- ↵Asghari H, Lin YY, Xu Y, Haghshenas E, Collins CC, Hach F. 2020. CircMiner: accurate and rapid detection of circular RNA through splice-aware pseudo-alignment scheme. Bioinformatics 36: 3703–3711. 10.1093/bioinformatics/btaa232
- ↵Clough E, Barrett T. 2016. The Gene Expression Omnibus database. Methods Mol Biol 1418: 93–110. 10.1007/978-1-4939-3578-9_5
- ↵Dobin A, Davis C, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras T. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635
- ↵The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247
- ↵Gao Y, Wang J, Zhao F. 2015. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 16: 4. 10.1186/s13059-014-0571-3
- ↵Gao Y, Wang J, Zheng Y, Zhang J, Chen S, Zhao F. 2016. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun 7: 12060. 10.1038/ncomms12060
- ↵Gao Y, Zhang J, Zhao F. 2018. Circular RNA identification based on multiple seed matching. Brief Bioinformatics 19: 803–810. 10.1093/bib/bbx014
- ↵Jeck WR, Sharpless NE. 2014. Detecting and characterizing circular RNAs. Nat Biotechnol 32: 453–461. 10.1038/nbt.2890
- ↵Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. 2013. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19: 141–157. 10.1261/rna.035667.112
- ↵Ji P, Wu W, Chen S, Zheng Y, Zhou L, Zhang J, Cheng H, Yan J, Zhang S, Yang P, 2019. Expanded expression landscape and prioritization of circular RNAs in mammals. Cell Rep 26: 3444–3460.e5. 10.1016/j.celrep.2019.02.078
- ↵Jia GY, Wang DL, Xue MZ, Liu YW, Pei YC, Yang YQ, Xu JM, Liang YC, Wang P. 2019. CircRNAFisher: a systematic computational approach for de novo circular RNA identification. Acta Pharmacol Sin 40: 55–63. 10.1038/s41401-018-0063-1
- ↵Kim D, Langmead B, Salzberg S. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12: 357–360. 10.1038/nmeth.3317
- ↵Kristensen LS, Jakobsen T, Hager H, Kjems J. 2022. The emerging roles of circRNAs in cancer and oncology. Nat Rev Clin Oncol 19: 188–206. 10.1038/s41571-021-00585-y
- ↵Leinonen R, Sugawara H, Shumway M; International Nucleotide Sequence Database Collaboration. 2011. The sequence read archive. Nucleic Acids Res 39: D19–D21. 10.1093/nar/gkq1019
- ↵Li X, Chu C, Pei J, Măndoiu I, Wu Y. 2018a. CircMarker: a fast and accurate algorithm for circular RNA detection. BMC Genomics 19: 572. 10.1186/s12864-018-4926-0
- ↵Li X, Yang L, Chen LL. 2018b. The biogenesis, functions, and challenges of circular RNAs. Mol Cell 71: 428–442. 10.1016/j.molcel.2018.06.034
- ↵Ma XK, Wang MR, Liu CX, Dong R, Carmichael GG, Chen LL, Yang L. 2019. CIRCexplorer3: a CLEAR pipeline for direct comparison of circular and linear RNA expression. Genomics Proteomics Bioinformatics 17: 511–521. 10.1016/j.gpb.2019.11.004
- ↵Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, 2013. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495: 333–338. 10.1038/nature11928
- ↵Nguyen DT, Trac QT, Nguyen TH, Nguyen HN, Ohad N, Pawitan Y, Vu TN. 2021. Circall: fast and accurate methodology for discovery of circular RNAs from paired-end RNA-sequencing data. BMC Bioinformatics 22: 495. 10.1186/s12859-021-04418-8
- ↵Rybak-Wolf A, Stottmeister C, Glažar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, 2015. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell 58: 870–885. 10.1016/j.molcel.2015.03.027
- ↵Stefanov SR, Meyer IM. 2023. CYCLeR: a novel tool for the full isoform assembly and quantification of circRNAs. Nucleic Acids Res 51: e10. 10.1093/nar/gkac1100
- ↵Suzuki H, Tsukahara T. 2014. A view of pre-mRNA splicing from RNase R resistant RNAs. Int J Mol Sci 15: 9331–9342. 10.3390/ijms15069331
- ↵Szabo L, Salzman J. 2016. Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 17: 679–692. 10.1038/nrg.2016.114
- ↵Venø MT, Hansen TB, Venø ST, Clausen BH, Grebing M, Finsen B, Holm IE, Kjems J. 2015. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol 16: 245. 10.1186/s13059-015-0801-3
- ↵Vo JN, Cieslik M, Zhang Y, Shukla S, Xiao L, Zhang Y, Wu YM, Dhanasekaran SM, Engelke CG, Cao X, 2019. The landscape of circular RNA in cancer. Cell 176: 869–881.e13. 10.1016/j.cell.2018.12.021
- ↵Vromman M, Anckaert J, Bortoluzzi S, Buratin A, Chen CY, Chu Q, Chuang TJ, Dehghannasiri R, Dieterich C, Dong X, 2023. Large-scale benchmarking of circRNA detection tools reveals large differences in sensitivity but not in precision. Nat Methods 20: 1159–1169. 10.1038/s41592-023-01944-6
- ↵Wang F, Nazarali AJ, Ji S. 2016. Circular RNAs as potential biomarkers for cancer diagnosis and therapy. Am J Cancer Res 6: 1167–1176.
- ↵Wu J, Li Y, Wang C, Cui Y, Xu T, Wang C, Wang X, Sha J, Jiang B, Wang K, 2019. CircAST: full-length assembly and quantification of alternatively spliced isoforms in circular RNAs. Genomics Proteomics Bioinformatics 17: 522–534. 10.1016/j.gpb.2019.03.004
- ↵Xin R, Gao Y, Gao Y, Wang R, Kadash-Edmondson KE, Liu B, Wang Y, Lin L, Xing Y. 2021. isoCirc catalogs full-length circular RNA isoforms in human transcriptomes. Nat Commun 12: 266. 10.1038/s41467-020-20459-8
- ↵Yang W, Du WW, Li X, Yee AJ, Yang BB. 2016. Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene 35: 3919–3931. 10.1038/onc.2015.460
- ↵Yu CY, Li TC, Wu YY, Yeh CH, Chiang W, Chuang CY, Kuo HC. 2017. The circular RNA circBIRC6 participates in the molecular circuitry controlling human pluripotency. Nat Commun 8: 1149. 10.1038/s41467-017-01216-w
- ↵Yu KHO, Shi CH, Wang B, Chow SHC, Chung GTY, Lung RWM, Tan KE, Lim YY, Tsang ACM, Lo KW, 2021. Quantifying full-length circular RNAs in cancer. Genome Res 31: 2340–2353. 10.1101/gr.275348.121
- ↵Zhang XO, Dong R, Zhang Y, Zhang JL, Luo Z, Zhang J, Chen LL, Yang L. 2016. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res 26: 1277–1287. 10.1101/gr.202895.115
- ↵Zhang J, Chen S, Yang J, Zhao F. 2020. Accurate quantification of circular RNAs identifies extensive circular isoform switching events. Nat Commun 11: 90. 10.1038/s41467-019-13840-9
- ↵Zhang J, Hou L, Zuo Z, Ji P, Zhang X, Xue Y, Zhao F. 2021. Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nat Biotechnol 39: 836–845. 10.1038/s41587-021-00842-6
- ↵Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G, 2016. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun 7: 11215. 10.1038/ncomms11215
- ↵Zheng Y, Ji P, Chen S, Hou L, Zhao F. 2019. Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med 11: 2. 10.1186/s13073-019-0614-1