
Abstract
Long-read sequencing technologies have improved the contiguity and, as a result, the quality of genome assemblies by generating reads long enough to span and resolve complex or repetitive regions of the genome. Several groups have shown the power of long reads in detecting thousands of genomic and epigenomic features that were previously missed by short-read sequencing approaches. While these studies demonstrate how long reads can help resolve repetitive and complex regions of the genome, they also highlight the throughput and coverage requirements needed to accurately resolve variant alleles across large populations using these platforms. At the time of this review, whole-genome long-read sequencing is more expensive than short-read sequencing on the highest throughput short-read instruments; thus, achieving sufficient coverage to detect low-frequency variants (such as somatic variation) in heterogenous samples remains challenging. Targeted sequencing, on the other hand, provides the depth necessary to detect these low-frequency variants in heterogeneous populations. Here, we review currently used and recently developed targeted sequencing strategies that leverage existing long-read technologies to increase the resolution with which we can look at nucleic acids in a variety of biological contexts.
There are several applications for which sequencing all the nucleic acids present in a sample is unnecessary and consumes resources. Targeted sequencing employs strategies that increase the proportion of specific DNA/RNA fragments in a sample, thus increasing the overall coverage of these regions of interest (ROIs). These approaches dramatically change our ability to study the genome by facilitating higher sample throughput than whole-genome sequencing, and improve accuracy by increasing the read depth coverage, which enhances the detection of potentially pathogenic alleles (Dapprich et al. 2016). Several methods have been developed for short-read target enrichment (Hodges et al. 2007; Turner et al. 2009; Mertes et al. 2011; Altmüller et al. 2014; Ballester et al. 2016) and typically rely on multiplexed PCR amplification (Jones et al. 2008; Tewhey et al. 2009) or oligonucleotide-based DNA hybridization capture (Albert et al. 2007; Hodges et al. 2007; Gnirke et al. 2009; Cao et al. 2013; Wang et al. 2015; Dapprich et al. 2016; Giolai et al. 2017). Since all these enrichment strategies were developed for short-read platforms, they involve fragmentation of genomic DNA (gDNA) before amplification, resulting in <1000 bp templates.
One of the main advantages of long-read technologies is its ability to generate highly contiguous sequences of large genomic regions, including complex and repetitive regions that are difficult to resolve using short-read technologies. This is particularly useful for applications such as structural variant (SV) detection, genome assembly, and haplotyping (Norris et al. 2016; Vu et al. 2017; Gong et al. 2018; Nattestad et al. 2018; Sedlazeck et al. 2018; Aganezov et al. 2022; Altemose et al. 2022; Nurk et al. 2022; Vollger et al. 2022). In complex samples, such as heterogeneous tumor samples, long reads are crucial for the discovery of low-frequency variants, such as somatic mutations. Newer long-read sequencing strategies also deliver highly accurate data, improving the detection of pathogenic single-nucleotide polymorphisms (SNPs) and copy number variation (CNV). Until recently, the lack of long-read compatible target enrichment strategies made it difficult to study specific ROIs on the existing long-read platforms. This especially limited researchers’ ability to fully resolve larger variants such as repeat expansions and SVs in regions of diagnostic or therapeutic interest. As long-read sequencing methods have developed, so too have methods for target enrichment using these platforms. Targeted long-read sequencing methods are powerful tools for studying genomic regions not readily accessible via short-read approaches. Targeted long reads provide enrichment for ROIs while maintaining variant positions within fragments up to 100 kb (Stangl et al. 2020; Iyer et al. 2022) as well as maintaining epigenetic modifications (Gilpatrick et al. 2020), provided PCR is not used. The preservation of long-range genomic structure is critical for the analysis of repeats, transposable elements, and regulatory elements (Gershman et al. 2022; Hoyt et al. 2022). This review will discuss targeted long-read strategies employed on the two main long-read platforms: Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio). It will focus on methods such as long-range PCR, hybridization, Cas9-mediated approaches, and on-instrument targeted sampling approaches (Table 1).
Summary of currently available targeted long-read sequencing approaches
| Method | DNA input mass per run/prep | Max. number of targets per run | Max. read length | Approx. depth per run/prepa | Platform compatibility | Time |
|---|---|---|---|---|---|---|
| Long-range PCR | ||||||
| ∼10 ng | ∼1000 | ∼10 kb | 10–1000× | ONT, PB | ++ | |
| Notes: Low cost, fast, removes base modifications, methylation marks not preserved | ||||||
| Hybridization methods | ||||||
| 1 µg | ∼20 k | 10 kb | 10–1000× | ONT, PB | +++ | |
| Notes: Probes are expensive, removes base modifications, methylation marks not preserved | ||||||
| CRISPR-Cas9-based approaches | ||||||
| CATCH | 2.75 × 105–1.5 × 106 cells | <10, fewer if targets are of varied sizes | 200 kb–1 Mb | 50–400× | ONT, PB | +++ |
| Notes: In-gel cell lysis and targeting, gives low yield for downstream sequencing, methylation marks preserved | ||||||
| nCATS | 3–10 µg | ≤25, Min. 4 crRNA guides/target, Max. 100 guides/rxn. | 30 kb | 20–850× | ONT | ++ |
| Notes: No background reduction, limited multiplexing options, methylation marks preserved | ||||||
| Single-ended cut | 1–5 µg (10 ng with WGA) | <10, depends on breakpoint/repeat/fusion size | 30 kb | 10–150× | ONT | ++ |
| Notes: Not suited for background reduction or tiling, limited multiplexing options, methylation marks preserved | ||||||
| Negative enrichment/CaBagE | 1–10 µg | ≤25, Min. 4 crRNA guides/target, Max. 100 guides/rxn. | 35 kb | 40–400× | ONT | +++ |
| Notes: Background reduction does not translate to higher coverage, not suited for single-ended cut, limited multiplexing options, methylation marks preserved | ||||||
| ACME | 5–20 µg | ≤25, Min. 4 crRNA guides/target, Max. 100 guides/rxn. | 100 kb | 35–1000× | ONT | ++ |
| Notes: Not suited for single-ended cut, limited multiplexing options, higher input mass required, methylation marks preserved | ||||||
| PureTarget | 2 µg per sample | 20 genes in the predesigned panel | 15 kb | 50–200× | PB | +++ |
| Notes: Limited to predesigned repeat expansion targets, methylation marks preserved | ||||||
| Adaptive sampling | ||||||
| readfish, UNCALLED, ONT built-in, BOSS-RUNS | 1–5 µg | Min. 0.1% Max. 10% of genome size, 1%–5% optimal | Dependent on input DNA size. 4 kb–10 kb | Dependent on target size relative to genome. 10–100× | ONT | + |
| Notes: Requires shearing, not effective on reads <3000 bp, needs many targets, methylation marks preserved | ||||||
[i] (PB) Pacific Biosciences, (ONT) Oxford Nanopore Technologies, (WGA) whole-genome amplification.
[ii] + 1–4 h; ++ 4–8 h; +++ >1 day of preparation.
[iii] aCoverage is highly dependent on the number/length of targets, level of multiplexing, and the type of sequencing run used. These numbers are approximate based on current use cases.
PCR sequencing
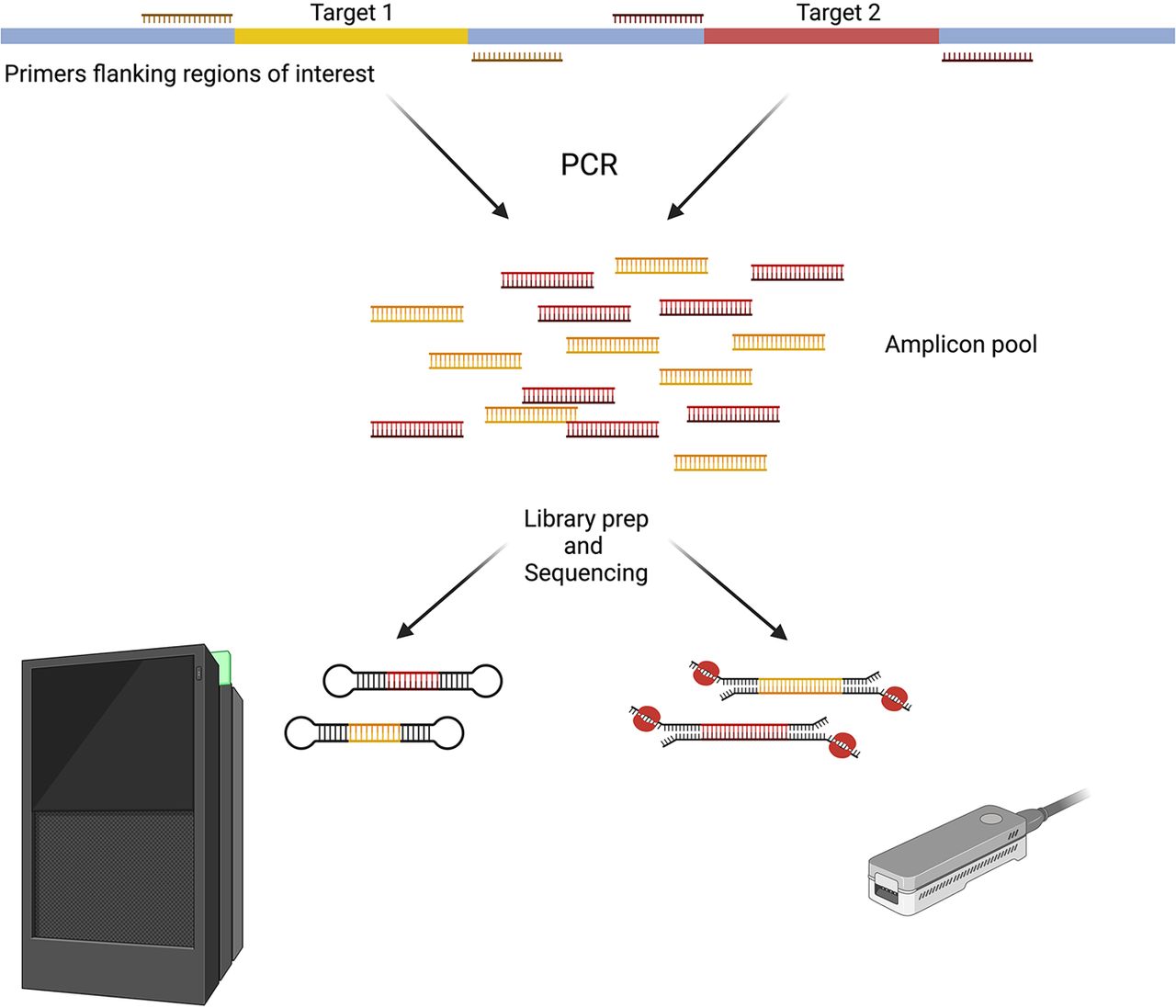
Before “next-generation” sequencing methods revolutionized genome science, Sanger sequencing of individually amplified targets was standard for pathogenic variant discovery (Schutte et al. 1996; Dunlop et al. 1997). Today, PCR methods are frequently used to generate the gene panels ubiquitously found in many clinical gene studies (Nagahashi et al. 2019). Amplification-based approaches using long-range PCR were, therefore, a logical application for early long-read sequencing methods. The typical workflow for long-range PCR long-read sequencing is as follows: High molecular weight (HMW) DNA is first isolated from the target tissue. While this DNA generally does not need to be ultra-long, it must be longer than the ROI. Then primers specific to the target regions are designed such that they anneal to the template upstream and downstream from the ROI. Once amplified, the target(s) can be barcoded if needed, and/or size-selected via gel, then sequenced on either ONT or PacBio instruments (Fig. 1). Long amplicons are quite beneficial for highly polymorphic or repetitive targets for which internal primers cannot be designed and long reads are needed to preserve intergenic variation (Cumming et al. 2018; Ciosi et al. 2021). CYP2D6, for example, is a highly polymorphic gene with different alleles displaying a wide spectrum of enzymatic activity related to drug metabolism. Charnaud et al. (2022) employed the PacBio platform to sequence and phase CYP2D6 alleles in an isolated population, revealing that as much as 7% of the population may be intermediate or poor metabolizers for the antimalarial drug primaquine.
Long-range PCR enrichment. Primers are designed to flank ROI. PCR can be carried out as single reactions for single targets or with multiple targets in a single PCR reaction. Amplified targets can be optionally size-selected via gel if the target size is known. Amplicons are pooled together before library preparation (prep). (Created with BioRender; https://www.biorender.com/)
Perhaps one of the most common uses of long-range PCR for targeted long-read sequencing is characterizing the human leukocyte antigen (HLA) region (Santamaria et al. 1993). The HLA loci play a critical role in the immune system's ability to recognize and respond to pathogens. It is known to be highly variable and complex, with high-resolution information about allelic variations and haplotypes (Rioux et al. 2009). Conventional methods tend to focus only on the variations in exons 2, 3, and 4, thus variations in the noncoding regions that regulate RNA expression are not explored (Ramsuran et al. 2015). HLA typing on the PacBio platform was demonstrated by Albrecht et al. by combining highly accurate MiSeq data with lower accuracy full-length RS II data to sequence a long HLA amplicon. The full-length data provided phasing information that was corrected with the MiSeq data and resulted in the identification of 606 novel alleles out of the 1056 genes fully sequenced, which were not previously characterized (Albrecht et al. 2017).
Like HLA sequences, 16S rDNA sequencing relies on an initial long-range PCR step to generate an amplicon target for long-read sequencing. The 16S rRNA gene is a conserved genetic marker that is present in nearly all bacteria and archaea, and its sequencing allows for the identification of microorganisms based on the sequence similarity of their 16S rRNA gene (Tringe and Hugenholtz 2008). Mitsuhashi et al. (2017) demonstrated that the ONT MinION had superior sensitivity to distinguish species level population in a mock microbial community—91% compared to 68% on an Ion PGM instrument. Additionally, as the accuracy of long-read methods has improved, the need to supplement with short reads has largely been eliminated. Long-range PCR-based approaches for characterizing single genes are now more tenable without the added cost of additional short-read data. Paired with low-cost Flongles from ONT, rapid screening for amplicon targets from pathogens like the monkeypox virus is becoming a more viable method (Israeli et al. 2022).
Transcript populations can also be explored with these methods as demonstrated by Adamopoulos et al. This group performed mRNA transcript-specific amplification for KRAS, NRAS, and HRAS, which are regulatory genes commonly mutated in various cancers, followed by MinION sequencing to characterize the diversity of transcripts in 40 human cell lines. The analysis of the full-length reads revealed 39 novel transcripts originating from previously uncharacterized alternative splicing junctions between the annotated exons of the genes (Adamopoulos et al. 2021).
Droplet-based PCR, which has been used to increase the number of targets that can be amplified in parallel (Tewhey et al. 2009), can also be used to enrich for target sequences via sorting. The Xdrop technology (Madsen et al. 2020), which uses a multiple displacement amplification (MDA) approach isolates DNA fragments up to 100 kb in size into individual droplets. Target-specific amplification is performed along with an intercalating dye to identify which droplets contain DNA fragments of interest. In one example, Grosso et al. (2021) employed Xdrop for contiguous sequencing of the FMR1 repeat expansion on the ONT platform. The FMR1 expansion is characterized by 5–200 CGG repeats and is indicated in more than 40 genetic human diseases (Paulson 2018). To evaluate this region, target-positive droplets were sorted using fluorescence-activated cell sorting (FACS). The sorted droplets were then broken, pooled, and re-encapsulated to allow for droplet-based MDA. After debranching, DNA molecules between 60 and 80 kb were sequenced on an ONT MinION device, achieving 200× enrichment over the background.
Long-range PCR is relatively straightforward and low cost with many established pipelines. However, while useful, these approaches can have significant limitations. Long-range PCR can be prone to generating chimeric sequences due to reduced specificity during the amplification of larger fragments. This raises the likelihood of amplifying nontarget sequences, resulting in off-target fragments. Another drawback is reduced amplification efficiency as specialized polymerases required for long-range PCR often have lower processivity and fidelity. This leads to lower amplification efficiency and yield, thus reducing the number of targets that can be included in a single run (Kanagawa 2003). Further, issues of lower polymerase fidelity can confound results especially when paired with lower accuracy long-read sequencing strategies (Ammar et al. 2015). While PCR can improve the accuracy of ONT-based long-read methods, by removing base modifications (Schatz 2017) it eliminates the ability to explore modified bases in a given study. Modification-aware basecallers abrogate this limitation somewhat, but accurate modeling of modifications is essential (Chiou et al. 2023). Long-range PCR also requires significant optimization for longer fragments, requiring users to test multiple enzymes and reaction conditions to achieve the longest amplicon lengths, which are generally just over 10 kb. Optimization is further complicated by the variable performance of each PCR reaction. While targeted regions can exceed 1 Mb (Tewhey et al. 2009), the variable performance of each amplicon results in uneven coverage across the target, requiring excess sequencing to obtain sufficient data from all amplicons (Mamanova et al. 2010). The need to optimize all amplicon reactions along with the relatively large DNA inputs needed to carry out many PCR reactions limits the breadth of targets that can be explored, with the most complex multiplex PCR panels limited to no more than 1000 targets (Khodakov et al. 2016). When paired with the number of samples needed for a meaningful study, the scalability of this approach is notably hampered.
Hybridization-based capture approaches
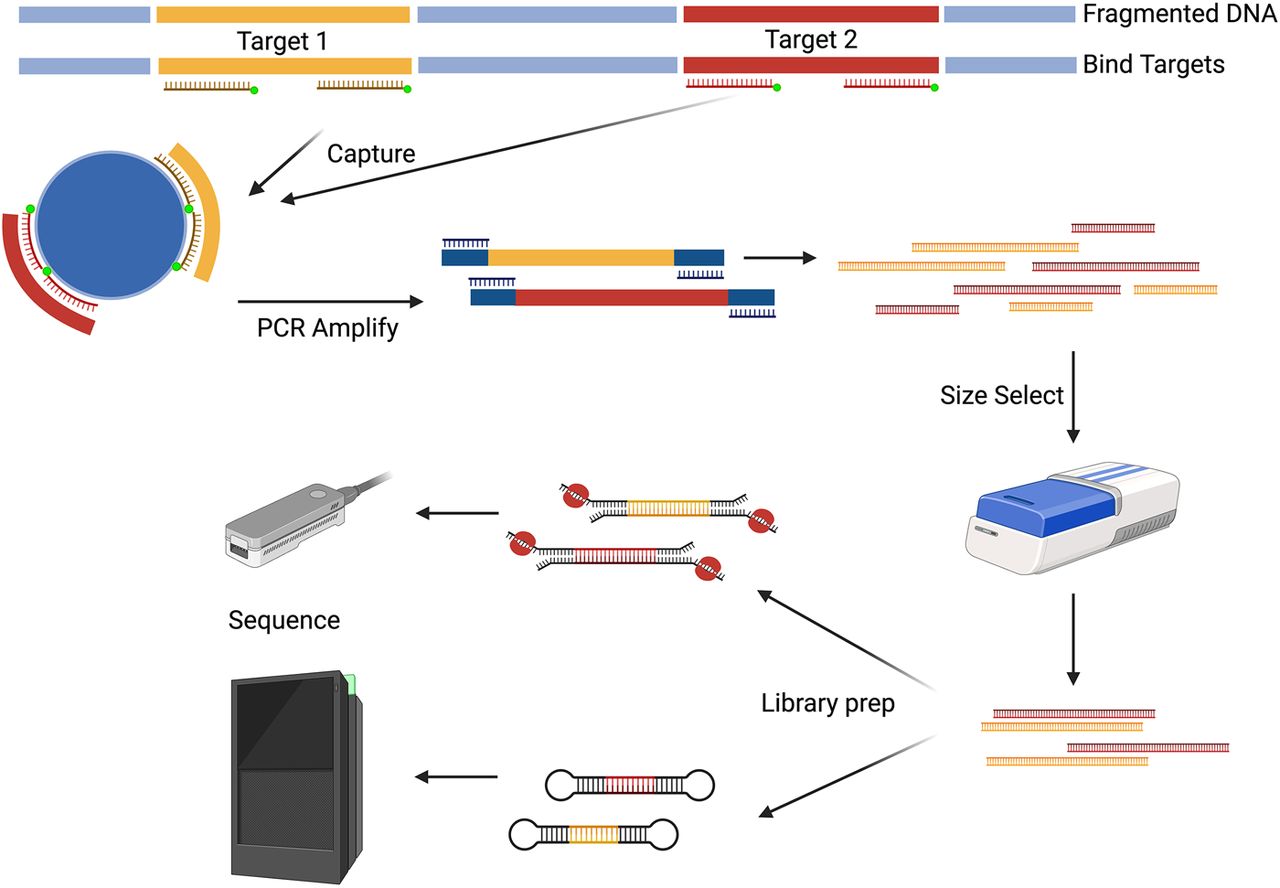
After PCR-based methods, hybridization-based approaches may be the next most widely used means of enriching targets. These approaches were derived from methods developed for short-read technologies like panel and exome capture (Hodges et al. 2007). Long-read capture approaches rely on the hybridization of complementary probes to ROIs followed by a bead-based pulldown before sequencing (Fig. 2). Most of these approaches rely on off-the-shelf or custom panel targets designed for short reads with few to no deviations from the manufacturers’ suggested hybridization protocol (Wang et al. 2015; Lagarde et al. 2017; Lefoulon et al. 2019; Schuele et al. 2020; Steiert et al. 2022). One such long-read hybridization method is PacBio-LITS, a large-insert targeted capture sequencing method (Wang et al. 2015), wherein DNA is first fragmented to between 1 and 8 kb. These fragments are then captured with a hybridization panel customized to the targets of interest. As with the short-read methods, the enriched targets require amplification, and these amplified fragments are then size-selected to enrich for the longest amplicons (∼4–6 kb) before PacBio library preparation.
Hybridization-based capture. Biotinylated DNA or RNA guides are designed to be complementary to the ROI. The DNA is fragmented to ∼10 kb and amplified if more mass is needed. Next, the probes bind to the denatured DNA. The probe–ROI complex undergoes a bead-based pulldown to separate the target regions from the rest of the genome. The enriched fragments are amplified and size-selected to maintain the target length. The amplicons are then prepared for long-read sequencing. (Created with BioRender; https://www.biorender.com/)
While many long-read capture methods use the same probes that would be used for short-read approaches, there is evidence that fewer probes are needed to effectively capture long DNA fragments. Dapprich et al. explored the density of probe placement and what effect more dispersed probes would have on long targets. This group proposed a method using Sanger sequencing called Region-Specific Extraction (RSE) and found that a spacing of ∼1 kb resulted in a 35-fold enrichment of target sequences over the background (Dapprich et al. 2016). This is quite different from short-read methods where probes are tiled completely across the target region to maximize the capture of small fragments ∼200 bp and improve on target rates (Parla et al. 2011).
Similar to the improved performance seen by increasing probe spacing, some researchers have capitalized on the nature of long DNA fragments to enrich for those regions for which specific probes cannot be designed. One example of this is work by Ramirez and colleagues looking at human papillomavirus (HPV) integration using PacBio sequencing. In this case, guides were designed specifically to the 8 kb HPV genome and not to the host. The authors demonstrated that by targeting HPV only, hundreds of DNA bases upstream and downstream from the integration site are also enriched. Analysis of the host flanking regions reveals a complex series of structural events induced by HPV integration (Ramirez et al. 2021).
Hybridization-based enrichment has also been used to characterize specific transcripts in single-cell applications. Singh et al. targeted T cell and B cell antigen receptor (TCR and BCR) genes in unfragmented 10x Genomics libraries derived from human lymphocytes. TCR and BCR genes undergo rearrangements to increase diversity to address various antigens. Due to the extraordinary diversity of these genes, substantial depth is needed to characterize the different isoforms, thus an unenriched single-cell library may not recapitulate the diversity present in the population. The authors enriched and sequenced these regions via a method called Repertoire and Gene Expression by Sequencing (RAGE-seq). This method splits a full-length cDNA pool into two, where one half undergoes short-read expression profiling, and one half undergoes hybridization capture of TCR and BCR cDNAs for long-read sequencing. The combination of these methods proved to be effective in characterizing somatic hypermutation in an immortalized B cell line (Singh et al. 2019).
With the success of various hybridization-based targeted long-read sequencing strategies, commercial manufacturers are now releasing predesigned panels for long-read sequencing. Notably, Twist has released the Twist Alliance Dark Genes Panel (TADGP) (Mahmoud et al. 2024) and the Twist Alliance Long-Read PGx panel. The TADGP was developed in collaboration with researchers to address tagged sequences of the medically relevant and complete autosomal genes. The 389 gene panel was extensively compared to whole-genome sequencing using 11 control samples. While the technical performance of the panel using PacBio yielded good results, the authors noted that 75% of the genes lacked ClinVar (Landrum et al. 2016) variants. The authors posit that the inherent complexity of the genes in this panel led to their underrepresentation in the short-read derived database; further supporting the need for targeted long-read methods.
Hybridization-based capture provides several benefits over strictly PCR-based approaches. Perhaps most notably, the number of targets for the hybridization-based approaches can be in millions compared to PCR's tens of thousands (Kozarewa et al. 2015). The DNA input mass required is also typically low since PCR is used to increase the abundance of preenriched targets, although PCR efficiency remains a limiting factor. The main limitations on the size of the regions targeted are the cost of the probes and finding unique sequences to which those probes can bind. Fidelity and amplicon length are still constrained by the enzymes used for long-range PCR, limiting most contiguous targets to ∼10 kb, and PCR will eliminate any epigenetic marks present on the source DNA. Moreover, long DNA fragments are prone to breakage during the various manipulations necessary while performing a hybrid-capture approach. Length limitations also impact the performance of hybrid approaches. Reads that are only a few kb long do not assemble with the same contiguity as reads larger than 10 kb (Lang et al. 2020). Capturing repetitive regions from satellite DNA (Vondrak et al. 2020) or telomeres (Bzikadze and Pevzner 2020), for example, can be quite problematic as each has repeats well above the capturable fragment length via hybridization, and it is extremely difficult, if not impossible, to accurately assemble DNA fragments <10 kb in regions like this.
CRISPR-Cas9-based enzymatic targeting approaches
The CRISPR–Cas system provides a specific, rapid, and flexible enzymology (Jinek et al. 2012; Doudna and Charpentier 2014; Rath et al. 2015; Sternberg et al. 2016) that has been successfully used to target and clone genomic sequences in vitro (Jiang et al. 2015; Lee et al. 2015). The flexibility in design, ease of use, and specificity of this system make it ideal for targeted long-read efforts (Sander and Joung 2014; Wu et al. 2014; Adli 2018; Loose 2018; Shola et al. 2020). In particular, this approach is superior to PCR due to the large fragment sizes that can be targeted and captured with very high depths, which aids in variant detection. Unlike hybridization approaches that tend to be lengthy and complex, Cas proteins require short incubation times at moderate temperatures (Schultzhaus et al. 2021). Compared to hybridization, these methods also offer more flexibility in targeting as only the flanking sequences are required to be known to target the whole region (Schultzhaus et al. 2021). CRISPR–Cas-based tools are constantly evolving with an expanding repertoire of Cas proteins and continuing efforts to minimize off-targets, making this an exciting avenue for sequence-specific targeting and enrichment (Adli 2018).
CRISPR-based long-read targeting strategies typically begin with HMW DNA extraction, followed by targeting and cleavage with Cas nuclease, followed by an optional long-target purification/separation/size-selection before library preparation for sequencing on long-read platforms. While methods vary in DNA extraction and library preparation, all approaches employ a nuclease (commonly Cas9), multiple target-specific CRISPR RNAs (crRNAs, also called guide RNAs), and a trans-activating crRNA (tracrRNA) that together form the RNP (ribonucleoprotein) complex—which acts as the targeting and cleavage unit. We have grouped the most widely used CRISPR-based enrichment strategies as below, highlighting their specific advantages and limitations for long-read target enrichment.
In-gel Cas9-cleavage and target-specific electrophoresis purification
One of the first methods to show the power of the CRISPR–Cas system for targeting long segments of the genome was the Cas9-Assisted Targeting of Chromosome segments (CATCH) wherein the authors performed Cas9 targeting and cleavage in agarose gel plugs to excise microbial genome segments of up to 100 kb for targeted cloning of gene clusters (Jiang et al. 2015; Jiang and Zhu 2016). CRISPR-mediated isolation of specific megabase-sized regions (CISMRs), developed by Bennett-Baker and Mueller (2017) further expanded CATCH's application from cloning to sequencing. Using the same principle, the authors targeted >2 Mb size regions in gel, separated it from the rest of the sample with pulsed-field gel electrophoresis (PFGE), excised the desired target band, and sequenced the purified DNA segments, reporting >100-fold enrichment of the targeted regions (Bennett-Baker and Mueller 2017). Though CISMR was used for short-read sequencing using Illumina, it helped set the stage for similar approaches to be adapted for long-read sequencing.
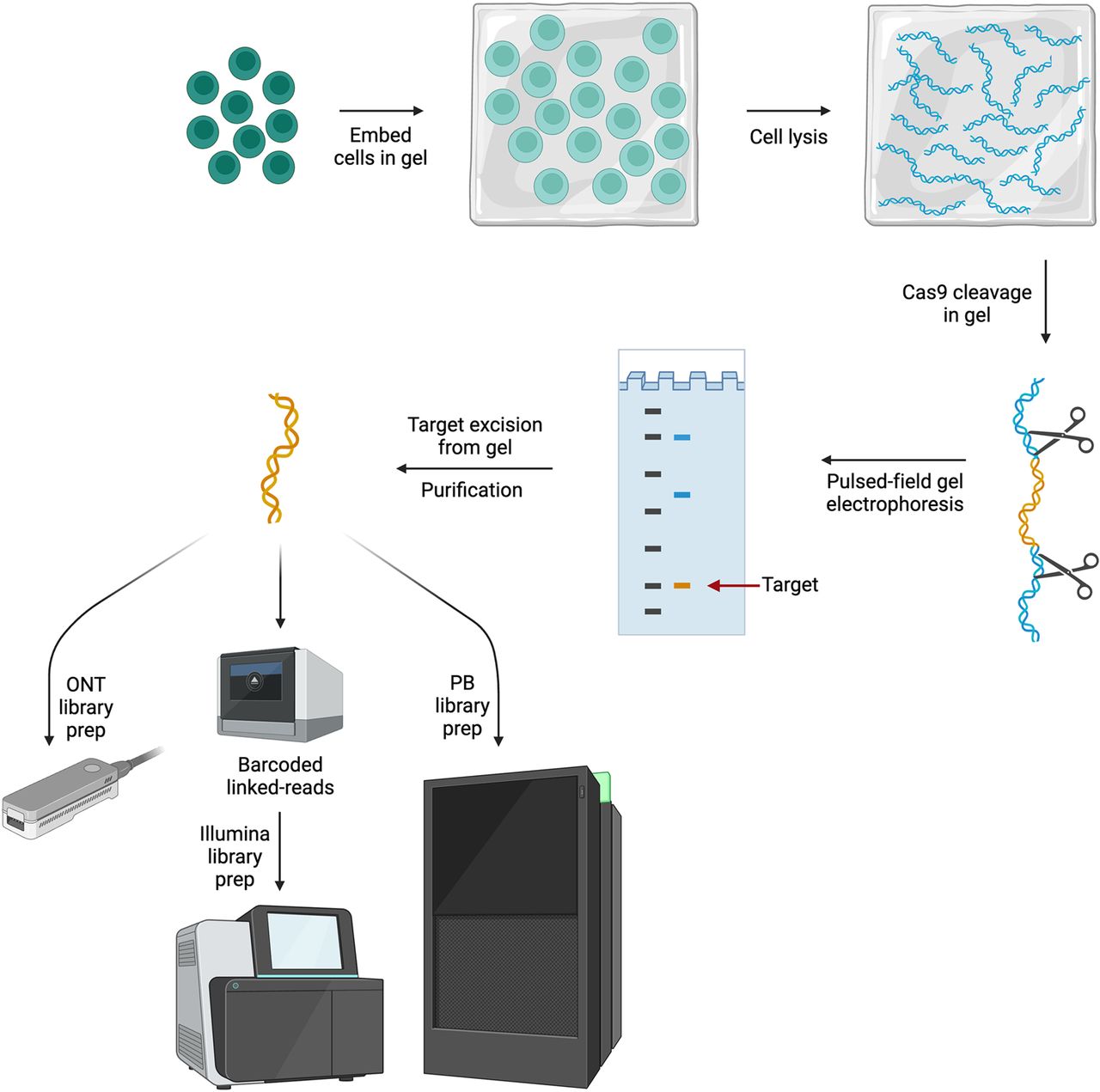
Gabrieli et al. (2018) extended CATCH and CISMR to enrich for a 200 kb region around the breast cancer gene BRCA1 in primary human peripheral blood cells for sequencing using the ONT platform. In this approach, cells are embedded in agarose plugs, with cell lysis and Cas9 cleavage of HMW DNA performed in the gel. Target DNA is separated using PFGE and since the size of the target is known a priori, the desired target band can be excised for library preparation (Fig. 3). Using this approach, Gabrieli et al. observed ∼240-fold enrichment and close to 70× coverage of BRCA1 from a single MinION flowcell, making this the first report of using CATCH for targeted long reads. Variations of this approach have been used by other groups to target large ROIs using a partially automated version of CATCH on the SageHLS (HMW Library System) instrument (Sage Science) for sequencing with the 10x Genomics Chromium platform (Shin et al. 2019) and the PacBio Sequel I instrument (Walsh et al. 2021). More recently CATCH was optimized to enrich extrachromosomal DNA to evaluate genetic variation as well as methylation in parallel with nanopore sequencing (Hung et al. 2022).
In-gel Cas9-cleavage and target-specific electrophoresis purification. Cells are embedded in agarose and lysed in gel, maintaining DNA fragment length. Cas9 cleavage is carried out in gel using guides specific to the ROI. PFGE is used to separate the target(s) from background DNA based on size, which is known a priori. The purified target is then prepared for sequencing on either of the long-read platforms using appropriate adapters/kits. (Created with BioRender; https://www.biorender.com/)
One major advantage of the in-gel targeting approaches is that the DNA is protected in the gel, reducing mechanical stress during the cleavage and purification steps, therefore, preserving molecule length. CATCH is amenable to large targets in the 100 kb–5 Mb range, with the SageHLS instrument offering a faster and partially automated option. However, since size-selection is a critical step in the enrichment process, this approach does not work well in cases where the target length is variable or unknown (e.g., repeat expansions). CATCH also takes longer than other Cas-based approaches (described below), typically taking 24–48 h from DNA extraction to loading on the sequencer, followed by sequencing time, greatly increasing the time-to-answer. The reliance on gel elution to retrieve desired fragments also limits the number and size of targets that can be grouped together for Cas9 cleavage, with sample multiplexing likely only possible during the postelution library preparation step. Lastly, it has been observed that though extremely large, the targets often constitute a very small fraction of the genome(s) and sample(s), resulting in low yields after cleavage and target separation. This typically necessitates a far higher starting material without which there is a risk of underloading the flowcell unless an amplification step is performed. It should also be noted that as of 2020, 10x Genomics has discontinued the sale of its Chromium Genome and Exome products. Since barcoded linked-reads are no longer available on this platform, it is not compatible with CATCH at present.
Cas9-based targeted sequencing on the nanopore platform
The next wave of CRISPR–Cas-based enrichment strategies focused on optimizing in-solution approaches that would provide more flexibility in the number and size of targets evaluated per reaction. Briefly, these approaches typically start with gel-free HMW DNA extraction, followed by dephosphorylating the gDNA to make fragment ends unavailable for sequencing adapter ligation. crRNA guides targeting upstream and/or downstream from the ROI are designed such that multiple guides flank the ROI to ensure optimal targeting through redundancy and reduce off-target enrichment. The resultant RNP complex facilitates the melting of duplex DNA and base-pairing of the crRNAs with the target sequence, followed by Cas9 cleavage around the ROI, freeing the target fragments and making their ends available for adapter ligation. Many different groups have devised methods for such Cas9-based enrichment, and since they are very similar in principle, we grouped these methods to better highlight the optimizations made for different types of research questions.
Double- and single-cut approaches
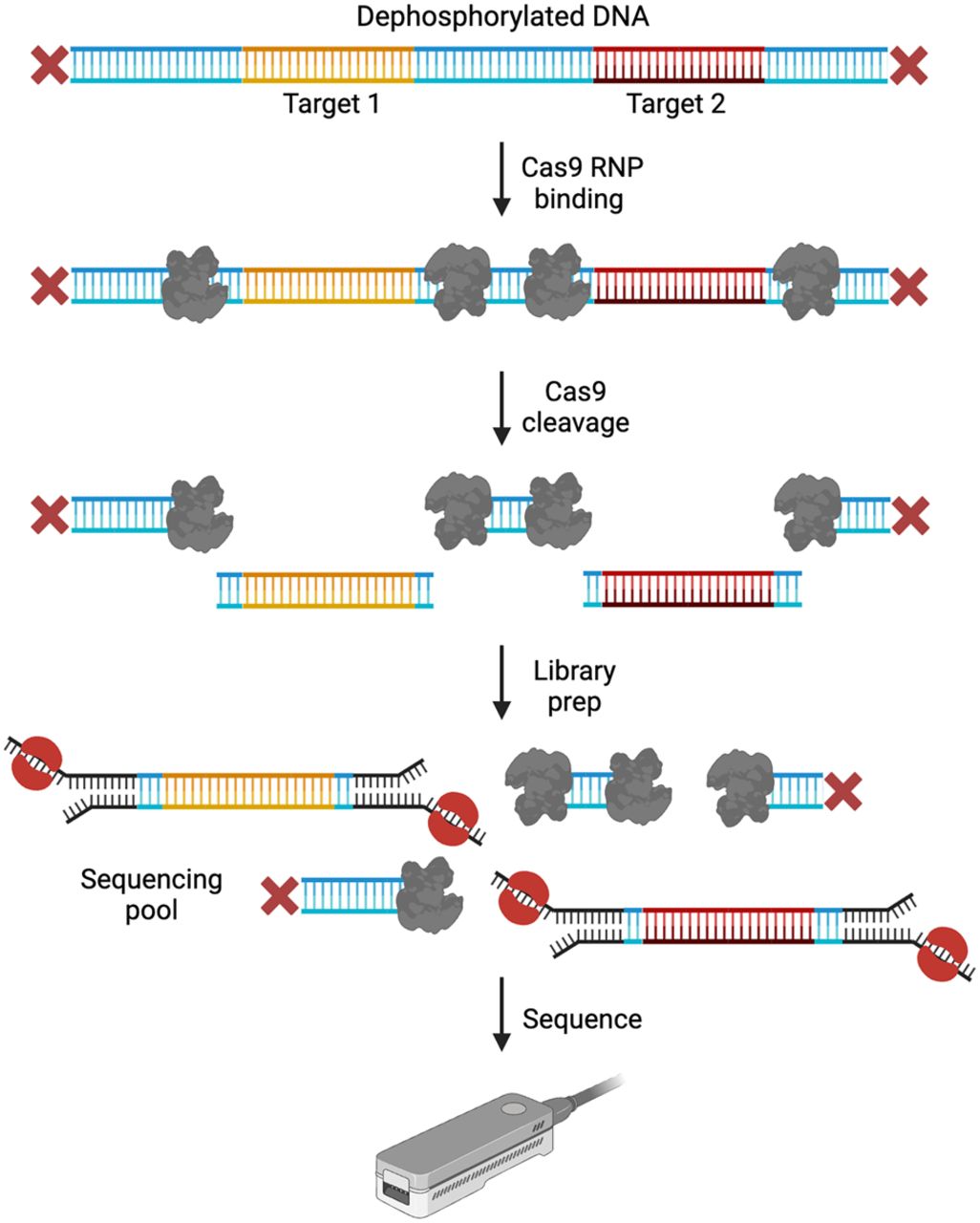
Initially developed to target short tandem repeat (STR) expansions, STR identification, quantification, and evaluation (STRique) was one of the first in-solution Cas-based excision approaches introduced. Through this work, the authors highlighted the benefits of an initial dephosphorylation step in Cas-based enrichment as well as the increased targeting efficiency of Cas9 over Cas12a (Giesselmann et al. 2019). Gilpatrick et al. (2020) in collaboration with ONT built upon this work and developed nanopore Cas9-targeted sequencing (nCATS), which was available as the SQK-CS9109 kit from ONT (discontinued early 2024) and is the most widely used Cas9-based excision approach. nCATS is an amplification-free approach wherein targets are excised from dephosphorylated gDNA with crRNA guides targeting flanking regions on both sides of the ROI (Fig. 4). The authors targeted 10 regions between 12 and 24 kb in size across different sample types—cell lines, cell-line-derived xenograft, normal and paired tumor/normal primary human breast tissue, and reported median coverage of 675×. Since its introduction, nCATS has been used by several different groups for genetic and/or epigenetic evaluation of ROIs across different samples and organisms (López-Girona et al. 2020; Wongsurawat et al. 2020; Bruijnesteijn et al. 2021; McDonald et al. 2021; Mizuguchi et al. 2021; van Haasteren et al. 2021; Alfano et al. 2022; Fiol et al. 2022; Kirov et al. 2022; Rubben et al. 2022; Vandiver et al. 2022; Bryant et al. 2023; Merkulov et al. 2023).
nCATS. DNA is dephosphorylated to prevent sequencing adapter ligation. Cas9 RNPs with guides specific to the ROI are used to cleave the DNA upstream and downstream from the targets. This exposes phosphate groups at the ends of the target strands to which sequencing adapters are then ligated. Targets are, therefore, preferentially sequenced from a sequencing pool consisting of adapter-bound targets and dephosphorylated nontarget DNA. (Created with BioRender; https://www.biorender.com/)
Many groups made modifications to this approach like tiling guides to cover larger target areas, modifying reagents, changing incubation times for Cas9 cleavage and sequencing adapter ligation, as well as including flowcell wash and reload steps (López-Girona et al. 2020; Bruijnesteijn et al. 2021; Mizuguchi et al. 2021; Alfano et al. 2022; Fiol et al. 2022; Rubben et al. 2022; Merkulov et al. 2023). Some groups have also adapted nCATS for unique applications by defining unconventional target regions through their crRNA guide designs (McDonald et al. 2021; van Haasteren et al. 2021; Vandiver et al. 2022; Bryant et al. 2023).
Fusion genes and large duplications, however, are tricky to characterize due to their variable configurations and breakpoint locations. In both cases, typically the sequence of only one flanking site (upstream or downstream) of an ROI is known with confidence, making it difficult to employ double-ended Cas9-cleavage enrichment methods that require the knowledge of flanking sequences on both ends. Two different groups have described a single-ended cleavage variation that leverages ONT sequencing's ability to read through long stretches of DNA fragments with an adapter molecule bound at only one end (Stangl et al. 2020; Watson et al. 2020). Since Cas9 protects the protospacer adjacent motif (PAM)-distal end after cleavage and exposes phosphate groups on the PAM-proximal end for sequencing adapter binding, crRNAs designed in a strand-directed manner help define sequencing directionality. For detecting duplications, Watson et al. designed plus-strand and minus-strand crRNA guides positioned within the duplicated sequence. Dephosphorylated DNA was split into two separate strand-specific cleavage reactions, which were then pooled, adapters ligated, and sequenced (Fig. 5). Using this approach, the authors reported close to 500-fold enrichment of their target regions, identifying a 200 kb duplication involving several exons in the DMD gene (Watson et al. 2020). Similarly, Stangl et al. designed crRNA guides to cut only at or near the known partner of a gene fusion in a method called FUDGE (Fusion Detection from Gene Enrichment), wherein dephosphorylated DNA is cut either upstream or downstream from the known gene. By directing reads upstream or downstream from the cut site, FUDGE facilitates sequencing of the suspected unknown fusion partner at either the 5′ or 3′ end of the known gene, with the authors reporting an average enrichment of 665× that helped successfully identify known and unknown fusion gene partners across different cancer cell lines and tumor samples (Stangl et al. 2020).
Single cut and read-out approaches. Dephosphorylated DNA is split into two separate reactions. crRNA guides are designed in a strand-directed manner with separate guide pools prepared for guides cutting upstream versus downstream from each target. Upstream and downstream guide pools are then used to cleave dephosphorylated DNA in separate reactions. After Cas9 cleavage, both reactions are pooled together, and sequencing adapters are ligated. The prepared library is loaded on nanopore flowcells and sequenced. (Created with BioRender; https://www.biorender.com/)
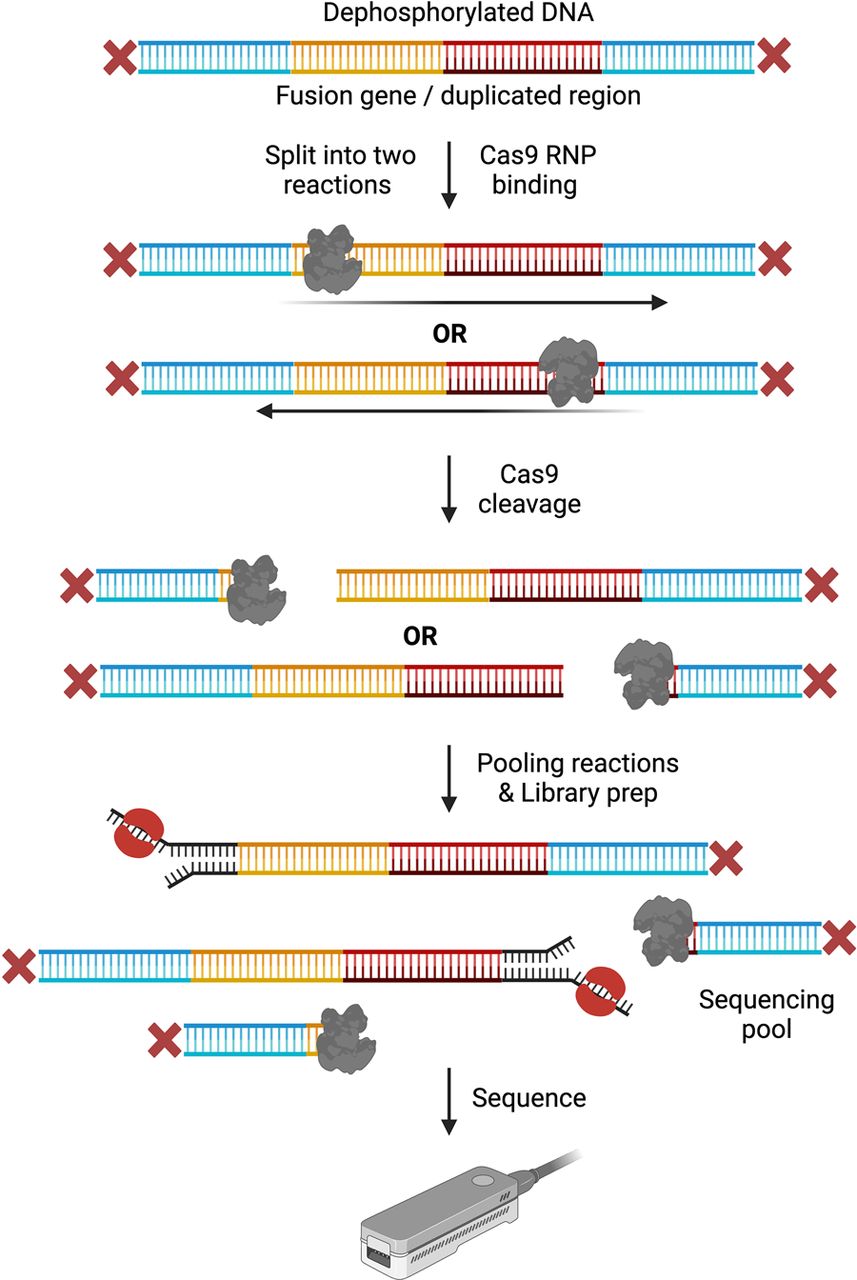
Generally, nCATS and most of its adaptations have been successful in enriching targets up to 30 kb in size. Typically, coverage is inversely proportional to target size, with shorter targets achieving much higher coverage than the longer ones. This is in part due to the presence of the entire sample DNA—consisting of adapter-bound targets, Cas9-bound nontargets, and completely dephosphorylated nontargets—in the final sequencing pool. The presence of a large proportion of competing background DNA can negatively impact not only the length of targets capturable, but also the overall coverage of (long) targets. Furthermore, it is possible that bound Cas9 could slip from the ends of the nontarget fragments during downstream processing of the cleaved product, exposing them to adapter ligation as well. Nicks and breaks in the background DNA could also lead to the sequencing of these regions unintentionally, as the exposed phosphate groups allow for adapter ligation. These points highlight that the lack of a background reduction step in nCATS could potentially be a major drawback in this approach. While single-ended cut approaches help address an important limitation of double-ended cut approaches, they are also limited by the availability of HMW DNA. While 200 kb duplications were successfully identified by Watson et al., Stangl et al. reported that for fusion detection, FUDGE had similar target length limitations as nCATS (<30 kb target-spanning read lengths), which could possibly be solved by tiling across larger breakpoint regions.
Approaches with background reduction
A common artifact observed in ONT sequencing is a strong bias to sequencing shorter fragments more efficiently (Ebbert et al. 2018; De Roeck et al. 2019; Giesselmann et al. 2019), the presence of which is likely due to the general difficulty in maintaining long fragments all the way to the end of library prep, as well as diffusion properties affecting how long fragments are delivered to the surface of the flowcell. Therefore, if shorter background DNA is not eliminated from the sequencing pool, it could compete with longer targets, reducing depth across the desired long fragments. Recognizing the detrimental role background DNA can play in Cas9-based target enrichment, three different groups leveraged the preferential binding of Cas9 for background reduction. Cas9 endonuclease forms a stable complex with its guide RNA and the corresponding target DNA, and remains tightly bound even when challenged with competing proteins (Clarke et al. 2018). The dissociation of Cas9 from its DNA target occurs only under harsh environmental conditions, and has a natural dissociation time of ∼6 h (Sternberg et al. 2014; Ma et al. 2016; Richardson et al. 2016).
The tight binding of Cas9 to its target was leveraged by two groups to protect targets of interest while employing exonucleases for background depletion. Exonucleases have been widely used for background elimination (Varley and Mitra 2008; Rossi et al. 2018) and can be used for extensive digestion of nontarget gDNA when coupled with Cas9 protection of ROI flanks, resulting in PCR-free enrichment. Unlike hybridization-based approaches, Cas9-based depletion does not exhibit sequence-specific bias, allowing for high-level multiplexing of targets. Additionally, since this strategy does not rely on DNA amplification or fragmentation, it can enrich for long, native DNA. The two approaches for Cas9-based depletion, while similar, differ primarily in the types of exonucleases used as well as incubation times for Cas9 RNP formation, Cas9 cleavage, and exonuclease degradation. Both approaches skip the dephosphorylation step and start directly with Cas9 cleavage of targets specified by crRNA guides that cut upstream and downstream from the ROIs. Steric inhibition from the Cas9/sgRNA complexes “shield” the ends of the DNA targets from exonuclease degradation, resulting in nontarget DNA depletion and enrichment of double-strand, nonamplified targets compatible with long-read and native-strand sequencing (Fig. 6A). Negative Enrichment (NE) developed by Stevens et al. (2019) achieved a 30- to 600-fold enrichment across their targets. Cas9-based Background Elimination (CaBagE) developed by Wallace et al. (2021) achieved threefold to 32-fold higher enrichment than NE, but this could be a function of the number and size of regions targeted per reaction as well as DNA input mass. Compared to nCATS, on-target coverage from CaBagE was approximately threefold to 11-fold lower for the same experimental conditions. Targeting ∼4 regions (4–20 kb) per reaction, the authors reported 39–416× depth. Both groups found no significant coverage difference when enriching for a single target per reaction versus multiplexing targets for the sample in a single reaction.
Approaches with background reduction. (A) NE and CaBagE. DNA is cleaved upstream and downstream from the ROI (dashed lines represent cut sites) using target-specific crRNPs. Immediately after Cas9 cleavage, 5′ and 3′ exonucleases are used to digest background DNA while target ends are protected by the bound Cas9. Heat incubation is used to dissociate Cas9 from the targets and inactivate the exonucleases before sequencing adapter ligation. The prepared library is loaded on nanopore flowcells and sequenced. (B) Affinity-based Cas9-mediated enrichment (ACME). DNA is dephosphorylated to prevent sequencing adapter ligation and Cas9 RNPs with guides specific to the ROI are used to cleave the DNA upstream and downstream from the target(s). After cleavage, Cas9 remains bound to the nontarget side of the cut sites (PAM-distal end). The Cas9 enzyme has a C-terminal 6 Histidine Tag. HisTag Dynabeads are used to pull down Cas9 and the nontarget fragments bound to it from the sequencing pool. Adapters are then ligated to the exposed phosphate groups at the ends of the target strand(s). The prepared library is sequenced on ONT flowcells. (Created with BioRender; https://www.biorender.com/)
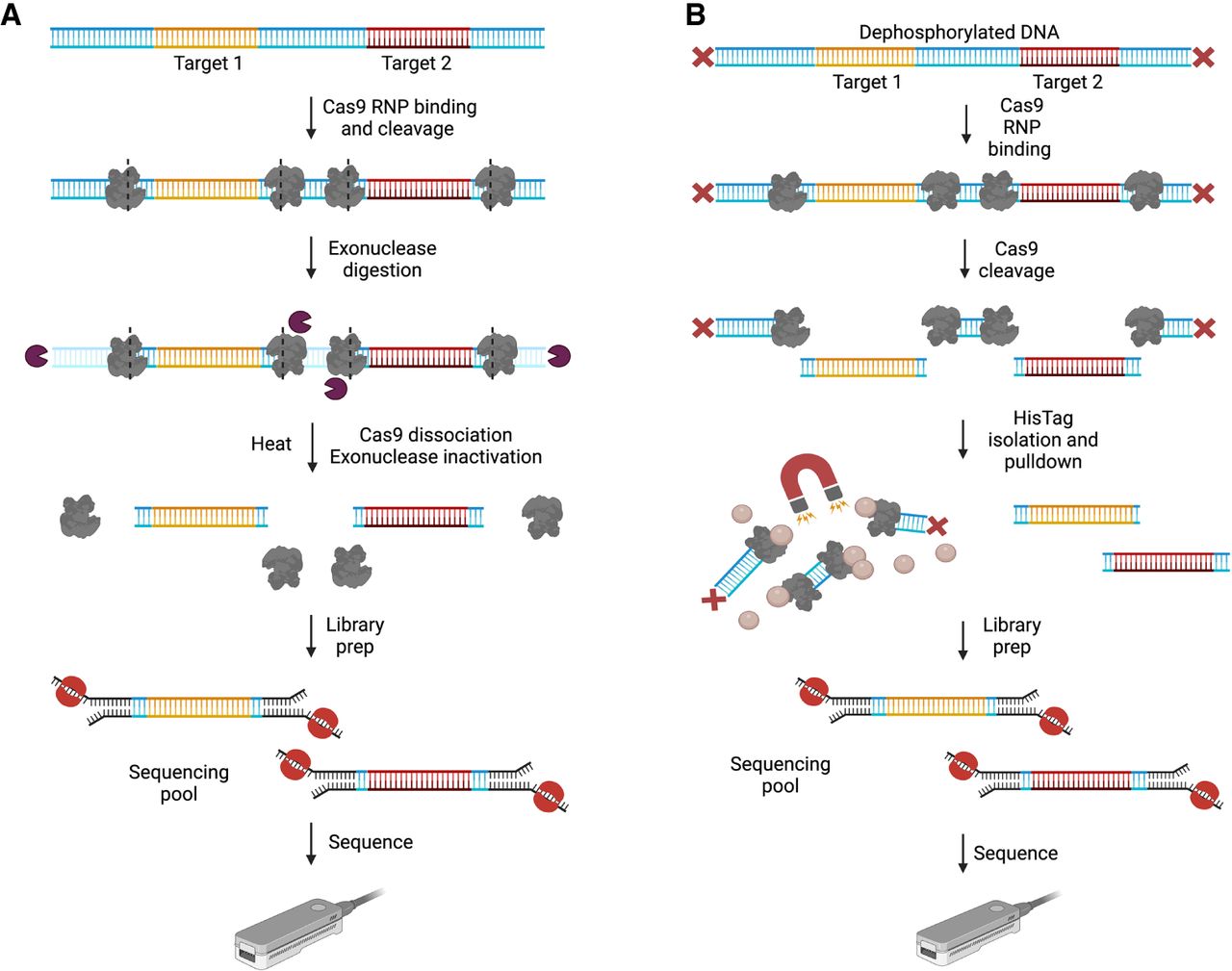
After cleavage, Cas9 tends to be strongly bound to the PAM-distal end of the cut site (Richardson et al. 2016), making it easy to direct its protective binding by defining directionality during crRNA guide design. Iyer et al. (2022) leveraged Cas9's preferential binding to develop ACME—an Affinity-based Cas9-Mediated Enrichment method that uses the C-terminal 6-Histidine tag present on the HiFi Cas9 nuclease to pull down Cas9-bound nontarget fragments from the sequencing pool. Through HisTag-based isolation and pulldown of Cas9-bound nontarget DNA after the Cas9 cleavage step, ACME physically reduces background DNA, allowing a higher proportion of target DNA to enter the flowcell (Fig. 6B). Compared to nCATS, ACME achieved a twofold to 25-fold increase in target coverage, increased target sizes capturable from 30 to 100 kb, and generated a threefold to sevenfold increase in the number of end-to-end target spanning reads. The authors reported >60-fold target enrichment, close to 70× coverage, and 3–20 end-to-end reads spanning a 95 kb target. The main advantage ACME offers over other Cas9-based nanopore targeting approaches is the ability to capture several large contiguous reads, up to 100 kb in size, that span the target from start to end. Since its development, ACME has been shown to perform on par with whole-genome long-read sequencing for SV detection (Iyer et al. 2022) and substitute bisulfite sequencing to determine methylation patterns across target promoter regions in acute myeloid leukemia (AML) cancer cell lines (Yang et al. 2021; Wei et al. 2022).
Compared to Cas9-based enrichment approaches like nCATS and ACME, depletion approaches like NE and CaBagE tend to take more time, cost more, and result in lower yield (Wallace et al. 2021). While lower yield could be attributed to inefficient exonuclease digestion, a more plausible explanation may be the increased sensitivity of these approaches to fragmentation between Cas9 binding sites i.e., within the targets. Any break in DNA or failure of Cas9 binding will result in target degradation by exonuclease. Furthermore, the sensitivity to such breakage will likely increase with increasing target sizes, imposing a limit on pursuing targets >30 kb. Although, this could be mitigated to an extent by ultra HMW (UHMW) DNA extraction and nick repair before exonuclease digestion. While ACME successfully increases end-to-end target sizes capturable from 30 to 100 kb, it struggles to deeply sequence targets >100 kb in size, with coverage dropouts observed in the center. As the developers of this approach, we note that one likely factor contributing to this dropout could be the DNA size, which may be improved by switching to UHMW DNA extraction methods. An important limiting factor in the wide application of ACME is input DNA mass, requiring at minimum 5 µg of starting mass, with close to 20 µg used for pooled preps that showed higher target depth. It is also important to note that while ACME takes nCATS a step further in several ways, it is not amenable to a “single cut and read out” approach, as the pulling down of Cas9-bound nontargets after a single-ended cut could result in other target regions being pulled out of solution as well.
Amplification-free targeted sequencing on the PacBio platform (PacBio PureTarget)
Like ONT, PacBio introduced a Cas9-mediated capture method called No-Amp Targeted Sequencing (Hafford-Tear et al. 2019; Wieben et al. 2019; DeJesus-Hernandez et al. 2021; Mangin et al. 2021). Since its introduction in 2019, the No-Amp method has been re-released as PureTarget; a predesigned panel of 20 genes known to harbor repeat expansions. Rather than relying on dephosphorylation to prevent off-target capture, the original method proposed in 2019 starts with the formation of circular libraries, called SMRTbells, from total gDNA. A single Cas9 guide is then used to create double-stranded breaks at a specific location within the SMRTbell, thus creating a new ligation site where a new SMRTbell adapter can be bound. The newly formed SMRTbell is then enriched via magnetic beads that carry a sequence complementary to the new adapter. Newer versions of this approach are more similar to the approaches employed by ONT, wherein DNA is initially dephosphorylated to prevent adapter ligation followed by Cas9 cleavage to expose ligation sites proximal to the target region. Once cleaved, SMRTbells are added to complete the library (Fig. 7; Tsai et al. 2022).
Amplification-free targeted sequencing on the PacBio platform (PacBio No-Amp). DNA is dephosphorylated to prevent sequencing adapter ligation. Cas9 RNPs with guides specific to the ROI are used to cleave the DNA upstream and downstream from the target(s). Sequencing adapters are ligated to the cleaved products. Since no background reduction has been performed yet, nontarget strands protected by Cas9 on both ends will also likely end up with SMRTbell adapters. Exonucleases are introduced to digest the rest of the background DNA. Only those fragments with SMRTbells attached on both sides survive the exonuclease digestion and make up the sequencing pool that is loaded on to PacBio flowcells. (Created with BioRender; https://www.biorender.com/)
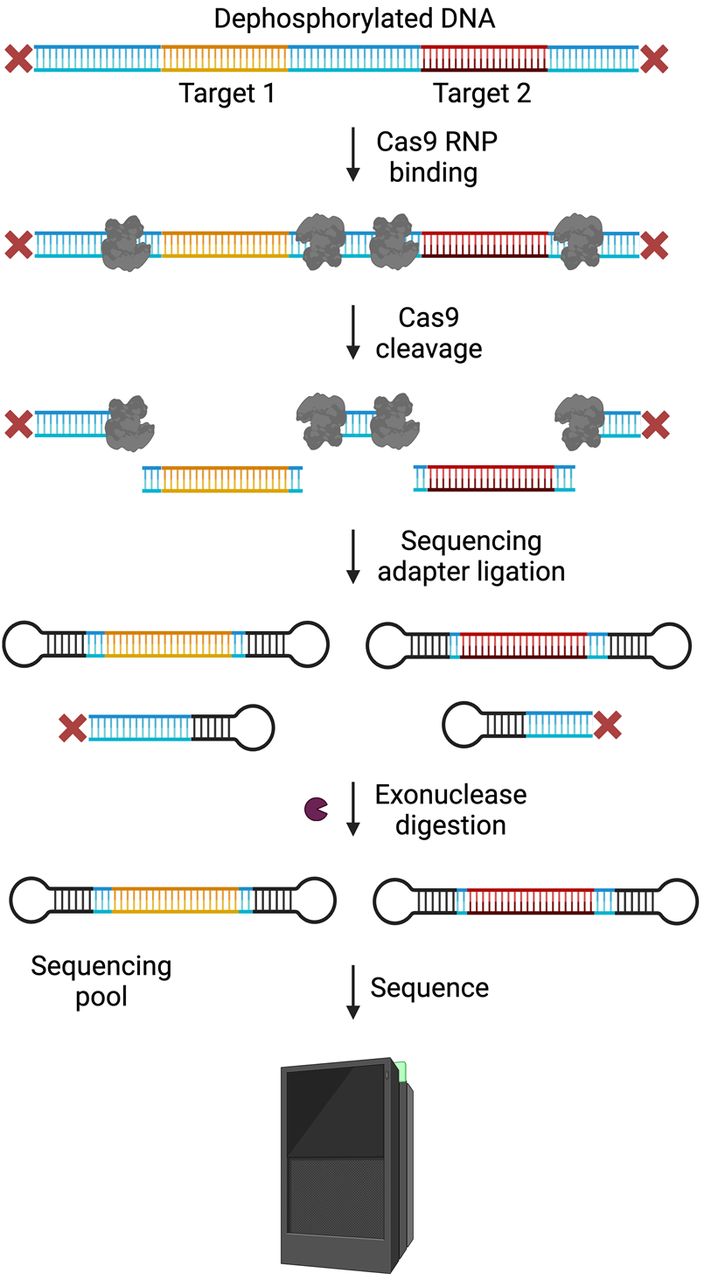
This method has been successfully used to study repeat expansions in the Huntington HTT gene (Höijer et al. 2018) and TCF4 (Hafford-Tear et al. 2019). The authors reported that the No-Amp method was effective in targeting regions of extreme GC content (<25%, >65%) as no PCR is required to enrich the target—an ideal situation for conditions caused by trinucleotide expansions (Loomis et al. 2013). The accuracy of PacBio sequencing is also an important feature in repeat expansion targeted sequencing. At >99.5% accuracy (Hon et al. 2020) PacBio has been shown to be more effective in sequencing through complex repeats than ONT, as demonstrated by work on the “GGGGCC” repeat found in many amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) cases (Ebbert et al. 2018).
Like the ONT-based Cas9 methods, the percent of total reads matching the target remains very low, typically <10%. It is important to note that these methods rely on the specificity and efficiency of the Cas9 and the guide RNA, which can be affected by factors such as guide RNA design, Cas9 activity levels, and the presence of off-target effects. While such approaches result in a many-fold enrichment of the targets over background, they are limited in the number of targets and multiplexed samples that can be sequenced in a single run. This issue is further confounded by existing technical limitations of the PacBio technology. In general, only a handful of targets can be enriched for at a time and the target length is limited by the ∼15 kb HiFi read lengths. Each Sequel II cell has 8M zero-mode waveguide (ZMW) sites where sequencing can occur and only one molecule can be sequenced in a ZMW—hence the PacBio label single molecule real-time (SMRT) sequencing. The SMRT Cell relies on diffusion loading of the ZMW, which is governed by Poisson statistics. Thus, while there are 8M possible sites for sequencing, only 4–6 M will generate data. Read length is also constrained by the ZMWs as larger DNA fragments diffuse slower and are too large to effectively fit into a single ZMW. These issues may be ameliorated by the newly available, at the time of writing, 25M ZMW Revio SMRT Cells. Similarly, the curated 20 gene panel that is part of the PureTarget product has optimized performance and multiplexing methods for repeat expansions thus improving the overall efficiency of this tool.
Summary of CRISPR-Cas9-based enzymatic targeting approaches
Cas9-based approaches are more sensitive than PCR and hybridization approaches as they rely on short (20–30 nt) sequences of complementarity and a PAM sequence for cut site detection; therefore, mismatches in the seed region adversely affect binding (Jiang and Doudna 2017). Furthermore, regions without adjacent PAM sites become difficult to target, requiring alternate target specifications, making guide design an important yet challenging feature of Cas9-based targeting. In addition to this, the binding efficiency of guide RNAs tends to be more variable than expected (Liu et al. 2020; Naim et al. 2020). While this has greater consequences for genome editing, off-target binding is not particularly detrimental for enrichment and can be mitigated by stringent guide selection based on GC content, self-complementarity, efficiency, and number of potentially mismatched binding sites prevalent in the genome. Lower binding/cleavage time, ideally 15 min, also greatly reduces the percentage of off-target binding and cleavage. Of all the approaches discussed in this section (CRISPR-Cas9-based enzymatic targeting approaches), the Cas9-based enrichment strategies offered on the ONT platform provide faster, flexible, and more economical options to obtain very high coverage of large targets of interest. However, all approaches discussed in this section are less applicable for large projects that survey 100s of targets across 100s of samples, as these approaches are not easily scalable. Alfano et al. (2022) evaluated a multiplex Cas9 enrichment approach using nCATS with ONT native barcoding (EXP-NBD104 kit) and reported >10-fold lower enrichment compared to a singleplex run, with 70% unclassified reads. These results were consistent with similar tests performed by us as well as other ONT users (https://community.nanoporetech.com/posts/high-of-unclassified-rea).
The main advantage of CRISPR-Cas9-based enrichment approaches is the very high depths (>100× for targets <30 kb, up to 60× for targets close to 100 kb in size) that are achievable for targets of interest, making it easier to resolve and identify low-frequency variants. Since these approaches are amplification-free and do not involve shearing of DNA, they allow for the targeting of very long contiguous regions, especially with approaches like CATCH and ACME. Currently, these methods are the main way to generate long (up to 100–200 kb), end-to-end target-spanning reads with high depth, provided sufficient DNA mass is available. The long lengths capturable through these approaches have a distinct advantage as they help reduce mapping errors due to SVs within targets, aiding in their detection.
Computational enrichment by adaptive sampling
While the CRISPR-based target enrichment strategies are biochemical approaches, adaptive sampling, also known as selective sequencing or “Read Until,” is a computational approach that “rejects” nontarget reads allowing only target reads to be sequenced fully (Loose et al. 2016; Edwards et al. 2019; Kovaka et al. 2021; Payne et al. 2021; Weilguny et al. 2023). At its core, adaptive sampling leverages a unique quality of ONT nanopore sequencing wherein pore current data can be assessed in real-time to make sequencing decisions for the same ongoing run. This is possible because nanopore devices collect data from all channels on a flowcell simultaneously and can communicate with each channel independently. Unwanted reads can, therefore, be physically ejected from the nanopores by reversing the driving voltage across them. Additionally, library preparation requires minimal specializations, and the rejected reads are unlikely to be resequenced since their motor proteins, located at the 5′ end of the molecules being sequenced, are no longer available once their respective strands are ejected.
ONT's Read Until application programming interface (API) was first developed into an implementable option by Loose et al. (2016) using dynamic time warping (DTW) to match reads to reference sequences. This early iteration was computationally extensive, especially for gigabase-size reference genomes, as it tried to match current trace (“squiggle”-space) data to a reference “squiggle” trace directly in order to bypass the slower speed basecallers of its time. As basecallers improved, Edwards et al. (2019) developed Read Until with Basecall and Reference-Informed Criteria (RUBRIC), which was computationally modest by working in the sequence-space, relying on real-time conventional basecalling using ONT's Nanonet basecaller and sequence-based alignment using LAST (Kiełbasa et al. 2011). This approach was faster, scalable, flexible, and efficient in filtering out unwanted reads. RUBRIC worked well for background depletion, but did not achieve significant absolute target enrichment.
Adaptive sampling approaches have consistently improved since these initial examples. Currently, there are four different ways to implement adaptive sampling—readfish, utility for nanopore current alignment to large expanses of DNA (UNCALLED), adaptive sampling directly through the ONT MinKNOW interface, and benefit-optimizing short-term strategy for read until nanopore sequencing (BOSS-RUNS) (Table 2).
Summary of currently available adaptive sampling approaches
| Features | readfish | UNCALLED | ONT built-in | BOSS-RUNS |
|---|---|---|---|---|
| Instrument compatibility | MinION, GridION, PromethION | MinION, GridION | MinION, GridION, PromethION | MinION, GridION, PromethION |
| Number of targets | Enrichment: Min. 0.1%–Max. 10% of genome size for targets within same genome, 1%–5% optimal Depletion: Variable based on proportion in sample | |||
| Customizable | ✓ | ✓ | ✓ | |
| Dynamic decisions | ✓ | ✓ | ||
| Setup | Requires GPU and considerable computational knowledge | Decent computational knowledge required | Not required | Requires GPU and considerable computational knowledge |
| Limitations | Nontrivial to install, requires reinstall for MinKNOW updates | Requires port codes for run initiation, nontrivial for GridION or PromethION | No dynamic decision capability | Nontrivial to install, requires reinstall for MinKNOW updates |
readfish and UNCALLED mainly differ in how they set two important parameters for adaptive sampling: speed of sequence identification–mapping–classification, and average read length of the prepared library. BOSS-RUNS takes readfish a step further by incorporating the ability to make sequencing decisions dynamically, based on the real-time performance of the run.
readfish
Payne et al. (2021) overcame the limitations of the first Read Until-based approach (Loose et al. 2016) by using the graphics processing unit (GPU)-based ONT basecalling software already integrated with the ONT sequencers (minIT, Mk1C, GridION, and PromethION). Target enrichment with readfish is achieved through real-time basecalling, followed by mapping reads as they are generated using minimap2 (Li 2018), based on which a decision on how to proceed with the read is made (reject, proceed, or stop receiving data), all within ∼1 sec of the read starting. readfish (https://www.github.com/looselab/readfish) is highly customizable, and configurations such as signal chunk durations and desired sample/target depth can be changed by the user to best suit their experimental goals. For example, the authors show a 1.6–4× enrichment of a low-abundance microbial subpopulation by setting a desired coverage of 40×, which prompted readfish to stop sequencing abundant populations once they met this depth and refocus sequencing on the low-abundance samples to improve their relative coverage. In human samples, the authors reported a 2.7× to 5.4× enrichment of exon targets (mean target depth ∼13–17×) for a search space of 176 Mb (5% of the genome) and a coverage of 30–40× when targeting the entire set of 717 Catalogue of Somatic Mutations in Cancer (COSMIC) genes (Tate et al. 2019) (search space 89.9 Mb, ∼2.7% of the genome). It was also observed that incorporating nuclease flush and reload steps further helps increase the overall yield as well as on-target coverage of readfish runs, with the increase proportional to the number of flushes and reloads. Since its introduction, readfish has been used successfully by different groups for adaptive sampling (Giannuzzi et al. 2021; Miller et al. 2021; Patel et al. 2022; Stevanovski et al. 2022).
While readfish is a comprehensive and flexible tool for adaptive sampling, it requires access to a reasonably powerful GPU or device capable of real-time basecalling. Furthermore, the installation of readfish is nontrivial, with several users, including us, reporting multiple issues with setup. Failed install attempts are common due to the constantly changing MinKNOW software and associated tool versions (https://github.com/LooseLab/readfish/issues). Similar issues are also bound to plague BOSS-RUNS (discussed below), which relies on readfish.
Utility for nanopore current alignment to large expanses of DNA (UNCALLED)
Basecallers require a large amount of input signal to generate sequence outputs and are typically devised to work on fully sequenced reads. Recognizing the additional computational burden imposed by basecalling reads first, Kovaka et al. (2021) developed a signal-based approach called UNCALLED (https://github.com/skovaka/UNCALLED), which is a nanopore current signal mapper. Using raw signals instead of mapped reads to make sequencing decisions, UNCALLED requires less computational power and time than readfish, achieving faster ejection of nontarget sequences and improved enrichment. With a lighter computational footprint than older signal-based methods, UNCALLED maps thousands of base pair signals to Mb-sized references per second, mapping ∼75% of reads within 1 sec (450 bp), making it faster than the Read Until API that provides signals at the rate of one chunk per second. The authors used UNCALLED to deplete known bacterial genomes from a mock microbial community to enrich for yeast genome sequences by mapping signal data to a 29 Mb reference containing seven bacterial genomes. UNCALLED retained >99% of yeast reads and ejected 90%–96% of bacterial reads, resulting in a 3.2- to 4.5-fold absolute enrichment of yeast sequences. Using UNCALLED to enrich for 148 human hereditary cancer genes (18.6 Mb), the authors mapped >90% of reads and reported a 29.6× coverage of the target sequences (Kovaka et al. 2021). UNCALLED also resulted in more sensitive and precise variant calling, with 100% concordance in SV calls, compared to whole-genome long-read sequencing. Like with readfish, the authors observed that nuclease flush improved UNCALLED yields substantially, underscoring the extent of pore blockage caused by the ejected DNA. The authors have since released UNCALLED4, which includes additional visualization and command line tools for nanopore signal-to-reference alignments.
Despite its lower computational footprint and faster decision time, UNCALLED is reported to still require considerable computational resources. While more effective with longer reads, UNCALLED runs with longer DNA fragment lengths are associated with lower yield. This approach is also limited in its search space, with a decline in performance observed as references become larger and/or include more repetitive sequences. At the time of writing, UNCALLED has only been adapted to run on the MinION instrument, with efforts to expand it to the GridION or PromethION instruments deemed nontrivial due to variable instrument port codes and slot specification issues.
Built-in adaptive sampling on the ONT MinKNOW interface
The Read Until programming interface (https://github.com/nanoporetech/read_until_api), while initially made available to third-party developers to devise implementation, was eventually implemented by ONT into its GridION control software in November 2020 as a user-selectable option. This has since been incorporated into the PromethION software as well. This built-in version of adaptive sampling opened up the approach to a much wider user base, who could now just upload a reference file and set whether to enrich for or deplete the genomes specified in the file. Furthermore, along with the reference file users also have the option to furnish a more specific target file with coordinates of multiple ROIs, such as genes or exons, to enrich within an uploaded reference genome. Like with readfish, reads are basecalled and mapped live, allowing for a decision to be made to either accept or reject the read within the first 1–2 sec. Since the adaptive sampling update has been made available on the ONT sequencers, many groups have reported successfully leveraging it for their targeting goals (Wanner et al. 2021; Marquet et al. 2022; Martin et al. 2022), with some reporting up to 5× enrichment of low abundance organisms (∼2% of total sample) (Martin et al. 2022). ONT's built-in tool is by far the easiest way to perform adaptive sampling, requiring no computational knowledge to set up and run apart from generating reference files or making BED files to specify targets.
While ONT's adaptive sampling tool functions as a convenient option for enriching or depleting sizable, predetermined target regions/genomes, it has limited options for customization, narrowing its scope to specific use case scenarios. At the time of writing, ONT's built-in adaptive sampling option does not support adjusting targets during the run by setting depth limits (like readfish) or making dynamic sequencing decisions (like BOSS-RUNS, discussed below).
Benefit-Optimising Short-term Strategy for Read Until Nanopore Sequencing (BOSS-RUNS)
BOSS-RUNS (https://github.com/goldman-gp-ebi/BOSS-RUNS) developed by Weilguny et al. (2023) is the latest adaptive sampling approach and takes readfish a step further in its optimization. A data-driven target enrichment approach, here sequencing decision strategies are dynamically updated in real time during the course of the run to better optimize flowcell use. BOSS-RUNS interacts with the ONT sequencing devices through the Read Until API and readfish (Payne et al. 2021). Like readfish, basecalled reads are mapped to reference genome(s) using minimap2 (Li 2018) to make a decision on how to proceed, but unlike readfish, BOSS-RUNS also has the ability to adapt the target set and change sequencing priorities throughout the run to maximize information gain and reduce uncertainty. BOSS-RUNS avoids wasteful data acquisition by assigning higher scores to poorly represented or ambiguous regions, therefore, prioritizing reads mapped to these loci over reads that do not add valuable/new information to the region being surveyed. Unlike other approaches wherein read acceptance/rejection is decided a priori, the dynamic decision strategy employed by BOSS-RUNS allows for coverage redistribution at any time during the run by changing what is sampled to positions of greatest value. Using BOSS-RUNS on a mock microbial community, the authors showed a boost in coverage of rare species with a more uniform coverage within each species, reducing low-coverage sites of low-abundance species (∼1% abundance) by 87.5%. BOSS-RUNS can be beneficial in pathogen surveillance or in a clinical setting due to its marked reduction in time-to-answer and coverage bias within or across genomes as well as improved confidence in genotype calls and variant calling.
While novel and beneficial in reducing time-to-answer in specific use cases, caution must be exercised when using a dynamic adaptive sampling approach like BOSS-RUNS, as it tends to skew relative coverages in a mixture. This can negatively impact the detection of CNVs due to the loss of underlying coverage information. BOSS-RUNS is also currently restricted to prokaryotic or small eukaryotic genomes due to the computational complexity involved in modeling every site of the genome. Furthermore, the current model does not account for low-frequency variants or complex variants (large insertions or deletions), limiting its use for such applications. The authors also reported the use of PCR-amplification during sample prep, which not only increases prep time, but also reduces average fragment length and removes native modification marks.
Summary of computational enrichment by adaptive sampling
Apart from the considerable computational knowledge required to set up and run most of the adaptive sampling approaches discussed in this section, there are several other factors that must be considered when choosing this approach for target enrichment. (1) The most widely used adaptive sampling approaches rely on live basecalling to make sequencing decisions; therefore, the speed of basecalling plays an important role in the accuracy of adaptive sampling calls. ONT currently offers three basecalling options—fast, high accuracy, and super high accuracy, of which the latter two models help reduce the likelihood of incorrect read rejections. (2) Pore blockages and burnouts are commonly observed in adaptive sampling due to the continuous ejection of nontarget reads. Pores that have been blocked cannot sequence reads for extended periods of time and can negatively impact overall yields and target enrichment (Payne et al. 2021). In extreme cases of pore burnout, seen especially if the flowcell is not washed during the run, the total yield of the targeted sequences can be lower in adaptive sampling runs than in a robust whole-genome run. Incorporating multiple nuclease flushes can help resolve these blocks, but will increase the prep time and input DNA required (Shafin et al. 2020). (3) Since adaptive sampling relies on fast decisions made by evaluating the first few hundred bases, the average length of the library is an important experimental consideration as it can influence overall yield as well as target enrichment. While longer fragment length is desirable to reduce mapping errors and sequence ambiguity, it reduces the overall throughput as the number of sequenceable molecules in the library is lower (Kovaka et al. 2021). Shorter molecules are detrimental to overall yield and contribute to faster pore burnouts as they either pass through the pore too quickly before a decision can be made, or, by the time the decision is made almost the entire read may need to be ejected, at which point sequencing the read is less time consuming than rejecting it (Martin et al. 2022). DNA sheared to 8–15 kb has been shown to be best for enrichment with adaptive sampling (Miller et al. 2021; Payne et al. 2021; Stevanovski et al. 2022); however, it can impact the ability to detect larger variants. (4) It is important to provide reference sequences that closely match the target since enrichment depends on sequencing decisions that rely heavily on this reference. Ideally, experiments should be designed such that target sequences make up 1%–5% of the sample genome. Depending on the research question, number of targets, target sizes, and the sample, this range can be expanded to 0.1%–10% of the genome (https://nanoporetech.com/document/adaptive-sampling). While this helps preserve pore activity to an extent by reducing ejections, the higher the proportion of target sequences to total sample size, the lower is the resultant coverage per target. Adaptive sampling approaches can realistically offer ∼20–30× coverage for most targets, which is greater than fivefold lower than depths achievable by chemical enrichment approaches.
The main advantage adaptive sampling offers is time-to-answer, especially in metagenomics and host DNA depletion to enrich for low abundance species. This approach is also ideal for the surveyance of 100s of targets in a single run and is capable of producing actionable coverage for detection of single-nucleotide variants, CNVs, and repeat expansions in human samples (Miller et al. 2021; Stevanovski et al. 2022). Unlike enzymatic enrichment, these approaches have no additional design, reagent, or equipment costs, and require minimal sample preparation, with ample flexibility in altering targets or experimental goals for future runs.
Conclusions
Target enrichment can be a highly effective way of reducing sequencing costs and saving sequencing time. As long-read sequencing strategies become more ubiquitous, the need to sequence more at cheaper costs per sample increases drastically. The choice of a long-read target enrichment strategy is heavily dependent on the exact nature of the experimental goals. Long-range PCR is already an established method for long-read target enrichment. As the cost of sequencing decreases, long-read gene panels based on fast and low-cost long-range PCR may become commonplace, particularly in the clinical and diagnostic realm. While a long-read exome pipeline may not be realistic nor needed, due in part to most exons being <200 bp (Sakharkar et al. 2004), hybridization methods may be important when only part of the target sequence is known such as in viral integration events (Ramirez et al. 2021) or transposon positioning (Hale et al. 2020) or to enrich for low abundance targets up to 20 kb long (Lagarde et al. 2017; Lefoulon et al. 2019). Cas9-based methods truly leverage the potential of long-read platforms by offering strategies to capture whole-target spanning reads with extremely high depths. Additionally, approaches on the ONT platform have the potential to enrich for targets >100 kb while also maintaining epigenetic modifications. While target and sample multiplexing options are limited, these approaches allow for accurate SV detection within extremely large regions by generating depths high enough to catch low-frequency variants in samples. The newest method presented here, computational enrichment through adaptive sampling, is potentially the lowest cost and offers the fastest time-to-answer. While generally subject to length limits (8–15 kb) and requiring computational resources, these approaches are highly flexible and dynamic, which is especially useful for metagenomic studies or in clinical settings when a whole catalog of ROIs needs to be surveyed rapidly.
Conversely, there are times when whole-genome sequencing (WGS) is preferable to targeted approaches. When the genetic component of a disease is largely unknown, as is in the case of suspected genetic disorders, a more comprehensive genetic picture may be warranted. While exome sequencing has widely been used for diagnosis, in such cases 50% of screened individuals show no variants via exome sequencing (Mastrorosa et al. 2023). Sequencing costs in general have decreased precipitously over the last 10 years (www.genome.gov/sequencingcostsdata). This shift has allowed researchers to leverage WGS in many more cases where it would have been previously cost prohibitive. Multiple reports have supported the notion that whole-genome long-read sequencing captures substantially more variation than short-read approaches (Audano et al. 2019; Nurk et al. 2022). As the costs of WGS decrease for both long and short reads, the overall cost of the experiment, in addition to the experimental goals should be considered. In nearly all cases, excluding adaptive sampling, the technical methods for target enrichment are more challenging than preparing a sample for WGS. Additionally, flowcell capacity and multiplexing limitations should also be considered. At some number of genomes, the technical challenges and baseline sequencing costs of targeted methods outweigh the costs of WGS. When designing experiments researchers should consider WGS when the sample size is small or consider if adding more samples to the study better leverages the power of targeted approaches.
As this field evolves, combinatorial strategies that leverage the strengths of more than one targeting approach could help build an efficient targeted long-read pipeline that checks the maximum boxes. For example, since enzymatic methods achieve sufficiently higher coverage compared to computational approaches while also maintaining target length, optimizing a combined enzymatic and computational enrichment approach may be beneficial for fast and optimal target enrichment. Interestingly, a couple of groups have tried such a combined approach with mixed reviews. OHMX.bio reported using a combined approach by first using nCATS during library preparation followed by further target enrichment with readfish (oral presentation, ONT London Calling Meeting—May 2022). With a search space covering the entire HLA complex (3–4 Mb), they showed improved coverage from the combined approach over readfish and nCATS individually. Rubben et al. (2022) tried the same approach to genotype the 28 kb CYP2D6 gene locus and found that the combined approach did not lead to higher on-target depth, which is consistent with our own results (SV Iyer, M Kramer, S Goodwin, et al., unpubl.) on testing combined ACME + UNCALLED, as well as, ACME + ONT built-in adaptive sampling approaches to target the same regions reported in Iyer et al. (2022). Some factors contributing to these mixed results may include (1) total size and spacing of regions being targeted—unlike the HLA region, which is sufficiently large, regions targeted by Rubben et al. as well as by us made up <0.1% of the genome, which is a known limiting factor for adaptive sampling. Furthermore, in the OHMX.bio study, the entire HLA complex was provided as a single contiguous target block for adaptive sampling even though nCATS guides were only designed for the genic regions. This inclusion of intergenic regions in the adaptive sampling search space may be more beneficial than using gene coordinates only, especially if the latter adds up to <0.1% of the genome. (2) Insufficient pore occupancy—typically only ∼10%–20% pore occupancy is observed when products of enzymatic enrichment are loaded on the flowcell. If pores are not adequately occupied, adaptive sampling is not optimally engaged. Multiplexing enzymatic enrichment products could help improve pore occupancy, but the effects of this on a combinatorial enrichment approach are yet to be explored.
In summary, there are several diverse options for targeted sequencing on the long-read platforms currently available for a variety of experimental goals. Given the rapid evolution of long-read technologies and the continual expansion of their applications, the targeted sequencing toolkit specific to these platforms is bound to grow, constantly adding to the wide array of research questions answerable in the near future.
Competing interest statement
W.R.M. is a founder, shareholder, and board member of Orion Genomics, which focuses on plant genomics. S.V.I. has received travel bursaries from Oxford Nanopore Technologies (ONT) to partially support travel to scientific conferences.
Acknowledgments
W.R.M. is the Davis Family Professor of Human Genetics at Cold Spring Harbor Laboratory (CSHL). S.V.I. was supported by the National Science Foundation (IOS 1758800) and the Davis Professor Endowment. S.G. was supported by the National Institutes of Health (5R50CA243890). This work was also supported by the CSHL Cancer Center (NIH 5P30CA045508).
Notes
[4] Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279168.124.
[5] Freely available online through the Genome Research Open Access option.
References
- ↵Adamopoulos PG, Tsiakanikas P, Boti MA, Scorilas A. 2021. Targeted long-read sequencing decodes the transcriptional atlas of the founding RAS gene family members. Int J Mol Sci 22: 13298. 10.3390/ijms222413298
- ↵Adli M. 2018. The CRISPR tool kit for genome editing and beyond. Nat Commun 9: 1911. 10.1038/s41467-018-04252-2
- ↵Aganezov S, Yan SM, Soto DC, Kirsche M, Zarate S, Avdeyev P, Taylor DJ, Shafin K, Shumate A, Xiao C, 2022. A complete reference genome improves analysis of human genetic variation. Science 376: eabl3533. 10.1126/science.abl3533
- ↵Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, Song X, Richmond TA, Middle CM, Rodesch MJ, Packard CJ, 2007. Direct selection of human genomic loci by microarray hybridization. Nat Methods 4: 903–905. 10.1038/nmeth1111
- ↵Albrecht V, Zweiniger C, Surendranath V, Lang K, Schöfl G, Dahl A, Winkler S, Lange V, Böhme I, Schmidt AH. 2017. Dual redundant sequencing strategy: full-length gene characterisation of 1056 novel and confirmatory HLA alleles. HLA 90: 79–87. 10.1111/tan.13057
- ↵Alfano M, De Antoni L, Centofanti F, Visconti VV, Maestri S, Degli Esposti C, Massa R, D'Apice MR, Novelli G, Delledonne M, 2022. Characterization of full-length CNBP expanded alleles in myotonic dystrophy type 2 patients by Cas9-mediated enrichment and nanopore sequencing. eLife 11: e80229. 10.7554/eLife.80229
- ↵Altemose N, Logsdon GA, Bzikadze AV, Sidhwani P, Langley SA, Caldas GV, Hoyt SJ, Uralsky L, Ryabov FD, Shew CJ, 2022. Complete genomic and epigenetic maps of human centromeres. Science 376: eabl4178. 10.1126/science.abl4178
- ↵Altmüller J, Budde BS, Nürnberg P. 2014. Enrichment of target sequences for next-generation sequencing applications in research and diagnostics. Biol Chem 395: 231–237. 10.1515/hsz-2013-0199
- ↵Ammar R, Paton TA, Torti D, Shlien A, Bader GD. 2015. Long read nanopore sequencing for detection of HLA and CYP2D6 variants and haplotypes. F1000Res 4: 17. 10.12688/f1000research.6037.2
- ↵Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AME, Dougherty ML, Nelson BJ, Shah A, Dutcher SK, 2019. Characterizing the major structural variant alleles of the human genome. Cell 176: 663–675.e19. 10.1016/j.cell.2018.12.019
- ↵Ballester LY, Luthra R, Kanagal-Shamanna R, Singh RR. 2016. Advances in clinical next-generation sequencing: target enrichment and sequencing technologies. Expert Rev Mol Diagn 16: 357–372. 10.1586/14737159.2016.1133298
- ↵Bennett-Baker PE, Mueller JL. 2017. CRISPR-mediated isolation of specific megabase segments of genomic DNA. Nucleic Acids Res 45: e165. 10.1093/nar/gkx749
- ↵Bruijnesteijn J, van der Wiel M, de Groot NG, Bontrop RE. 2021. Rapid characterization of complex killer cell immunoglobulin-like receptor (KIR) regions using Cas9 enrichment and nanopore sequencing. Front Immunol 12: 722181. 10.3389/fimmu.2021.722181
- ↵Bryant WB, Yang A, Griffin SH, Zhang W, Rafiq AM, Han W, Deak F, Mills MK, Long X, Miano JM. 2023. CRISPR-Cas9 long-read sequencing for mapping transgenes in the mouse genome. CRISPR J 6: 163–175. 10.1089/crispr.2022.0099
- ↵Bzikadze AV, Pevzner PA. 2020. Automated assembly of centromeres from ultra-long error-prone reads. Nat Biotechnol 38: 1309–1316. 10.1038/s41587-020-0582-4
- ↵Cao H, Wu J, Wang Y, Jiang H, Zhang T, Liu X, Xu Y, Liang D, Gao P, Sun Y, 2013. An integrated tool to study MHC region: accurate SNV detection and HLA genes typing in human MHC region using targeted high-throughput sequencing. PLoS One 8: e69388. 10.1371/journal.pone.0069388
- ↵Charnaud S, Munro JE, Semenec L, Mazhari R, Brewster J, Bourke C, Ruybal-Pesántez S, James R, Lautu-Gumal D, Karunajeewa H, 2022. Pacbio long-read amplicon sequencing enables scalable high-resolution population allele typing of the complex CYP2D6 locus. Commun Biol 5: 168. 10.1038/s42003-022-03102-8
- ↵Chiou C-S, Chen B-H, Wang Y-W, Kuo N-T, Chang C-H, Huang Y-T. 2023. Correcting modification-mediated errors in nanopore sequencing by nucleotide demodification and reference-based correction. Commun Biol 6: 1215. 10.1038/s42003-023-05605-4
- ↵Ciosi M, Cumming SA, Chatzi A, Larson E, Tottey W, Lomeikaite V, Hamilton G, Wheeler VC, Pinto RM, Kwak S, 2021. Approaches to sequence the HTT CAG repeat expansion and quantify repeat length variation. J Huntingt Dis 10: 53–74. 10.3233/JHD-200433
- ↵Clarke R, Heler R, MacDougall MS, Yeo NC, Chavez A, Regan M, Hanakahi L, Church GM, Marraffini LA, Merrill BJ. 2018. Enhanced bacterial immunity and mammalian genome editing via RNA polymerase-mediated dislodging of Cas9 from double strand DNA breaks. Mol Cell 71: 42–55.e8. 10.1016/j.molcel.2018.06.005
- ↵Cumming SA, Hamilton MJ, Robb Y, Gregory H, McWilliam C, Cooper A, Adam B, McGhie J, Hamilton G, Herzyk P, 2018. De novo repeat interruptions are associated with reduced somatic instability and mild or absent clinical features in myotonic dystrophy type 1. Eur J Hum Genet 26: 1635–1647. 10.1038/s41431-018-0156-9
- ↵Dapprich J, Ferriola D, Mackiewicz K, Clark PM, Rappaport E, D'Arcy M, Sasson A, Gai X, Schug J, Kaestner KH, 2016. The next generation of target capture technologies - large DNA fragment enrichment and sequencing determines regional genomic variation of high complexity. BMC Genomics 17: 486. 10.1186/s12864-016-2836-6
- ↵DeJesus-Hernandez M, Aleff RA, Jackson JL, Finch NA, Baker MC, Gendron TF, Murray ME, McLaughlin IJ, Harting JR, Graff-Radford NR, 2021. Long-read targeted sequencing uncovers clinicopathological associations for C9orf72-linked diseases. Brain 144: 1082–1088. 10.1093/brain/awab006
- ↵De Roeck A, De Coster W, Bossaerts L, Cacace R, De Pooter T, Van Dongen J, D'Hert S, De Rijk P, Strazisar M, Van Broeckhoven C, 2019. Nanosatellite: accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biol 20: 239. 10.1186/s13059-019-1856-3
- ↵Doudna JA, Charpentier E. 2014. The new frontier of genome engineering with CRISPR-Cas9. Science 346: 1258096. 10.1126/science.1258096
- ↵Dunlop MG, Farrington SM, Carothers AD, Wyllie AH, Sharp L, Burn J, Liu B, Kinzler KW, Vogelstein B. 1997. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet 6: 105–110. 10.1093/hmg/6.1.105
- ↵Ebbert MTW, Farrugia SL, Sens JP, Jansen-West K, Gendron TF, Prudencio M, McLaughlin IJ, Bowman B, Seetin M, DeJesus-Hernandez M, 2018. Long-read sequencing across the C9orf72 ‘GGGGCC’ repeat expansion: implications for clinical use and genetic discovery efforts in human disease. Mol Neurodegener 13: 46. 10.1186/s13024-018-0274-4
- ↵Edwards HS, Krishnakumar R, Sinha A, Bird SW, Patel KD, Bartsch MS. 2019. Real-time selective sequencing with RUBRIC: read until with basecall and reference-informed criteria. Sci Rep 9: 11475. 10.1038/s41598-019-47857-3
- ↵Fiol A, Jurado-Ruiz F, López‐Girona E, Aranzana MJ. 2022. An efficient CRISPR-Cas9 enrichment sequencing strategy for characterizing complex and highly duplicated genomic regions. A case study in the Prunus salicina LG3-MYB10 genes cluster. Plant Methods 18: 105. 10.1186/s13007-022-00937-4
- ↵Gabrieli T, Sharim H, Fridman D, Arbib N, Michaeli Y, Ebenstein Y. 2018. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Res 46: e87. 10.1093/nar/gky411
- ↵Gershman A, Sauria MEG, Guitart X, Vollger MR, Hook PW, Hoyt SJ, Jain M, Shumate A, Razaghi R, Koren S, 2022. Epigenetic patterns in a complete human genome. Science 376: eabj5089. 10.1126/science.abj5089
- ↵Giannuzzi G, Logsdon GA, Chatron N, Miller DE, Reversat J, Munson KM, Hoekzema K, Bonnet-Dupeyron M-N, Rollat-Farnier P-A, Baker CA, 2021. Alpha satellite insertion close to an ancestral centromeric region. Mol Biol Evol 38: 5576–5587. 10.1093/molbev/msab244
- ↵Giesselmann P, Brändl B, Raimondeau E, Bowen R, Rohrandt C, Tandon R, Kretzmer H, Assum G, Galonska C, Siebert R, 2019. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat Biotechnol 37: 1478–1481. 10.1038/s41587-019-0293-x
- ↵Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, Downs B, Sukumar S, Sedlazeck FJ, Timp W. 2020. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol 38: 433–438. 10.1038/s41587-020-0407-5
- ↵Giolai M, Paajanen P, Verweij W, Witek K, Jones JDG, Clark MD. 2017. Comparative analysis of targeted long read sequencing approaches for characterization of a plant's immune receptor repertoire. BMC Genomics 18: 564. 10.1186/s12864-017-3936-7
- ↵Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, 2009. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 27: 182–189. 10.1038/nbt.1523
- ↵Gong L, Wong C-H, Cheng W-C, Tjong H, Menghi F, Ngan CY, Liu ET, Wei C-L. 2018. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nat Methods 15: 455–460. 10.1038/s41592-018-0002-6
- ↵Grosso V, Marcolungo L, Maestri S, Alfano M, Lavezzari D, Iadarola B, Salviati A, Mariotti B, Botta A, D'Apice MR, 2021. Characterization of FMR1 repeat expansion and intragenic variants by indirect sequence capture. Front Genet 12: 743230. 10.3389/fgene.2021.743230
- ↵Hafford-Tear NJ, Tsai Y-C, Sadan AN, Sanchez-Pintado B, Zarouchlioti C, Maher GJ, Liskova P, Tuft SJ, Hardcastle AJ, Clark TA, 2019. CRISPR/Cas9-targeted enrichment and long-read sequencing of the Fuchs endothelial corneal dystrophy-associated TCF4 triplet repeat. Genet Med 21: 2092–2102. 10.1038/s41436-019-0453-x
- ↵Hale H, Gardner EM, Viruel J, Pokorny L, Johnson MG. 2020. Strategies for reducing per-sample costs in target capture sequencing for phylogenomics and population genomics in plants. Appl Plant Sci 8: e11337. 10.1002/aps3.11337
- ↵Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, Smith SW, Middle CM, Rodesch MJ, Albert TJ, Hannon GJ, 2007. Genome-wide in situ exon capture for selective resequencing. Nat Genet 39: 1522–1527. 10.1038/ng.2007.42
- ↵Höijer I, Tsai Y, Clark TA, Kotturi P, Dahl N, Stattin E, Bondeson M, Feuk L, Gyllensten U, Ameur A. 2018. Detailed analysis of HTT repeat elements in human blood using targeted amplification-free long-read sequencing. Hum Mutat 39: 1262–1272. 10.1002/humu.23580
- ↵Hon T, Mars K, Young G, Tsai Y-C, Karalius JW, Landolin JM, Maurer N, Kudrna D, Hardigan MA, Steiner CC, 2020. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci Data 7: 399. 10.1038/s41597-020-00743-4
- ↵Hoyt SJ, Storer JM, Hartley GA, Grady PGS, Gershman A, de Lima LG, Limouse C, Halabian R, Wojenski L, Rodriguez M, 2022. From telomere to telomere: the transcriptional and epigenetic state of human repeat elements. Science 376: eabk3112. 10.1126/science.abk3112
- ↵Hung KL, Luebeck J, Dehkordi SR, Colón CI, Li R, Wong IT-L, Coruh C, Dharanipragada P, Lomeli SH, Weiser NE, 2022. Targeted profiling of human extrachromosomal DNA by CRISPR-CATCH. Nat Genet 54: 1746–1754. 10.1038/s41588-022-01190-0
- ↵Israeli O, Guedj-Dana Y, Shifman O, Lazar S, Cohen-Gihon I, Amit S, Ben-Ami R, Paran N, Schuster O, Weiss S, 2022. Rapid amplicon nanopore sequencing (RANS) for the differential diagnosis of monkeypox virus and other vesicle-forming pathogens. Viruses 14: 1817. 10.3390/v14081817
- ↵Iyer SV, Kramer M, Goodwin S, McCombie WR. 2022. ACME: an affinity-based Cas9 mediated enrichment method for targeted nanopore sequencing. bioRxiv 10.1101/2022.02.03.478550
- ↵Jiang F, Doudna JA. 2017. CRISPR–Cas9 structures and mechanisms. Annu Rev Biophys 46: 505–529. 10.1146/annurev-biophys-062215-010822
- ↵Jiang W, Zhu TF. 2016. Targeted isolation and cloning of 100-kb microbial genomic sequences by Cas9-assisted targeting of chromosome segments. Nat Protoc 11: 960–975. 10.1038/nprot.2016.055
- ↵Jiang W, Zhao X, Gabrieli T, Lou C, Ebenstein Y, Zhu TF. 2015. Cas9-Assisted Targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nat Commun 6: 8101. 10.1038/ncomms9101
- ↵Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. 10.1126/science.1225829
- ↵Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321: 1801–1806. 10.1126/science.1164368
- ↵Kanagawa T. 2003. Bias and artifacts in multitemplate polymerase chain reactions (PCR). J Biosci Bioeng 96: 317–323. 10.1016/S1389-1723(03)90130-7
- ↵Khodakov D, Wang C, Zhang DY. 2016. Diagnostics based on nucleic acid sequence variant profiling: PCR, hybridization, and NGS approaches. Adv Drug Deliv Rev 105: 3–19. 10.1016/j.addr.2016.04.005
- ↵Kiełbasa SM, Wan R, Sato K, Horton P, Frith MC. 2011. Adaptive seeds tame genomic sequence comparison. Genome Res 21: 487–493. 10.1101/gr.113985.110
- ↵Kirov I, Polkhovskaya E, Dudnikov M, Merkulov P, Vlasova A, Karlov G, Soloviev A. 2022. Searching for a needle in a haystack: Cas9-targeted nanopore sequencing and DNA methylation profiling of full-length glutenin genes in a Big cereal genome. Plants 11: 5. 10.3390/plants11010005
- ↵Kovaka S, Fan Y, Ni B, Timp W, Schatz MC. 2021. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat Biotechnol 39: 431–441. 10.1038/s41587-020-0731-9
- ↵Kozarewa I, Armisen J, Gardner AF, Slatko BE, Hendrickson CL. 2015. Overview of target enrichment strategies. Curr Protoc Mol Biol 112: 7.21.1–7.21.23. 10.1002/0471142727.mb0721s112
- ↵Lagarde J, Uszczynska-Ratajczak B, Carbonell S, Pérez-Lluch S, Abad A, Davis C, Gingeras TR, Frankish A, Harrow J, Guigo R, 2017. High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nat Genet 49: 1731–1740. 10.1038/ng.3988
- ↵Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, 2016. Clinvar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44: D862–D868. 10.1093/nar/gkv1222
- ↵Lang D, Zhang S, Ren P, Liang F, Sun Z, Meng G, Tan Y, Li X, Lai Q, Han L, 2020. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. GigaScience 9: giaa123. 10.1093/gigascience/giaa123
- ↵Lee NCO, Larionov V, Kouprina N. 2015. Highly efficient CRISPR/Cas9-mediated TAR cloning of genes and chromosomal loci from complex genomes in yeast. Nucleic Acids Res 43: e55. 10.1093/nar/gkv112
- ↵Lefoulon E, Vaisman N, Frydman HM, Sun L, Voland L, Foster JM, Slatko BE. 2019. Author correction: large enriched fragment targeted sequencing (LEFT-SEQ) applied to capture of Wolbachia genomes. Sci Rep 9: 20184. 10.1038/s41598-019-55305-5
- ↵Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191
- ↵Liu G, Zhang Y, Zhang T. 2020. Computational approaches for effective CRISPR guide RNA design and evaluation. Comput Struct Biotechnol J 18: 35–44. 10.1016/j.csbj.2019.11.006
- ↵Loomis EW, Eid JS, Peluso P, Yin J, Hickey L, Rank D, McCalmon S, Hagerman RJ, Tassone F, Hagerman PJ. 2013. Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res 23: 121–128. 10.1101/gr.141705.112
- ↵Loose M. 2018. Finding the needle: targeted nanopore sequencing and CRISPR-Cas9. CRISPR J 1: 265–267. 10.1089/crispr.2018.29028.mlo
- ↵Loose M, Malla S, Stout M. 2016. Real-time selective sequencing using nanopore technology. Nat Methods 13: 751–754. 10.1038/nmeth.3930
- ↵López-Girona E, Davy MW, Albert NW, Hilario E, Smart MEM, Kirk C, Thomson SJ, Chagné D. 2020. CRISPR-Cas9 enrichment and long read sequencing for fine mapping in plants. Plant Methods 16: 121. 10.1186/s13007-020-00661-x
- ↵Ma H, Tu L-C, Naseri A, Huisman M, Zhang S, Grunwald D, Pederson T. 2016. CRISPR-Cas9 nuclear dynamics and target recognition in living cells. J Cell Biol 214: 529–537. 10.1083/jcb.201604115
- ↵Madsen EB, Höijer I, Kvist T, Ameur A, Mikkelsen MJ. 2020. Xdrop: targeted sequencing of long DNA molecules from low input samples using droplet sorting. Hum Mutat 41: 1671–1679. 10.1002/humu.24063
- ↵Mahmoud M, Harting J, Corbitt H, Chen X, Jhangiani SN, Doddapaneni H, Meng Q, Han T, Lambert C, Zhang S, 2024. Closing the gap: solving complex medically relevant genes at scale. medRxiv 10.1101/2024.03.14.24304179
- ↵Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J, Turner DJ. 2010. Target-enrichment strategies for next-generation sequencing. Nat Methods 7: 111–118. 10.1038/nmeth.1419
- ↵Mangin A, de Pontual L, Tsai Y-C, Monteil L, Nizon M, Boisseau P, Mercier S, Ziegle J, Harting J, Heiner C, 2021. Robust detection of somatic mosaicism and repeat interruptions by long-read targeted sequencing in myotonic dystrophy type 1. Int J Mol Sci 22: 2616. 10.3390/ijms22052616
- ↵Marquet M, Zöllkau J, Pastuschek J, Viehweger A, Schleußner E, Makarewicz O, Pletz MW, Ehricht R, Brandt C. 2022. Evaluation of microbiome enrichment and host DNA depletion in human vaginal samples using Oxford Nanopore's adaptive sequencing. Sci Rep 12: 4000. 10.1038/s41598-022-08003-8
- ↵Martin S, Heavens D, Lan Y, Horsfield S, Clark MD, Leggett RM. 2022. Nanopore adaptive sampling: a tool for enrichment of low abundance species in metagenomic samples. Genome Biol 23: 11. 10.1186/s13059-021-02582-x
- ↵Mastrorosa FK, Miller DE, Eichler EE. 2023. Applications of long-read sequencing to Mendelian genetics. Genome Med 15: 42. 10.1186/s13073-023-01194-3
- ↵McDonald TL, Zhou W, Castro CP, Mumm C, Switzenberg JA, Mills RE, Boyle AP. 2021. Cas9 targeted enrichment of mobile elements using nanopore sequencing. Nat Commun 12: 3586. 10.1038/s41467-021-23918-y
- ↵Merkulov P, Gvaramiya S, Dudnikov M, Komakhin R, Omarov M, Kocheshkova A, Konstantinov Z, Soloviev A, Karlov G, Divashuk M, 2023. Cas9-targeted nanopore sequencing rapidly elucidates the transposition preferences and DNA methylation profiles of mobile elements in plants. J Integr Plant Biol 65: 2242–2261. 10.1111/jipb.13555
- ↵Mertes F, Elsharawy A, Sauer S, van Helvoort JMLM, van der Zaag PJ, Franke A, Nilsson M, Lehrach H, Brookes AJ. 2011. Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief Funct Genomics 10: 374–386. 10.1093/bfgp/elr033
- ↵Miller DE, Sulovari A, Wang T, Loucks H, Hoekzema K, Munson KM, Lewis AP, Fuerte EPA, Paschal CR, Walsh T, 2021. Targeted long-read sequencing identifies missing disease-causing variation. Am J Hum Genet 108: 1436–1449. 10.1016/j.ajhg.2021.06.006
- ↵Mitsuhashi S, Kryukov K, Nakagawa S, Takeuchi JS, Shiraishi Y, Asano K, Imanishi T. 2017. A portable system for rapid bacterial composition analysis using a nanopore-based sequencer and laptop computer. Sci Rep 7: 5657. 10.1038/s41598-017-05772-5
- ↵Mizuguchi T, Toyota T, Miyatake S, Mitsuhashi S, Doi H, Kudo Y, Kishida H, Hayashi N, Tsuburaya RS, Kinoshita M, 2021. Complete sequencing of expanded SAMD12 repeats by long-read sequencing and Cas9-mediated enrichment. Brain 144: 1103–1117. 10.1093/brain/awab021
- ↵Nagahashi M, Shimada Y, Ichikawa H, Kameyama H, Takabe K, Okuda S, Wakai T. 2019. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci 110: 6–15. 10.1111/cas.13837
- ↵Naim F, Shand K, Hayashi S, O'Brien M, McGree J, Johnson AAT, Dugdale B, Waterhouse PM. 2020. Are the current gRNA ranking prediction algorithms useful for genome editing in plants? PLoS One 15: e0227994. 10.1371/journal.pone.0227994
- ↵Nattestad M, Goodwin S, Ng K, Baslan T, Sedlazeck FJ, Rescheneder P, Garvin T, Fang H, Hutton E, Tseng E, 2018. Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res 28: 1126–1135. 10.1101/gr.231100.117
- ↵Norris AL, Workman RE, Fan Y, Eshleman JR, Timp W. 2016. Nanopore sequencing detects structural variants in cancer. Cancer Biol Ther 17: 246–253. 10.1080/15384047.2016.1139236
- ↵Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, 2022. The complete sequence of a human genome. Science 376: 44–53. 10.1126/science.abj6987
- ↵Parla JS, Iossifov I, Grabill I, Spector MS, Kramer M, McCombie WR. 2011. A comparative analysis of exome capture. Genome Biol 12: R97. 10.1186/gb-2011-12-9-r97
- ↵Patel A, Dogan H, Payne A, Krause E, Sievers P, Schoebe N, Schrimpf D, Blume C, Stichel D, Holmes N, 2022. Rapid-CNS2: rapid comprehensive adaptive nanopore-sequencing of CNS tumors, a proof-of-concept study. Acta Neuropathol (Berl) 143: 609–612. 10.1007/s00401-022-02415-6
- ↵Paulson H. 2018. Repeat expansion diseases. Handb Clin Neurol 147: 105–123. 10.1016/B978-0-444-63233-3.00009-9
- ↵Payne A, Holmes N, Clarke T, Munro R, Debebe BJ, Loose M. 2021. Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat Biotechnol 39: 442–450. 10.1038/s41587-020-00746-x
- ↵Ramirez R, van Buuren N, Gamelin L, Soulette C, May L, Han D, Yu M, Choy R, Cheng G, Bhardwaj N, 2021. Targeted long-read sequencing reveals comprehensive architecture, burden, and transcriptional signatures from hepatitis B virus-associated integrations and translocations in hepatocellular carcinoma cell lines. J Virol 95: e00299–21. 10.1128/JVI.00299-21
- ↵Ramsuran V, Kulkarni S, O'huigin C, Yuki Y, Augusto DG, Gao X, Carrington M. 2015. Epigenetic regulation of differential HLA-A allelic expression levels. Hum Mol Genet 24: 4268–4275. 10.1093/hmg/ddv158
- ↵Rath D, Amlinger L, Rath A, Lundgren M. 2015. The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie 117: 119–128. 10.1016/j.biochi.2015.03.025
- ↵Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. 2016. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol 34: 339–344. 10.1038/nbt.3481
- ↵Rioux JD, Goyette P, Vyse TJ, Hammarström L, Fernando MMA, Green T, De Jager PL, Foisy S, Wang J, de Bakker PIW, 2009. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc Natl Acad Sci 106: 18680–18685. 10.1073/pnas.0909307106
- ↵Rossi MJ, Lai WKM, Pugh BF. 2018. Simplified ChIP-exo assays. Nat Commun 9: 2842. 10.1038/s41467-018-05265-7
- ↵Rubben K, Tilleman L, Deserranno K, Tytgat O, Deforce D, Nieuwerburgh FV. 2022. Cas9 targeted nanopore sequencing with enhanced variant calling improves CYP2D6-CYP2D7 hybrid allele genotyping. PLoS Genet 18: e1010176. 10.1371/journal.pgen.1010176
- ↵Sakharkar MK, Chow VTK, Kangueane P. 2004. Distributions of exons and introns in the human genome. In Silico Biol 4: 387–393.
- ↵Sander JD, Joung JK. 2014. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32: 347–355. 10.1038/nbt.2842
- ↵Santamaria P, Lindstrom AL, Boyce-Jacino MT, Myster SH, Barbosa JJ, Faras AJ, Rich SS. 1993. HLA class I sequence-based typing. Hum Immunol 37: 39–50. 10.1016/0198-8859(93)90141-M
- ↵Schatz MC. 2017. Nanopore sequencing meets epigenetics. Nat Methods 14: 347–348. 10.1038/nmeth.4240
- ↵Schuele L, Cassidy H, Lizarazo E, Strutzberg-Minder K, Schuetze S, Loebert S, Lambrecht C, Harlizius J, Friedrich AW, Peter S, 2020. Assessment of viral targeted sequence capture using nanopore sequencing directly from clinical samples. Viruses 12: 1358. 10.3390/v12121358
- ↵Schultzhaus Z, Wang Z, Stenger D. 2021. CRISPR-based enrichment strategies for targeted sequencing. Biotechnol Adv 46: 107672. 10.1016/j.biotechadv.2020.107672
- ↵Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, 1996. DPC4 gene in various tumor types. Cancer Res 56: 2527–2530.
- ↵Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, Schatz MC. 2018. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15: 461–468. 10.1038/s41592-018-0001-7
- ↵Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, Bosworth C, Armstrong J, Tigyi K, Maurer N, Koren S, 2020. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat Biotechnol 38: 1044–1053. 10.1038/s41587-020-0503-6
- ↵Shin GW, Greer SU, Xia LC, Lee HJ, Zhou J, Boles TC, Ji HP. 2019. Targeted short read sequencing and assembly of re-arrangements and candidate gene loci provide megabase diplotypes. Nucleic Acids Res 47: e115. 10.1093/nar/gkz661
- ↵Shola DTN, Yang C, Kewaldar V-S, Kar P, Bustos V. 2020. New additions to the CRISPR toolbox: CRISPR-CLONInG and CRISPR-CLIP for donor construction in genome editing. CRISPR J 3: 109–122. 10.1089/crispr.2019.0062
- ↵Singh M, Al-Eryani G, Carswell S, Ferguson JM, Blackburn J, Barton K, Roden D, Luciani F, Giang Phan T, Junankar S, 2019. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat Commun 10: 3120. 10.1038/s41467-019-11049-4
- ↵Stangl C, de Blank S, Renkens I, Westera L, Verbeek T, Valle-Inclan JE, González RC, Henssen AG, van Roosmalen MJ, Stam RW, 2020. Partner independent fusion gene detection by multiplexed CRISPR-Cas9 enrichment and long read nanopore sequencing. Nat Commun 11: 2861. 10.1038/s41467-020-16641-7
- ↵Steiert TA, Fuß J, Juzenas S, Wittig M, Hoeppner MP, Vollstedt M, Varkalaite G, ElAbd H, Brockmann C, Görg S, 2022. High-throughput method for the hybridisation-based targeted enrichment of long genomic fragments for PacBio third-generation sequencing. NAR Genom Bioinform 4: lqac051. 10.1093/nargab/lqac051
- ↵Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. 2014. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507: 62–67. 10.1038/nature13011
- ↵Sternberg SH, Richter H, Charpentier E, Qimron U. 2016. Adaptation in CRISPR-Cas systems. Mol Cell 61: 797–808. 10.1016/j.molcel.2016.01.030
- ↵Stevanovski I, Chintalaphani SR, Gamaarachchi H, Ferguson JM, Pineda SS, Scriba CK, Tchan M, Fung V, Ng K, Cortese A, 2022. Comprehensive genetic diagnosis of tandem repeat expansion disorders with programmable targeted nanopore sequencing. Sci Adv 8: 17. 10.1126/sciadv.abm5386
- ↵Stevens RC, Steele JL, Glover WR, Sanchez-Garcia JF, Simpson SD, O'Rourke D, Ramsdell JS, MacManes MD, Thomas WK, Shuber AP. 2019. A novel CRISPR/Cas9 associated technology for sequence-specific nucleic acid enrichment. PLoS One 14: e0215441. 10.1371/journal.pone.0215441
- ↵Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, 2019. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res 47: D941–D947. 10.1093/nar/gky1015
- ↵Tewhey R, Warner JB, Nakano M, Libby B, Medkova M, David PH, Kotsopoulos SK, Samuels ML, Hutchison JB, Larson JW, 2009. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat Biotechnol 27: 1025–1031. 10.1038/nbt.1583
- ↵Tringe SG, Hugenholtz P. 2008. A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol 11: 442–446. 10.1016/j.mib.2008.09.011
- ↵Tsai Y-C, Zafar F, McEachin ZT, McLaughlin I, Van Blitterswijk M, Ziegle J, Schüle B. 2022. Multiplex CRISPR/Cas9-guided No-Amp targeted sequencing panel for spinocerebellar ataxia repeat expansions. In Genomic structural variants in nervous system disorders. Neuromethods (ed. Proukakis C), pp. 95–120. Springer, New York.
- ↵Turner EH, Ng SB, Nickerson DA, Shendure J. 2009. Methods for genomic partitioning. Annu Rev Genomics Hum Genet 10: 263–284. 10.1146/annurev-genom-082908-150112
- ↵Vandiver AR, Pielstick B, Gilpatrick T, Hoang AN, Vernon HJ, Wanagat J, Timp W. 2022. Long read mitochondrial genome sequencing using Cas9-guided adaptor ligation. Mitochondrion 65: 176–183. 10.1016/j.mito.2022.06.003
- ↵van Haasteren J, Munis AM, Gill DR, Hyde SC. 2021. Genome-wide integration site detection using Cas9 enriched amplification-free long-range sequencing. Nucleic Acids Res 49: e16. 10.1093/nar/gkaa1152
- ↵Varley KE, Mitra RD. 2008. Nested patch PCR enables highly multiplexed mutation discovery in candidate genes. Genome Res 18: 1844–1850. 10.1101/gr.078204.108
- ↵Vollger MR, Guitart X, Dishuck PC, Mercuri L, Harvey WT, Gershman A, Diekhans M, Sulovari A, Munson KM, Lewis AP, 2022. Segmental duplications and their variation in a complete human genome. Science 376: eabj6965. 10.1126/science.abj6965
- ↵Vondrak T, Ávila Robledillo L, Novák P, Koblížková A, Neumann P, Macas J. 2020. Characterization of repeat arrays in ultra-long nanopore reads reveals frequent origin of satellite DNA from retrotransposon-derived tandem repeats. Plant J 101: 484–500. 10.1111/tpj.14546
- ↵Vu T, Davidson S-L, Borgesi J, Maksudul M, Jeon T-J, Shim J. 2017. Piecing together the puzzle: nanopore technology in detection and quantification of cancer biomarkers. RSC Adv 7: 42653–42666. 10.1039/C7RA08063H
- ↵Wallace AD, Sasani TA, Swanier J, Gates BL, Greenland J, Pedersen BS, Varley KE, Quinlan AR. 2021. CaBagE: a Cas9-based background elimination strategy for targeted, long-read DNA sequencing. PLoS One 16: e0241253. 10.1371/journal.pone.0241253
- ↵Walsh T, Casadei S, Munson KM, Eng M, Mandell JB, Gulsuner S, King M-C. 2021. CRISPR–Cas9/long-read sequencing approach to identify cryptic mutations in BRCA1 and other tumour suppressor genes. J Med Genet 58: 850–852. 10.1136/jmedgenet-2020-107320
- ↵Wang M, Beck CR, English AC, Meng Q, Buhay C, Han Y, Doddapaneni HV, Yu F, Boerwinkle E, Lupski JR, 2015. PacBio-LITS: a large-insert targeted sequencing method for characterization of human disease-associated chromosomal structural variations. BMC Genomics 16: 214. 10.1186/s12864-015-1370-2
- ↵Wanner N, Larsen PA, McLain A, Faulk C. 2021. The mitochondrial genome and Epigenome of the Golden lion Tamarin from fecal DNA using Nanopore adaptive sequencing. BMC Genomics 22: 726. 10.1186/s12864-021-08046-7
- ↵Watson CM, Crinnion LA, Hewitt S, Bates J, Robinson R, Carr IM, Sheridan E, Adlard J, Bonthron DT. 2020. Cas9-based enrichment and single-molecule sequencing for precise characterization of genomic duplications. Lab Invest 100: 135–146. 10.1038/s41374-019-0283-0
- ↵Wei Y, Huang Y-H, Skopelitis DS, Iyer SV, Costa ASH, Yang Z, Kramer M, Adelman ER, Klingbeil O, Demerdash OE, 2022. SLC5A3-dependent myo-inositol auxotrophy in acute myeloid leukemia. Cancer Discov 12: 450–467. 10.1158/2159-8290.CD-20-1849
- ↵Weilguny L, De Maio N, Munro R, Manser C, Birney E, Loose M, Goldman N. 2023. Dynamic, adaptive sampling during nanopore sequencing using Bayesian experimental design. Nat Biotechnol 41: 1018–1025. 10.1038/s41587-022-01580-z
- ↵Wieben ED, Aleff RA, Basu S, Sarangi V, Bowman B, McLaughlin IJ, Mills JR, Butz ML, Highsmith EW, Ida CM, 2019. Amplification-free long-read sequencing of TCF4 expanded trinucleotide repeats in Fuchs Endothelial Corneal Dystrophy. PLoS One 14: e0219446. 10.1371/journal.pone.0219446
- ↵Wongsurawat T, Jenjaroenpun P, De Loose A, Alkam D, Ussery DW, Nookaew I, Leung YK, Ho SM, Day JD, Rodriguez A. 2020. A novel Cas9-targeted long-read assay for simultaneous detection of IDH1/2 mutations and clinically relevant MGMT methylation in fresh biopsies of diffuse glioma. Acta Neuropathol Commun 8: 87. 10.1186/s40478-020-00963-0
- ↵Wu X, Kriz AJ, Sharp PA. 2014. Target specificity of the CRISPR-Cas9 system. Quant Biol 2: 59–70. 10.1007/s40484-014-0030-x
- ↵Yang Z, Wu XS, Wei Y, Polyanskaya SA, Iyer SV, Jung M, Lach FP, Adelman ER, Klingbeil O, Milazzo JP, 2021. Transcriptional silencing of ALDH2 confers a dependency on Fanconi anemia proteins in acute myeloid leukemia. Cancer Discov 11: 2300–2315. 10.1158/2159-8290.CD-20-1542