
Abstract
Cross-talk between DNA methylation and histone modifications drives the establishment of composite epigenetic signatures and is traditionally studied using correlative rather than direct approaches. Here, we present sequential ChIP-bisulfite-sequencing (ChIP-BS-seq) as an approach to quantitatively assess DNA methylation patterns associated with chromatin modifications or chromatin-associated factors directly. A chromatin-immunoprecipitation (ChIP)-capturing step is used to obtain a restricted representation of the genome occupied by the epigenetic feature of interest, for which a single-base resolution DNA methylation map is then generated. When applied to H3 lysine 27 trimethylation (H3K27me3), we found that H3K27me3 and DNA methylation are compatible throughout most of the genome, except for CpG islands, where these two marks are mutually exclusive. Further ChIP-BS-seq-based analysis in Dnmt triple-knockout (TKO) embryonic stem cells revealed that total loss of CpG methylation is associated with alteration of H3K27me3 levels throughout the genome: H3K27me3 in localized peaks is decreased while broad local enrichments (BLOCs) of H3K27me3 are formed. At an even broader scale, these BLOCs correspond to regions of high DNA methylation in wild-type ES cells, suggesting that DNA methylation prevents H3K27me3 deposition locally and at a megabase scale. Our strategy provides a unique way of investigating global interdependencies between DNA methylation and other chromatin features.
Epigenetic regulation, involving DNA methylation and histone modifications, is fundamental to a multitude of biological processes such as transcription, DNA replication, and repair. The different modifications do not act independently of each other. Instead, cross-talk between different modifications plays an important role in establishment of chromatin diversity within the genome. Interdependent deposition and mutual exclusion of various marks result in complex modification patterns with different functional outcomes (Fischle 2008; Cedar and Bergman 2009; Lee et al. 2010). Classically, such patterns are determined by parallel genomic mapping of the various modifications within the same samples, using chromatin immunoprecipitation and deep sequencing (ChIP-seq) (Barski et al. 2007; Mikkelsen et al. 2007). However, the analysis of cross-talk through independent profiling experiments is complicated by cell population heterogeneity, cell cycle effects, and allele-specific marking of chromatin such as in imprinting or X-inactivation.
Here, we present a method for the integrated analysis of histone modification or transcription factor deposition patterns and the underlying DNA methylation. In our approach, termed ChIP-BS-seq, ChIP capturing is followed by bisulfite conversion and deep sequencing to directly assess DNA methylation levels in captured chromatin fragments. While the use of whole-genome bisulfite shotgun sequencing is limited by the cost of the required sequencing depth, a restricted genomic representation obtained by ChIP capturing allows one to reach adequate coverage at routine-scale sequencing, providing increased quantitative accuracy of DNA methylation measurements within captured regions.
We used ChIP-BS-seq to study the global cross-talk between H3K27me3 and DNA methylation, which are both linked to repression. Polycomb Repressive Complex 2 (PRC2) catalyzes H3K27me3 methylation via the SET domain of EZH2, while Polycomb Repressive Complex 1 (PRC1) is recruited to the H3K27me3 mark and is involved in gene silencing (Simon and Kingston 2009). The DNA methyltransferases DNMT3A/B and DNMT1 are responsible for the establishment and maintenance of the DNA methylation mark, respectively (Cedar and Bergman 2009). The interplay between DNA methylation and H3K27me3/Polycomb has been subject to extensive studies, and different phenomena have been described (Cedar and Bergman 2009). Direct interactions between Polycomb and the DNA methylation machinery have been reported, suggesting that H3K27me3 and DNA methylation co-occur (Viré et al. 2006). Co-occurrence was further supported by ChIP experiments analyzing DNA hypermethylated promoters in cancer cells (McGarvey et al. 2006; Schlesinger et al. 2007). Whereas H3K27me3 has been shown to “prime” gene promoters for later DNA methylation (Ohm et al. 2007; Schlesinger et al. 2007; Widschwendter et al. 2007; Gal-Yam et al. 2008; Mohn et al. 2008), several reports have shown antagonism or mutual exclusiveness between H3K27me3 and DNA methylation (Kondo et al. 2008; Lindroth et al. 2008; Bartke et al. 2010; Wu et al. 2010). Although the different observations are not necessarily incompatible, the co-occurrence of both marks is still a subject of debate. Using ChIP-BS-seq, we addressed the overlap of H3K27me3 and DNA methylation directly on a genome-wide scale. Our results unequivocally show that DNA methylation and H3K27me3 generally co-occur but are mutually exclusive in CpG-dense regions. This mutual exclusivity is found in both a cancer cell line as well as in mouse ES cells. Loss of DNA methylation in ES cells is associated with the formation of H3K27me3 patterns previously described as broad local enrichments (BLOCs), and, at an even larger scale, H3K27me3 appears in megabase-sized regions that were marked by high DNA-methylation in wild-type cells.
Results
Establishment of the strategy
An outline of our strategy, which we named ChIP-BS-seq, is shown in Figure 1A. ChIP is used to capture a genomic subfraction associated with a specific histone modification or transcription factor. Similarly, such subfraction can be obtained by capture of methylated DNA using a methyl-CpG binding domain (MBD). Captured DNA fragments are subjected to end-repair, adapter ligation using methylated adapters, bisulfite conversion, PCR-amplification, and deep sequencing (Fig. 1A). In this way, the levels of DNA methylation can be quantitatively assessed at base-resolution within the genomic subfraction of interest.
Integration of capturing methods and bisulfite-deep sequencing: ChIP-BS-seq and MethylCap-BS-seq. (A) Schematic outline of the ChIP-BS-seq and MethylCap-BS-seq procedures. Capturing of genomic regions of interest is achieved by the MethylCap procedure or ChIP with an antibody of interest. Captured DNA is processed as indicated. Shown intermediate products and final PCR fragments indicate the fate of unmethylated as well as methylated cytosines throughout the procedure. (B) Examples of conventional MethylCap-seq and MethylCap-BS-seq data of normal (N) and tumor (T) colon tissues. For each covered CpG, percentage methylation as derived from the MethylCap-BS-seq data is indicated by color (yellow, 0%; blue, 100%). (Green) CpG islands and a CpG density profile (CpG/bp). (C) Average profiles of DNA methylation and coverage in MethylCap peaks of normal colon tissue, as determined by MethylCap-BS-seq. (Blue) Percentage DNA methylation; (magenta) CpG coverage; (brown) read density. (D) MethylCap-BS-seq analysis of differentially methylated regions from normal/tumor colon tissue. Regions that gain DNA methylation in tumor tissue show increased CpG coverage (x-axis) and read-density (y-axis). Color-code depicts absolute changes in percent methylation of these regions, as determined by bisulfite sequencing. (Blue) increase; (yellow) decrease.
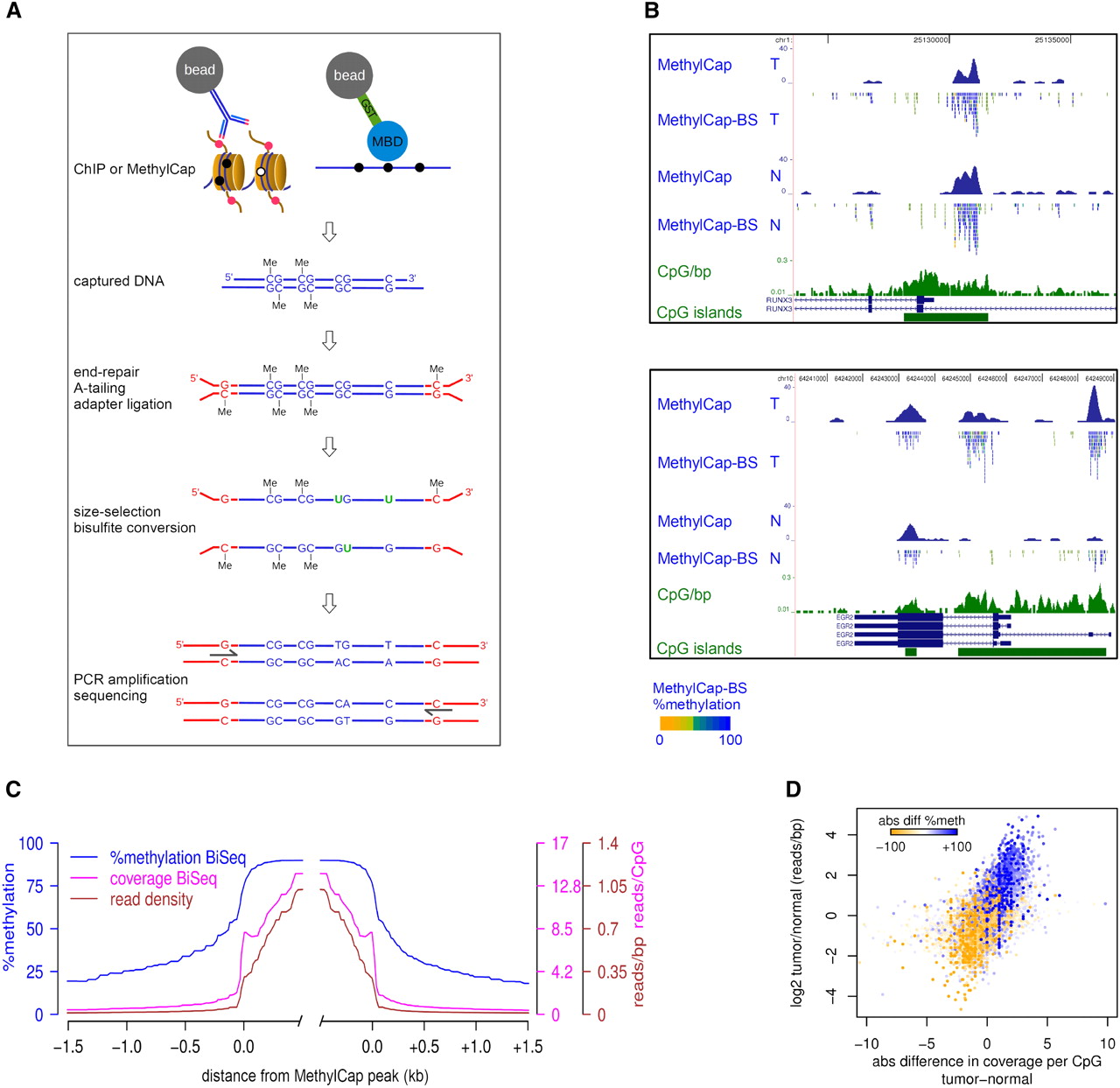
To provide technical proof-of-principle of our strategy, we used capturing of methylated DNA by MethylCap (Bock et al. 2010; Brinkman et al. 2010), followed by bisulfite-deep sequencing (MethylCap-BS-seq). In MethylCap, an MBD domain is used to capture methylated DNA (Brinkman et al. 2010). Genomic DNA isolated from normal and tumor colon tissues was used for MethylCap-BS-seq as well as for conventional MethylCap-seq experiments. Sequence reads were mapped as described in Methods (see also Supplemental Table S1). Read densities across the entire genome of MethylCap and MethylCap-BS-seq experiments correlated well (Pearson R = 0.833) (Supplemental Fig. S1A), which is comparable to technical replicates of conventional MethylCap samples (Pearson R ≤ 0.85) (AB Brinkman and HG Stunnenberg, unpubl.). This indicated that the same genomic subfraction was captured and sequenced in both procedures. Within each sequencing read, cytosines within a CpG context were scored for their methylation status by counting the percentage of bisulfite-induced mutations. As a control for bisulfite conversion efficiency, we assessed DNA methylation within mitochondrial DNA which was present at low levels due to its cellular abundance. Mitochondrial DNA is known to be completely unmethylated, and conversion was calculated to be 99.91%. In addition, efficient conversion of 99.7% was found in genomic CHG/CHH context.
Next, we focused on the captured genomic subfraction. Visual inspection of individual peaks showed that they were mostly hypermethylated (Fig. 1B; Supplemental Fig. S1B). This was confirmed on a global scale (Fig. 1C); mean methylation was 87% within MethylCap peaks. At boundaries, methylation showed a sharp drop that continued further with increasing distance from peaks. This was accompanied by decreases in read densities and, thus, CpG coverage. When focused exclusively on regions inside the MethylCap peaks, we found that 85% and 79% of the CpGs were at least 80% methylated in normal and tumor, respectively (Supplemental Fig. S1C).
Differential methylation between normal and tumor as observed by conventional MethylCap-seq was confirmed by MethylCap-BS-seq; differentially methylated regions showed corresponding alterations in CpG coverage and absolute CpG methylation (Fig. 1B, bottom panel; Supplemental Fig. S1B, middle panel). The same was observed on a global scale: Regions that gained DNA methylation in tumor compared to normal tissue (color-coded in Fig. 1D) showed increased coverage and methylation levels in MethylCap-BS-seq, whereas the opposite was observed for regions that lost methylation. Taken together, MethylCap-BS-seq showed hypermethylation of MethylCap-captured DNA, and differences observed between normal and tumor tissue could be corroborated and extended using MethylCap-BS-seq. These experiments demonstrate the successful integration of capturing experiments with bisulfite-deep sequencing.
H3K27me3-ChIP-BS-seq
We next applied our approach to ChIP-captured DNA (ChIP-BS-seq) for analysis of DNA methylation patterns associated with specific chromatin modifications. H3K27me3 and DNA methylation are both involved in gene silencing, but their interplay is under debate and has not been directly investigated. We performed ChIP-BS-seq on H3K27me3, using HCT116 colon carcinoma cells. For comparison, we also generated conventional ChIP-seq profiles for H3K27me3. Read densities of the conventional H3K27me3 ChIP-seq and ChIP-BS-seq experiments correlated well (Pearson R = 0.854) (Supplemental Fig. S2A), showing that the bisulfite/mapping procedure did not alter the H3K27me3 patterns. The H3K27me3 genome-wide profile showed H3K27me3 enrichments over broad regions, comprising genes and intergenic regions (Fig. 2A; Supplemental Figs. S2C, S3C). This pattern resembled the H3K27me3 “BLOCs” profile in mouse and human fibroblast cells (Pauler et al. 2009; Hawkins et al. 2010) and differs from H3K27me3 patterns in mouse ES cells, where the mark is present in focal areas at silent promoters (Mikkelsen et al. 2007; Pan et al. 2007; Zhao et al. 2007; Marks et al. 2009) (see also below). As described before, genes that were located within the H3K27me3 BLOCs were generally silent, whereas genes outside BLOCs had a significantly higher average expression level (Supplemental Fig. S3A).
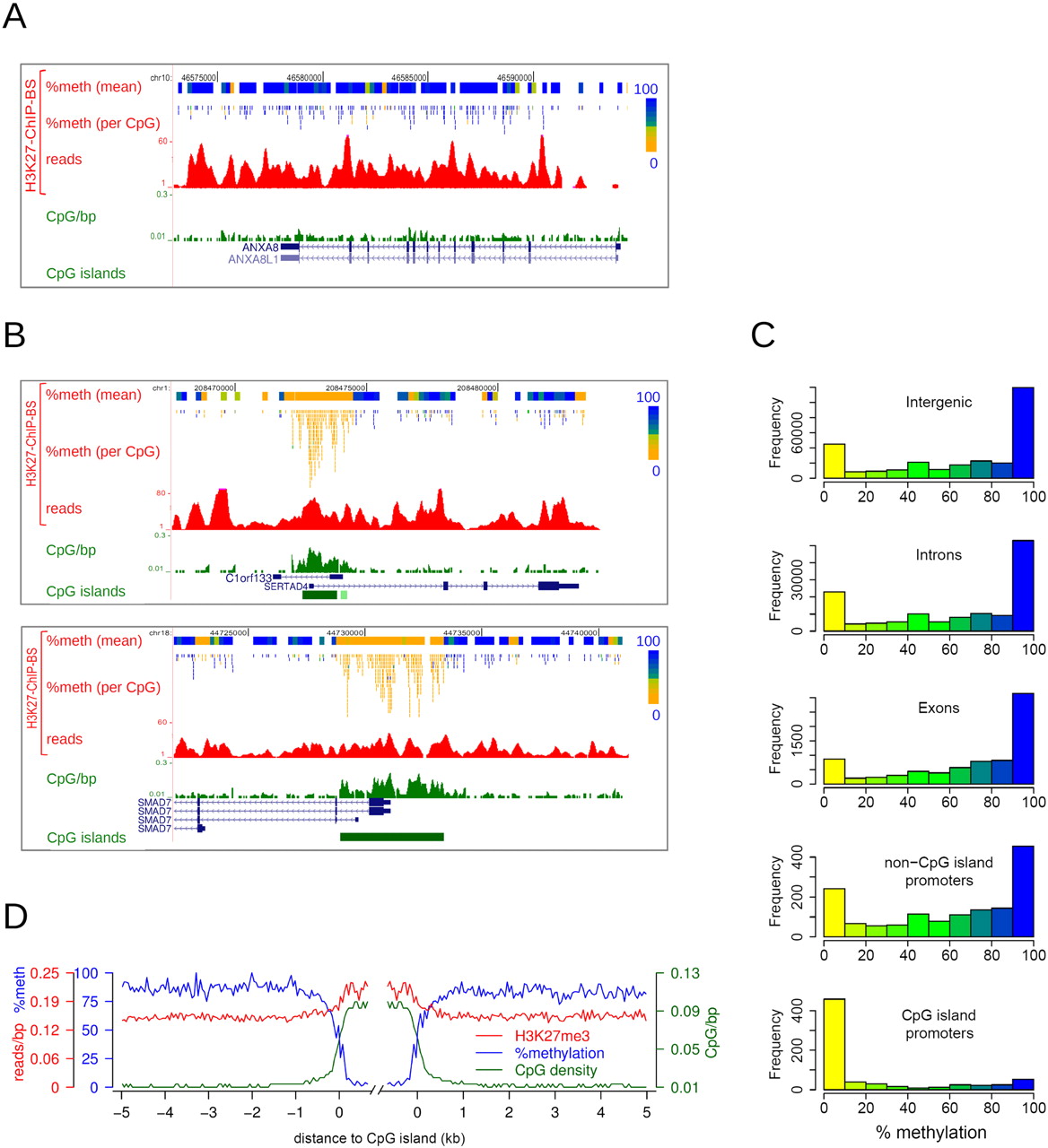
H3K27me3-enriched CpG island promoters are devoid of DNA methylation. (A,B) Examples of H3K27me3 ChIP-BS-seq data of HCT116 cells. H3K27me3 ChIP-BS-seq profiles are shown, as well as the derived DNA methylation data per covered CpG and per 200-bp window. Percentage methylation is color-coded as in Figure 1B. (C) Histograms showing the distribution of mean methylation in 300-bp windows throughout H3K27me3-enriched regions, as derived from H3K27me3 ChIP-BS-seq results. Windows were categorized according to functional genomic elements (intergenic, intron, exon, non-CpG island promoter, or CpG island promoter). (D) Average profiles of DNA methylation (blue), H3K27me3 (red), and CpG density (green) in regions flanking H3K27me3-enriched CpG islands, as determined from H3K27me3 ChIP-BS-seq data.
To rule out that crosslinking/decrosslinking of chromatin interfered with bisulfite conversion, we performed bisulfite sequencing on 12 independent PCR fragments amplified from genomic DNA isolated directly or after crosslinking/decrosslinking (Supplemental Fig. S2B). None of the fragments showed altered DNA methylation patterns after crosslinking/decrosslinking, showing that ChIP-derived DNA did not affect bisulfite conversion.
DNA hypomethylation in H3K27me3-enriched high CpG density regions
We then interrogated the DNA methylation status of captured H3K27me3-marked chromatin. H3K27me3-marked chromatin coincided mostly with fully methylated CpGs, with smaller patches of lower methylation occurring in between (Fig. 2A; Supplemental Fig. S2C). Strikingly, CpG-rich promoter regions marked with H3K27me3 contained exclusively unmethylated CpGs. For example, the SERTAD4, SMAD7, and OVOL2 genes were located within H3K27me3 BLOCs (Fig. 2B; Supplemental Fig. S2D) and were mostly DNA methylated, except for the CpG islands encompassing their gene promoters; these were completely hypomethylated.
To analyze DNA methylation of the H3K27me3-enriched fraction on a genome-wide scale, DNA methylation was determined in 300-bp windows throughout H3K27me3 BLOCs, and windows were subsequently categorized according to their genomic function (intergenic, intron, exon, non-CpG island promoter, CpG island promoter). Histograms displaying DNA methylation levels are shown in Figure 2C. Within a window size of 300 bp, the DNA methylation pattern was clearly bimodal, as shown previously (Meissner et al. 2008). Genes, intergenic regions, and non-CpG island promoters enriched for H3K27me3 generally contained unmethylated as well as methylated DNA, although methylated DNA was prevalent. In contrast, CpG island promoters marked with H3K27me3 contained only hypomethylated DNA. Since this exclusively applied to CpG island containing promoters, we also categorized windows according to CpG density (Supplemental Fig. S2E). While low-CpG-dense windows (<0.06 CpGs/bp) contained mostly DNA hypermethylation, high-CpG-density windows (>0.06 CpGs/bp) were exclusively hypomethylated. We next focused in more detail on H3K27me3-captured CpG islands. Median DNA methylation from 5 kb outside to 0.5kb within these CpG islands was plotted along with CpG-density and H3K27me3 levels (Fig. 2D). A clear and sharp decrease in DNA methylation took place at CpG island borders, resulting in an almost perfect inverse correlation between DNA methylation and CpG density. Inside CpG islands, H3K27me3 was consolidated. Taken together, our results show that H3K27me3 and DNA methylation generally co-occur in low- CpG-density regions, i.e., the bulk of the human genome. Within H3K27me3-marked regions of high-CpG-density, such as CpG islands, DNA is exclusively hypomethylated. We conclude that H3K27me3 and DNA methylation are mutually exclusive in CpG-dense regions.
Hypermethylated CpG islands show local H3K27me3 depletion within H3K27me3 BLOCs
The results obtained by H3K27me3 ChIP-BS-seq clearly demonstrate the existence of mutual exclusiveness of H3K27me3 with DNA methylation in CpG islands. To further confirm the relevance of this observation, we analyzed genome-wide profiles for H3K27me3 and DNA methylation generated by conventional ChIP-seq and MethylCap-seq. Using this approach, we could test whether DNA methylation also excludes H3K27me3. Obviously, the latter could not be directly shown by ChIP-BS-seq because regions lacking H3K27me3 were not captured and, thus, not available for DNA methylation measurements.
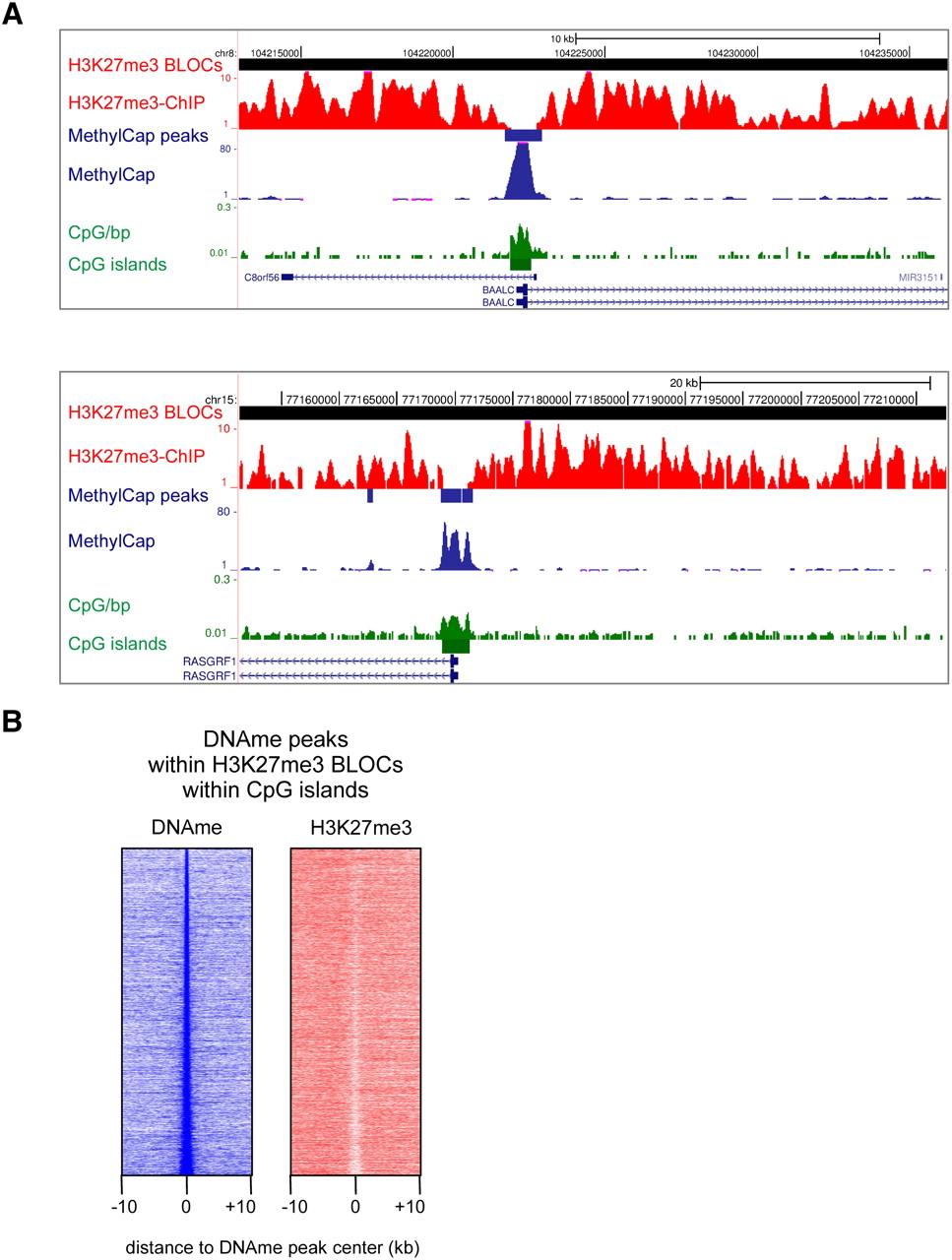
To test our hypothesis, we examined DNA-methylated CpG islands within H3K27me3 BLOCs. We detected 6456 H3K27me3 BLOCs with a median length of 77 kb (Supplemental Fig. S3B). Thirty-one percent, or 8781 out of the total 28,226 annotated CpG islands (http://genome.ucsc.edu/) were located within H3K27me3 BLOCs. Of these CpG islands, 6473 contained MethylCap peaks. Visual inspection of such methylated CpG islands within H3K27me3 BLOCs revealed local depletions of H3K27me3 (Fig. 3A; Supplemental Fig. S3C). To assess this at a genome-wide scale, we generated density maps representing read densities of MethylCap and H3K27me3 ChIP around BLOCs-contained methylated CpG islands (Fig. 3B). A clear local depletion of H3K27me3 was evident over methylated CpG islands. To exclude that this local depletion of H3K27me3 was due to a local drop in nucleosome density, we used publicly available DNaseI hypersensitivity data from HCT116 cells (ENCODE). MethylCap peaks were not enriched for DNaseI hypersensitivity and are, thus, unlikely to represent nucleosome-depleted regions (Supplemental Fig. S3D). Taken together, our data show that DNA-hypermethylated CpG islands present within H3K27me3 BLOCs are locally depleted for H3K27me3. This corroborates and extends the above findings on mutual exclusiveness of DNA methylation and H3K27me3 in regions of high CpG density.
H3K27me3 is locally depleted at hypermethylated CpG islands. (A) Examples of the genome-wide H3K27me3 ChIP-seq and MethylCap-seq data, demonstrating hypermethylated CpG islands within H3K27me3 BLOCs, and concomitant local depletion of H3K27me3. H3K27me3-enriched BLOCs and MethylCap peaks are indicated as red and blue rectangles, respectively. (B) Density maps of MethylCap-seq and H3K27me3 ChIP-seq read densities in 20-kb regions surrounding MethylCap peaks that reside in H3K27me3 BLOCs and overlap with CpG islands.
Mutual exclusiveness of H3K27me3 and DNA methylation in mouse ES cells
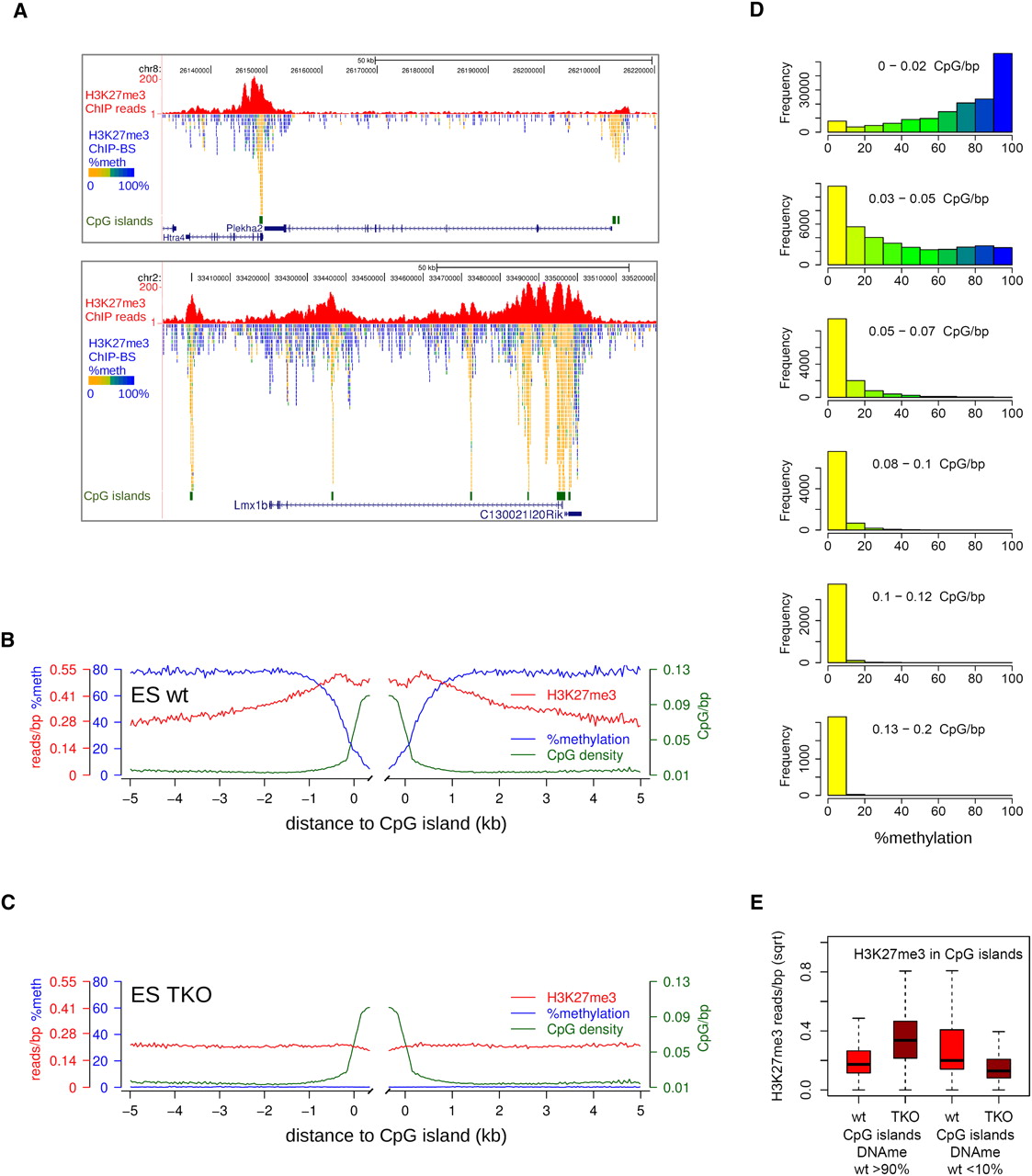
To extend our observations to noncancerous cells, we applied our H3K27me3-ChIP-BS-seq strategy to mouse embryonic stem (mES) cells. Regions of H3K27me3 enrichment were smaller than in HCT116, representing the typical, more peak-like mES pattern described before (Fig. 4A; Supplemental Fig. S4A; Mikkelsen et al. 2007; Pan et al. 2007; Zhao et al. 2007; Marks et al. 2009). About one third of these H3K27me3 peaks overlap with CpG islands or transcription start sites encompassing CpG islands (32%) (Supplemental Fig. S4B). Importantly and as in HCT116 cells, these CpG-rich regions contained exclusively unmethylated CpGs. For example, the Htra4/Plekha2 and Lmx1b genes were mostly DNA methylated, except for the CpG islands underneath the H3K27me3 peaks (Fig. 4A). We generated average profiles for H3K27me3 peaks over CpG islands and accompanying DNA methylation from H3K27me3-ChIP-BS-seq (Fig. 4B). Whereas DNA methylation on the flanks of CpG islands was 80%, this dropped to zero in CpG islands. This decrease appeared to be instigated at least 0.5 kb away from CpG islands, where CpG density is elevated but, by far, not maximal. To further relate DNA methylation–H3K27me3 mutual exclusiveness to high CpG density, we applied a 300-bp sliding window approach over all H3K27me3 peaks, categorized these windows according to their CpG density, and inferred their DNA methylation status from the ChIP-BS-seq data (Fig. 4D). Up to a CpG density of 0.05 CpG/bp, H3K27me3 and DNA methylation co-occurred within the same windows. This CpG density corresponds to that encountered at the borders (“shores”) of CpG islands (http://genome.ucsc.edu/). Within windows of higher CpG density (>0.05 CpG/bp), DNA methylation was virtually absent, confirming the antagonism between H3K27me3 and DNA methylation. Together, these data clearly confirm and extend our findings on mutual exclusiveness of DNA methylation and H3K27me3 in regions of high CpG density to mES cells.
Localized peaks of H3K27me3 in wild-type mES cells over CpG islands. (A) Screenshots of localized H3K27me3 peaks (red). Per covered CpG, percentage methylation as derived from the H3K27me3-BS-seq data is indicated in color. (B,C) Average profiles of DNA methylation (blue), H3K27me3 (red), and CpG density (green) in H3K27me3 peaks over CpG islands, as determined from H3K27me3-ChIP-BS-seq. (D) Histograms showing the distribution of methylation in 300-bp windows through H3K27me3 peaks, as inferred from H3K27me3 ChIP-BS-seq. Windows were categorized according to CpG density. (E) H3K27me3 changes in CpG islands that were either hypermethylated (>90%, left) or hypomethylated (<10%, right) in wild-type mES cells. Read counts shown are from conventional H3K27me3 ChIP.
As an extension of our observations with H3K27me3, we performed ChIP-BS-seq using H3K9me3, a repressive mark that has classically been linked to DNA hypermethylation. Indeed, examples of H3K9me3 peaks, such as in the imprinting control region (ICR) upstream of H19 (Bell and Felsenfeld 2000; Hark et al. 2000; Kanduri et al. 2000), showed that this mark was associated with hypermethylated DNA in mES cells (Supplemental Fig. S5A). Of all detected H3K9me3 peaks, over 90% of them were associated with hypermethylated DNA (Supplemental Fig. S5B). This is in sharp contrast to H3K27me3 peaks, under which there is a more equal subdivision of hypomethylated and hypermethylated DNA (Supplemental Fig. S5B), corresponding to high-CpG-density and low-CpG-density sequences, respectively (see Fig. 4D). These data show that histone marks other than H3K27me3 may display different DNA methylation properties, which extends and confirms the validity of the ChIP-BS-seq strategy.
H3K27me3 changes upon loss of DNA methylation
We next addressed the question of what happens to the H3K27me3 distribution upon removal of DNA methylation. Therefore, we performed H3K27me3-ChIP-BS-seq on Dnmt[1kd,3a−/−,3b−/−] (TKO) mES cells (Meissner et al. 2005). DNA methylation in virtually all of the captured/sequenced CpGs had disappeared (Supplemental Fig. S5B).
We first focused on CpG islands and created average profiles for H3K27me3 peaks over CpG islands (Fig. 4C). DNA methylation had disappeared, and the H3K27me3 signal over CpG islands that was generally high in wild-type cells had also decreased in TKO cells. It should be noted that hypermethylated CpG islands in wild-type mES cells are scarce and not captured by H3K27me3 ChIP due to the observed antagonism, so their contribution to these average profiles is almost zero. To enable analysis of H3K27me3 changes in hypermethylated CpG islands, we subselected such CpG islands using publicly available data (Stadler et al. 2011) and plotted the changes in H3K27me3 from conventional ChIP data. In case of antagonism between DNA methylation and H3K27me3, we expected that hypermethylated CpG islands would gain H3K27me3 in TKO cells, which was, indeed, the case (Fig. 4E). The opposite effect, although to a lesser magnitude, occurred at hypomethylated CpG islands. These CpG islands—containing high H3K27me3—displayed a loss in H3K27me3 in TKO cells (Fig. 4E). Thus, antagonism between DNA methylation and H3K27me3 was clearly observed in hypermethylated CpG islands but appeared to be absent in unmethylated CpG islands, where loss of DNA methylation caused a concomitant decrease of H3K27me3.
The H3K27me3 changes described above concern mainly the sharp and localized peaks of H3K27me3 typical of mES cells. An additional and striking observation in TKO cells was the appearance of large regions of H3K27me3 enrichment, resembling the typical BLOCs (Fig. 5A). To analyze the co-occurring changes of peaks and BLOCs in more detail, we plotted the fold-change of H3K27me3 BLOCs against the fold-change of the localized peaks within these BLOCs (Fig. 5B). In 43% of the cases, the increase in H3K27me3 in BLOCs was accompanied by a decrease of H3K27me3 in localized peaks within the same BLOCs (Fig. 5B, quadrant II). In another 18% of the cases, BLOCs appeared, but the peaks of H3K27me3 in these BLOCs were maintained (Fig. 5B, quadrant I). The observed changes of H3K27me3 in TKO cells—decrease in peaks and accumulation in BLOCs—could be clearly confirmed using targeted ChIP-qPCR. Ten out of the 12 tested peaks showed a decrease, and eight out of the nine tested BLOCs showed an increase (Supplemental Fig. S6). These results not only validated our genome-wide analyses but also excluded the possibility that a decrease of H3K27me3 peaks was a technical artifact due to a higher complexity of the TKO sequencing libraries by accumulation of H3K27me3 throughout a larger part of the genome.
Changes in H3K27me3 patterns upon loss of DNA methylation in Dnmt triple-knockout (TKO) mES cells. (A) Examples of H3K27me3 BLOCs appearing in TKO cells, and concomitant loss of H3K27me3 in localized peaks. Per covered CpG, percentage methylation as derived from H3K27me3-ChIP-BS-seq is indicated in color. (B) Scatterplot of H3K27me3 changes in peaks (x-axis) vs. BLOCs (y-axis). H3K27me3 peaks were matched against BLOCs in which they reside. I–IV indicate the four quadrants of the plot, as determined by log2 fold changes deviating from zero. (C) Histograms of percent DNA methylation in the BLOCs of each quadrant of B, as deduced from H3K27me3-ChIP-BS-seq in wild-type mES cells. (D) Density maps of H3K27me3 through BLOC transition regions detected in TKO mES cells. Average profiles are shown on top.
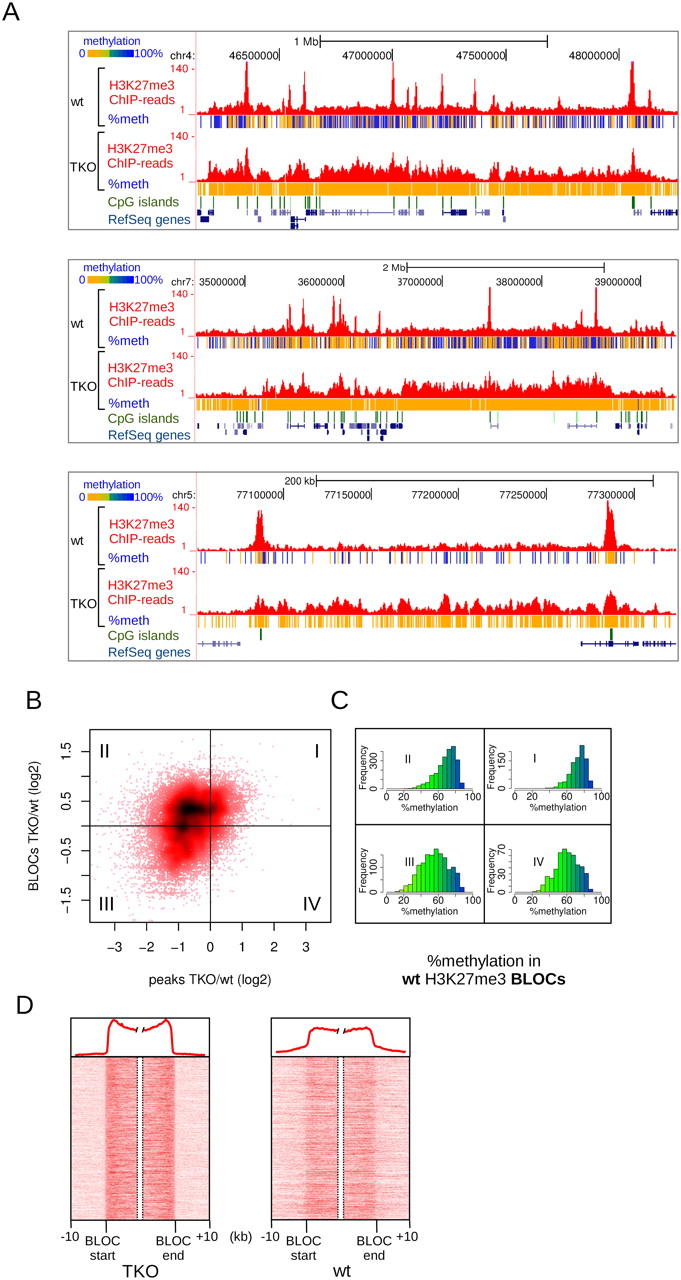
A possible explanation for accumulation of H3K27me3 BLOCs could be a compensatory repressive effect instigated by the loss of DNA methylation. If this were the case, H3K27me3 BLOCs elevated in TKO cells are expected to represent genomic regions with high DNA methylation in wild-type cells. We made use of our H3K27me3-ChIP-BS-seq data to assess the DNA methylation status in TKO-BLOC regions in wild-type cells (Fig. 5C). Indeed, in cases where BLOCs became more prominent (quadrants I & II) (Fig. 5C), DNA methylation in wild-type mES cells was significantly higher compared to regions where H3K27me3 was lost (quadrants I vs. IV, P = 2.8 × 10−68; quadrants II vs. III, P = 1.5 × 10−137, Mann–Whitney U-test) (Fig. 5C). These data suggest that large chromosomal regions with high DNA methylation become more susceptible for accumulation of H3K27me3 upon removal of DNA methylation. To analyze H3K27me3 changes instigated by loss of DNA methylation at an even larger scale and independent of BLOC-like patterns, we generated a MethylCap DNA methylation profile for wild-type mES cells. A sliding window of 0.5 Mb was applied (Supplemental Fig. S7A). We found that H3K27me3 in TKO cells resembled the MethylCap profile of wild type (Supplemental Fig. S7B). Correlation between H3K27me3 and MethylCap profiles increased from 0.41 in wild type to 0.71 in TKO (Pearson correlation) (Supplemental Fig. S7C).
To analyze the consequence—if any—of H3K27me3 accumulation in BLOCs at the level of gene expression, we categorized RefSeq genes according to their position within or outside BLOCs and plotted their expression levels in wild-type and TKO cells (RNA-seq data from Karimi et al. 2011). Genes located outside BLOCs were expressed at higher levels than genes located within BLOCs (Supplemental Fig. S8A), which is in line with observations made by others (Pauler et al. 2009) and by us in HCT116 cells (see Supplemental Fig. S3A). Strikingly, in wild-type mES cells, these differences were already evident, even though BLOCs are less prominent than in TKO cells. As shown before (Karimi et al. 2011), loss of DNA methylation did not cause massive deregulation, and the expression levels of most genes were stably maintained. Only 190 transcripts were found to be deregulated (FDR of 0.05, minimal twofold change) (Supplemental Fig. S8B).
A closer inspection of H3K27me3 patterns in wild-type mES cells revealed that BLOCs of TKO cells could already be distinguished in wild-type cells, although signals were much weaker (Fig. 5A). This was confirmed by plotting density maps of all BLOC transition regions in both wild-type and TKO cells (Fig. 5D): the same BLOC boundaries were present in wild-type cells as in TKO cells. Taken together, total removal of DNA methylation caused an accumulation of H3K27me3 signal in BLOC-like patterns with boundaries that had been set in wild-type cells. This suggests that besides the local antagonism between the two marks at high-CpG-dense regions, there is also antagonism between the two marks at a much larger scale in the genome.
Discussion
Various methods for the generation of genome-wide DNA methylation maps exist. An extensive comparison of the most frequently used bisulfite- and enrichment-based technologies has recently been performed (Bock et al. 2010; Harris et al. 2010). In our strategy, we integrated MethylCap and ChIP-capturing procedures with bisulfite-based DNA methylation analysis. Although similar strategies have been used to interrogate DNA methylation within a limited number of preselected regions (Kagey et al. 2010; Thomson et al. 2010), our strategy allows us to obtain base-resolution DNA methylation information within all fragments obtained by a capturing step. Any subset of the genome occupied by a specific feature can be directly assessed for DNA methylation, provided that the feature of interest can be enriched for or captured. Not only does this open up the possibility to assess cross-talk between DNA methylation and other chromatin features, it also allows for the analysis of DNA methylation within binding sites of transcription factors that are dependent on or excluded by DNA methylation. In addition, it allows for detection of allele-specific marking of chromatin such as in imprinting or X-inactivation.
We used MethylCap-BS-seq as a proof-of-principle for our strategy and showed that DNA fragments obtained by MethylCap represent a highly methylated fraction of the genome. In addition, differences in methylation between normal and tumor tissue as detected by conventional MethylCap-seq were corroborated and extended by MethylCap-BS-seq. ChIP-BS-seq was successfully established using H3K27me3 and H3K9me3 as the epigenetic marks of interest. Depending on CpG density, H3K27me3 and DNA methylation co-occur or are mutual exclusive, whereas H3K9me3 and DNA methylation coincide.
Upon loss of DNA methylation in TKO cells, we observed two notable effects: (1) accumulation of H3K27me3 in BLOC patterns; and (2) a decrease (flattening) of sharp localized H3K27me3 peaks. Thus, depending on whether one looks at BLOCs or smaller peaks, the effects of DNA methylation loss appear to be different. Our data strongly suggest that the increase in H3K27me3 BLOCs is related to antagonism between H3K27me3 and DNA methylation, since regions where H3K27me3 accumulates in BLOC patterns have elevated methylation already in wild-type mES cells. A decrease of H3K27me3 in peaks occurs mostly in unmethylated CpG islands; likely, surrounding DNA methylation normally constrains H3K27me3 to unmethylated CpG islands in wild-type cells, a constraint which is lost in TKO cells. This constraint probably relates to the same antagonism observed elsewhere in the genome and, as such, limits the spread of H3K27me3 into neighboring chromatin. Thus, although the H3K27me3 changes observed in BLOCs and peaks are different, they may result from the same antagonism between DNA methylation and H3K27me3. We have ruled out that the decrease in peaks is a technical artifact caused by increased complexity of sequencing libraries because a larger part of the genome is enriched for H3K27me3 (Supplemental Fig. S6).
How the observed mutual exclusiveness of H3K27me3 and DNA methylation within regions of high CpG density is achieved mechanistically is not yet clear, but several studies have described an antagonism between H3K27me3 and DNA methylation. In SILAC nucleosome affinity purifications, DNA methylation of nucleosome positioning sequences impeded PRC2 binding to H3K27me3-modified nucleosomes (Bartke et al. 2010). The CpG density of the nucleosome positioning sequences (“601” and “603”) was 0.09 CpG/bp. At this density, we observed mutual exclusiveness of the H3K27me3 and DNA methylation marks in both HCT116 as well as in mES cells (see Supplemental Fig. S2E; Fig. 4D). Therefore, one explanation for our observations may be obstruction of PRC2 recruitment at DNA methylated CpG-dense regions. Such obstruction may also be involved in epigenetic switching as described for prostate cancer cells (Gal-Yam et al. 2008). Genes initially silenced by Polycomb in normal prostate cells acquired DNA methylation and lost H3K27me3 in the PC3 cancer cells.
The appearance of H3K27me3 BLOCs in TKO cells suggests that DNA methylation more globally antagonizes accumulation of H3K27me3, which is alleviated in TKO cells. Still, the resulting BLOCs occur in specific genomic regions that, importantly, are preset in wild-type mES cells, indicating that there are positional restraints on the deposition of H3K27me3. It is possible that these regions and their boundaries are characterized by other (epi)genetic features such as overrepresentation of genomic elements like certain repeat classes, CpG islands, gene density, lamina-associated domains (Guelen et al. 2008), or transcriptional factor binding. We anticipate that the two-dimensional information obtained using ChIP-BS-seq will provide new insights into the composition of different types of chromatin and their biological roles.
Methods
ChIP-seq, MethylCap-seq, and RNA-seq
HCT116 cells were cultured in McCoy's 5A medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (all Gibco/Invitrogen) at 37°C in 5% CO2 atmosphere. Mouse ES cells were cultured as described in Meissner et al. 2005). Chromatin harvesting and ChIPs were performed as described (Denissov et al. 2007). The following antibodies were used: anti-H3K27me3 (07-449, Millipore) (Peters et al. 2003) and anti-H3K4me3 (Diagenode). MethylCap-seq was performed as described (Brinkman et al. 2010). For RNA-seq, total RNA was isolated using Trizol (Invitrogen) according to the manufacturer's recommendations. 100 μg total RNA was subjected to two rounds of poly(A) selection (Oligotex mRNA Mini Kit, QIAGEN), followed by DNaseI treatment (QIAGEN). 100 ng mRNA was fragmented by hydrolysis (5× fragmentation buffer: 200 mM Tris acetate, pH 8.2, 500 mM potassium acetate, and 150 mM magnesium acetate) at 94°C for 90 sec and purified (RNeasy MinElute Cleanup Kit, QIAGEN). cDNA was synthesized using 5 μg random hexamers by Superscript III Reverse Transcriptase (Invitrogen). ds-cDNA synthesis was performed in second strand buffer (Invitrogen) according to the manufacturer's recommendations and purified (MinElute Reaction Cleanup Kit, QIAGEN). ChIP and MethylCap DNA and ds-cDNA were prepared for Illumina sequencing according to the manufacturer's protocols (Illumina).
ChIP-BS-seq and MethylCap-BS-seq
100 ng ChIP DNA or 10 ng MethylCap DNA from the HIGH fraction (Brinkman et al. 2010) was prepared for bisulfite-deep sequencing. DNA was first subjected to end-repair in a 30-μl reaction containing 6 units T4 DNA polymerase, 2.5 units DNA Polymerase I (Large Klenow Fragment), 20 units T4 Polynucleotide Kinase (all New England Biolabs), dATP, dCTP, dGTP, and dTTP (0.125 mM each), and 1× T4 Ligase buffer with ATP for 30 min at 20°C. Illumina sequencing generates sequences corresponding to the 5′ ends of the input DNA fragments (see also Figure 1A). Therefore, fill-in of 5′ overhangs did not alter sequenced DNA methylation patterns. Purification was performed using a standard phenol:chloroform:isoamyl alcohol (25:24:1) protocol and ethanol precipitation as described previously (Smith et al. 2009). DNA was then adenylated in a 20-μl reaction containing 10 units Klenow Fragment (3′→5′ exo-) (New England Biolabs), 0.5 mM dATP and 1× NEB buffer 2 for 30 min at 37°C. After phenol extraction and ethanol precipitation, Illumina genomic DNA adapters containing 5-methylcytosine instead of cytosine (ATDBio), preventing deamination during bisulfite conversion, were ligated. In a 20-μl reaction, DNA was incubated with 1.5 μM preannealed adapters, 2000 units T4 DNA Ligase (New England Biolabs), and 1× T4 Ligase buffer with ATP, for 16–20 h at 16°C.
Adapter-ligated DNA fragments were subsequently purified by phenol extraction and ethanol precipitation and size-selected on gel. 50 ng sheared and dephosphorylated Escherichia coli K12 genomic DNA was added to adapter-ligated DNA as carrier during size-selection and bisulfite conversion. DNA was run on 2.5% Nusieve 3:1 Agarose (Lonza) gels. Lanes containing marker (50 bp ladder; New England Biolabs) were stained with SYBR Green (Invitrogen), and size regions to be excised were marked with toothpicks. To obtain 80- to 280-bp (low) and 280- to 430-bp (high) insert sizes, adapter-ligated DNA fragments from 200–400 and 400–550 bp, respectively, were excised. The Illumina adapters cause the fragments to run slower, presumably due to the forked structure, as has been described before (Smith et al. 2009). Note that, after PCR, dsDNA libraries appear at 140–340 bp and 340–490 bp, in accordance with exact sizes. DNA was isolated from gel using the MinElute Gel Extraction kit (QIAGEN). The low and high libraries were kept separate in subsequent steps.
Analytical PCRs were performed to check ligation efficiency and sizes of the libraries. Amplifications were performed in 10-μl reactions containing 0.3 μl template DNA (from 20 μl eluted after size-selection), 0.5 units Pfu Turbo Cx Hotstart DNA Polymerase (Stratagene), Illumina primers LPX 1.1 and 2.1 (0.3 μM each), dNTPs (0.25 mM each), and 1× Turbo Cx buffer under the following thermocycler conditions: 94°C for 5 min, n cycles (94°C for 30 sec, 65°C for 30 sec, 72°C for 1 min), and 72°C for 7 min. We tested three different cycle numbers (n)—10, 15, and 20—and analyzed PCR products on 4%–20% TBE Criterion precast gels (BioRad) using SYBR Green staining.
Adapter-ligated and size-selected DNA was subjected to two subsequent 5-h bisulfite treatments using the EpiTect Bisulfite kit (QIAGEN) following the manufacturer's protocol for DNA isolated from FFPE tissue samples. After bisulfite conversion, analytical PCRs were performed as before to determine the minimum number of cycles required in the final amplification step for each sample. In this case, tested cycle numbers were several cycles higher than before bisulfite conversion: 15, 19, and 22 PCR cycles were performed. The minimum cycle number for final large-scale amplification was determined as the lowest cycle number that generated enough PCR product of the desired size range to be visualized on analytical gels as above. Large-scale amplification was performed in eight reactions of 25 μl, each containing 3 μl DNA (from 40 μl bisulfite-converted DNA; the remainder was stored at −80°C as back-up), 1.25 units Pfu Turbo Cx Hotstart DNA Polymerase (Stratagene), primer LPX 1.1 and 2.1 (0.3 μM each), dNTPs (0.25 mM each), 1× Turbo Cx buffer, and thermocycler conditions as above. Amplified libraries were purified with the MinElute PCR Purification kit (QIAGEN) and subsequently purified from gel essentially as described above; whole gels were stained with SYBR Green, and no carrier DNA was added. Final libraries were analyzed on analytical 4%–20% TBE Criterion precast gels (BioRad), and measured by Quant-iT dsDNA HS Assays (Invitrogen). The protocol for preparation of captured DNA for bisulfite-deep sequencing was adapted from Smith et al. (2009) and Gu et al. (2010, 2011). Sequence reads were generated on the Illumina Genome Analyzer IIx or the HiSeq2000 using a standard 36-base protocol. After sequencing, low and high library reads were combined.
Bisulfite sequencing of PCR fragments
Cultured cells (SKNO-1) were either untreated or crosslinked directly in culture medium by the addition of 1% formaldehyde for 15 min. at room temperature. Chromatin harvesting, decrosslinking, and DNA isolation were done as described previously (Denissov et al. 2007). Bisulfite conversion was performed using the EpiTect Bisulfite kit (QIAGEN) following the manufacturer's standard protocol. For each sample, PCR amplicons of the appropriate size were excised from agarose gel, pooled, and subjected to end-repair as above. Pooled fragments were subsequently concatamerized by ligation in the presence of 17% PEG3350 and sonicated using a Bioruptor (Diagenode) at high power for 30 min. in a final volume of 300 μl. The obtained DNA fragments were subjected to library preparation according to the standard procedure (Illumina). Index sequences were introduced by using in-house generated single-read adapters that contained a six-base barcode directly after the sequence primer binding site. The two samples (untreated and crosslinked/decrosslinked) were pooled and sequenced on the Illumina Genome Analyzer IIx using a standard, 36-base protocol.
ChIP-seq and MethylCap-seq data analysis
MethylCap peaks in HCT116 and H3K27me3 peaks in mES cells were called by MACS (Zhang et al. 2008), with mfold = 4 and P-value = 1 × 10−06 and 1 × 10−10, respectively. MethylCap LOW, MEDIUM, and HIGH fractions (Brinkman et al. 2010) were used individually for peak calling, after which peaks were merged. Identification of H3K27me3-enriched regions (BLOCs) was performed using the RSEG algorithm (Song and Smith 2011), which models the read counts with a negative binomial distribution after correcting for the effect of genomic dead zones. Subsequently, it uses a two-state HMM for segmentation of the genome into foreground domains and background domains. We used a bin size of 500 bp in combination with default RSEG settings. These include that the posterior probability of each bin obtained by HMM decoding is larger than 0.95 and that the mean of read counts within a region is above the top 90th percentile of foreground emission distribution. BLOCs within 20-kb proximity were merged, and BLOCs smaller than 20 kb were discarded.
ChIP-BS-seq and MethylCap-BS-seq data analysis
Initial data processing and base-calling was done using the Illumina pipeline software. Mapping of bisulfite-converted sequence reads was done using a custom-made pipeline using a strategy similar to that in Lister et al. (2009). To reduce PCR artifacts, a maximum of three identical sequence reads was allowed. To perform mapping independently of DNA methylation status, sequence reads were in silico bisulfite-converted (C to T) and subsequently mapped to two different in silico converted hg18 genome sequences; one C to T converted genome and one G to A converted genome. Reads mapping to both genomes were discarded, which typically represented a very minor fraction of all reads. Mapping was done using the Burrows-Wheeler Aligner (BWA) (Li and Durbin 2010), with default settings allowing a single mismatch. Percentages of uniquely mapped reads ranged from 78% to 50% for the 80- to 280-bp and 280- to 430-bp libraries, respectively (see Supplemental Table S1 for details). The obtained mapping positions were used to align unconverted sequence reads with their corresponding unconverted genomic sequence and to subsequently determine the methylation status of each sequenced cytosine within a CpG context, both on the forward strand as well as on the reverse strand. The mapping and CpG methylation scoring procedure was driven by a custom-generated Perl script. Further data analysis was done using in-house generated scripts written in LINUX shell, Python, Perl, and R. Gene annotations were based on RefSeq (hg18); CpG islands annotations were based on UCSC (http://genome.ucsc.edu/)
Data access
The data generated for this work have been deposited in the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE28254.
Acknowledgments
We thank E.M. Janssen-Megens, Y. Tan, and K.-J. Francoijs for technical support. This work was supported by the Dutch Cancer Foundation (KWF) grant KUN 2008-4130, NWO-VIDI-864-05-002, the CancerDIP EU Collaborative project HEALTH-F2-2007-200620, the Harvard Stem Cell Institute, and the U.S. National Institutes of Health Roadmap Initiative on Epigenomics (U01ES017155).
References
- ↵A Barski, S Cuddapah, K Cui, T-Y Roh, DE Schones, Z Wang, G Wei, I Chepelev, K Zhao. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837.
- ↵T Bartke, M Vermeulen, B Xhemalce, SC Robson, M Mann, T Kouzarides. 2010. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143: 470–484.
- ↵AC Bell, G Felsenfeld. 2000. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405: 482–485.
- ↵C Bock, EM Tomazou, AB Brinkman, F Müller, F Simmer, H Gu, N Jäger, A Gnirke, HG Stunnenberg, A Meissner. 2010. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 28: 1106–1114.
- ↵AB Brinkman, F Simmer, K Ma, A Kaan, J Zhu, HG Stunnenberg. 2010. Whole-genome DNA methylation profiling using MethylCap-seq. Methods 52: 232–236.
- ↵H Cedar, Y Bergman. 2009. Linking DNA methylation and histone modification: Patterns and paradigms. Nat Rev Genet 10: 295–304.
- ↵S Denissov, M van Driel, R Voit, M Hekkelman, T Hulsen, N Hernandez, I Grummt, R Wehrens, H Stunnenberg. 2007. Identification of novel functional TBP-binding sites and general factor repertoires. EMBO J 26: 944–954.
- ↵W Fischle. 2008. Talk is cheap–cross-talk in establishment, maintenance, and readout of chromatin modifications. Genes Dev 22: 3375–3382.
- ↵EN Gal-Yam, G Egger, L Iniguez, H Holster, S Einarsson, X Zhang, JC Lin, G Liang, PA Jones, A Tanay. 2008. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci 105: 12979–12984.
- ↵H Gu, C Bock, TS Mikkelsen, N Jäger, ZD Smith, E Tomazou, A Gnirke, ES Lander, A Meissner. 2010. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat Methods 7: 133–136.
- ↵H Gu, ZD Smith, C Bock, P Boyle, A Gnirke, A Meissner. 2011. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc 6: 468–481.
- ↵L Guelen, L Pagie, E Brasset, W Meuleman, MB Faza, W Talhout, BH Eussen, A de Klein, L Wessels, W de Laat, . 2008. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948–951.
- ↵AT Hark, CJ Schoenherr, DJ Katz, RS Ingram, JM Levorse, SM Tilghman. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405: 486–489.
- ↵RA Harris, T Wang, C Coarfa, RP Nagarajan, C Hong, SL Downey, BE Johnson, SD Fouse, A Delaney, Y Zhao, . 2010. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 28: 1097–1105.
- ↵RD Hawkins, GC Hon, LK Lee, Q Ngo, R Lister, M Pelizzola, LE Edsall, S Kuan, Y Luu, S Klugman, . 2010. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6: 479–491.
- ↵JD Kagey, P Kapoor-Vazirani, MT McCabe, DR Powell, PM Vertino. 2010. Long-term stability of demethylation after transient exposure to 5-aza-2′-deoxycytidine correlates with sustained RNA polymerase II occupancy. Mol Cancer Res 8: 1048–1059.
- ↵C Kanduri, V Pant, D Loukinov, E Pugacheva, CF Qi, A Wolffe, R Ohlsson, VV Lobanenkov. 2000. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr Biol 10: 853–856.
- ↵MM Karimi, P Goyal, IA Maksakova, M Bilenky, D Leung, JX Tang, Y Shinkai, DL Mager, S Jones, M Hirst, . 2011. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8: 676–687.
- ↵Y Kondo, L Shen, AS Cheng, S Ahmed, Y Boumber, C Charo, T Yamochi, T Urano, K Furukawa, B Kwabi-Addo, . 2008. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 40: 741–750.
- ↵J-S Lee, E Smith, A Shilatifard. 2010. The language of histone crosstalk. Cell 142: 682–685.
- ↵H Li, R Durbin. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595.
- ↵AM Lindroth, YJ Park, CM McLean, GA Dokshin, JM Persson, H Herman, D Pasini, X Miró, ME Donohoe, JT Lee, . 2008. Antagonism between DNA and H3K27 methylation at the imprinted Rasgrf1 locus. PLoS Genet 4: e1000145. doi: 10.1371/journal.pgen.1000145.
- ↵R Lister, M Pelizzola, RH Dowen, RD Hawkins, G Hon, J Tonti-Filippini, JR Nery, L Lee, Z Ye, Q-M Ngo, . 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322.
- ↵H Marks, JC Chow, S Denissov, K-J Françoijs, N Brockdorff, E Heard, HG Stunnenberg. 2009. High-resolution analysis of epigenetic changes associated with X inactivation. Genome Res 19: 1361–1373.
- ↵KM McGarvey, JA Fahrner, E Greene, J Martens, T Jenuwein, SB Baylin. 2006. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res 66: 3541–3549.
- ↵A Meissner, A Gnirke, GW Bell, B Ramsahoye, ES Lander, R Jaenisch. 2005. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 33: 5868–5877.
- ↵A Meissner, TS Mikkelsen, H Gu, M Wernig, J Hanna, A Sivachenko, X Zhang, BE Bernstein, C Nusbaum, DB Jaffe, . 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770.
- ↵TS Mikkelsen, M Ku, DB Jaffe, B Issac, E Lieberman, G Giannoukos, P Alvarez, W Brockman, T-K Kim, RP Koche, . 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560.
- ↵F Mohn, M Weber, M Rebhan, TC Roloff, J Richter, MB Stadler, M Bibel, D Schübeler. 2008. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30: 755–766.
- ↵JE Ohm, KM McGarvey, X Yu, L Cheng, KE Schuebel, L Cope, HP Mohammad, W Chen, VC Daniel, W Yu, . 2007. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39: 237–242.
- ↵G Pan, S Tian, J Nie, C Yang, V Ruotti, H Wei, GA Jonsdottir, R Stewart, JA Thomson. 2007. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1: 299–312.
- ↵FM Pauler, MA Sloane, R Huang, K Regha, MV Koerner, I Tamir, A Sommer, A Aszodi, T Jenuwein, DP Barlow. 2009. H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome Res 19: 221–233.
- ↵AHFM Peters, S Kubicek, K Mechtler, RJ O'Sullivan, AAHA Derijck, L Perez-Burgos, A Kohlmaier, S Opravil, M Tachibana, Y Shinkai, . 2003. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12: 1577–1589.
- ↵Y Schlesinger, R Straussman, I Keshet, S Farkash, M Hecht, J Zimmerman, E Eden, Z Yakhini, E Ben-Shushan, BE Reubinoff, . 2007. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 39: 232–236.
- ↵JA Simon, RE Kingston. 2009. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat Rev Mol Cell Biol 10: 697–708.
- ↵ZD Smith, H Gu, C Bock, A Gnirke, A Meissner. 2009. High-throughput bisulfite sequencing in mammalian genomes. Methods 48: 226–232.
- ↵Q Song, AD Smith. 2011. Identifying dispersed epigenomic domains from ChIP-Seq data. Bioinformatics 27: 870–871.
- ↵MB Stadler, R Murr, L Burger, R Ivanek, F Lienert, A Schöler, C Wirbelauer, EJ Oakeley, D Gaidatzis, VK Tiwari, . 2011. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495.
- ↵JP Thomson, PJ Skene, J Selfridge, T Clouaire, J Guy, S Webb, ARW Kerr, A Deaton, R Andrews, KD James, . 2010. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 464: 1082–1086.
- ↵E Viré, C Brenner, R Deplus, L Blanchon, M Fraga, C Didelot, L Morey, A Van Eynde, D Bernard, J-M Vanderwinden, . 2006. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439: 871–874.
- ↵M Widschwendter, H Fiegl, D Egle, E Mueller-Holzner, G Spizzo, C Marth, DJ Weisenberger, M Campan, J Young, I Jacobs, . 2007. Epigenetic stem cell signature in cancer. Nat Genet 39: 157–158.
- ↵H Wu, V Coskun, J Tao, W Xie, W Ge, K Yoshikawa, E Li, Y Zhang, YE Sun. 2010. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 329: 444–448.
- ↵Y Zhang, T Liu, CA Meyer, J Eeckhoute, DS Johnson, BE Bernstein, C Nusbaum, RM Myers, M Brown, W Li, . 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. doi: 10.1186/gb-2008-9-9-r137.
- ↵XD Zhao, X Han, JL Chew, J Liu, KP Chiu, A Choo, YL Orlov, W-K Sung, A Shahab, VA Kuznetsov, . 2007. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1: 286–298.
Footnotes
[1] [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.133728.111.