
Abstract
Beginning with precursor lesions, aberrant DNA methylation marks the entire spectrum of prostate cancer progression. We mapped the global DNA methylation patterns in select prostate tissues and cell lines using MethylPlex–next-generation sequencing (M-NGS). Hidden Markov model–based next-generation sequence analysis identified ∼68,000 methylated regions per sample. While global CpG island (CGI) methylation was not differential between benign adjacent and cancer samples, overall promoter CGI methylation significantly increased from ∼12.6% in benign samples to 19.3% and 21.8% in localized and metastatic cancer tissues, respectively (P-value < 2 × 10−16). We found distinct patterns of promoter methylation around transcription start sites, where methylation occurred not only on the CGIs, but also on flanking regions and CGI sparse promoters. Among the 6691 methylated promoters in prostate tissues, 2481 differentially methylated regions (DMRs) are cancer-specific, including numerous novel DMRs. A novel cancer-specific DMR in the WFDC2 promoter showed frequent methylation in cancer (17/22 tissues, 6/6 cell lines), but not in the benign tissues (0/10) and normal PrEC cells. Integration of LNCaP DNA methylation and H3K4me3 data suggested an epigenetic mechanism for alternate transcription start site utilization, and these modifications segregated into distinct regions when present on the same promoter. Finally, we observed differences in repeat element methylation, particularly LINE-1, between ERG gene fusion-positive and -negative cancers, and we confirmed this observation using pyrosequencing on a tissue panel. This comprehensive methylome map will further our understanding of epigenetic regulation in prostate cancer progression.
CpG residues, the targets of DNA methylation, have an asymmetric distribution in mammalian genomes and are often found in small clusters termed CpG islands (CGIs) (Bird 2002). Approximately 60% of all human gene promoters overlap with CGIs (Illingworth and Bird 2009), and accumulation of promoter DNA methylation is associated with gene silencing (Jones and Baylin 2007). Previously, DNA methylation studies in prostate cancer have used methodologies of variable scale, focusing on either a few promoters (Li et al. 2005) or several thousand genomic regions with a CpG island array (Kron et al. 2009). Alternatively, functional approaches that monitored gene expression changes after treatment with the demethylating agent 5-aza-2′-deoxycytidine (5-Aza) have also been used (Yegnasubramanian et al. 2004, 2008). However, to date, only 115 genes are reported as methylation targets in prostate cancer, 85 of which are listed in the Pubmeth database (http://www.pubmeth.org) (Ongenaert et al. 2008).
The advent of next-generation sequencing (NGS) now presents a novel approach to assess genome-wide epigenetic changes without the limitations of probe-based microarray platforms. MethylC-seq, a bisulfite conversion approach, was previously used to analyze the methylome at single-base resolution for Arabidopsis (Cokus et al. 2008) and recently for human H1 embryonic stem cells and fetal lung fibroblasts (Harris et al. 2010). Meissner et al. (2008) produced methylation maps by reduced representation bisulfite sequencing of MspI-digested, genomic DNA from pluripotent and differentiated cells, and the same method was used by Gu et al. (2010) more recently on colon cancer samples. Alternatively, several groups enriched methylated fragments based on their affinities to anti-5′-methylcytosine antibody (Weber et al. 2005; Down et al. 2008; Feber et al. 2011) and methylated DNA binding protein Mbd2b (Rauch and Pfeifer 2009), susceptibility to methylation-sensitive restriction enzymes (Brunner et al. 2009), or capture technology (Weber et al. 2005; Hodges et al. 2009) before sequencing.
While the enrichment-based and bisulfite conversion methods identified largely comparable methylation events, variation was observed in CpG coverage, resolution, quantitative accuracy, and other measures (Bock et al. 2010; Harris et al. 2010). We used a novel MethylPlex technology described here to enrich methylated regions present in genomic DNA from LNCaP prostate cancers, normal prostate epithelial cells (PrEC), and clinical prostate specimens (n = 17). Massively parallel sequencing of the enriched product identified differentially methylated regions (DMRs) and revealed novel insights regarding the genomic placement and functional consequences of DNA methylation in cancer.
Results
Characterization of DNA methylation by M-NGS in prostate cells
To perform a genome-wide analysis of DNA methylation in prostate cancer, we used MethylPlex–next generation sequencing (M-NGS) methodology, which enriches methylated DNA using restriction enzymes and requires minimal input genomic DNA (i.e., 50 ng). The ability of M-NGS to identify methylated genomic regions was first evaluated in a prostate cancer cell line, LNCaP, and normal PrEC cells. A schematic describing sequencing library generation is provided in Supplemental Figure 1. Briefly, MethylPlex libraries were constructed by digesting input genomic DNA isolated from samples with a cocktail of methylation-sensitive restriction enzymes, followed by ligation of adaptors containing universal primers sequences and PCR-based amplification. A second round of enzymatic treatment depleted non-GC-rich sequences, followed by an additional amplification step to ensure enrichment of highly methylated DNA fragments. The amplification adaptors were enzymatically removed prior to NGS library preparation (Supplemental Fig. 1). The MethylPlex libraries described above were constructed through the commercial service provided by Rubicon Genomics Inc.
For initial standardization, we used two different concentrations (1 and 5 μg) of each MethylPlex sample from LNCaP and PrEC cells as input DNA to obtain single-read sequencing on the Illumina Genome Analyzer II (for protocol details, see Methods). For each cell type (LNCaP and PrEC), a total of four sequencing libraries were prepared corresponding to 200- and 400-bp size selections of 1 μg and 5 μg of MethylPlex product. We obtained an average of 5 million mappable reads for each M-NGS sample (Supplemental Table 1). CG dinucleotides were enriched by the MethylPlex procedure up to threefold in mapped reads from M-NGS compared to previously obtained control ChIP-sequencing data, namely, pan-histone ChIP-seq (Supplemental Table 1; Yu et al. 2010).
To demonstrate experimental consistency, a comparative analysis of data from 1 and 5 μg of MethylPlex DNA exhibited high correlation both for reads mapping to chromosome 21 and for reads mapping to all CpG islands (Supplemental Fig. 2). Data from 400 bp–5 μg were most enriched for CG-rich sequences (Supplemental Table 1) and showed maximum overlap (∼70%) with methylation identified by hybridizing the MethylPlex product to a CpG island array (Supplemental Fig. 3A; Supplemental Table 2). We therefore selected these data for further analysis.
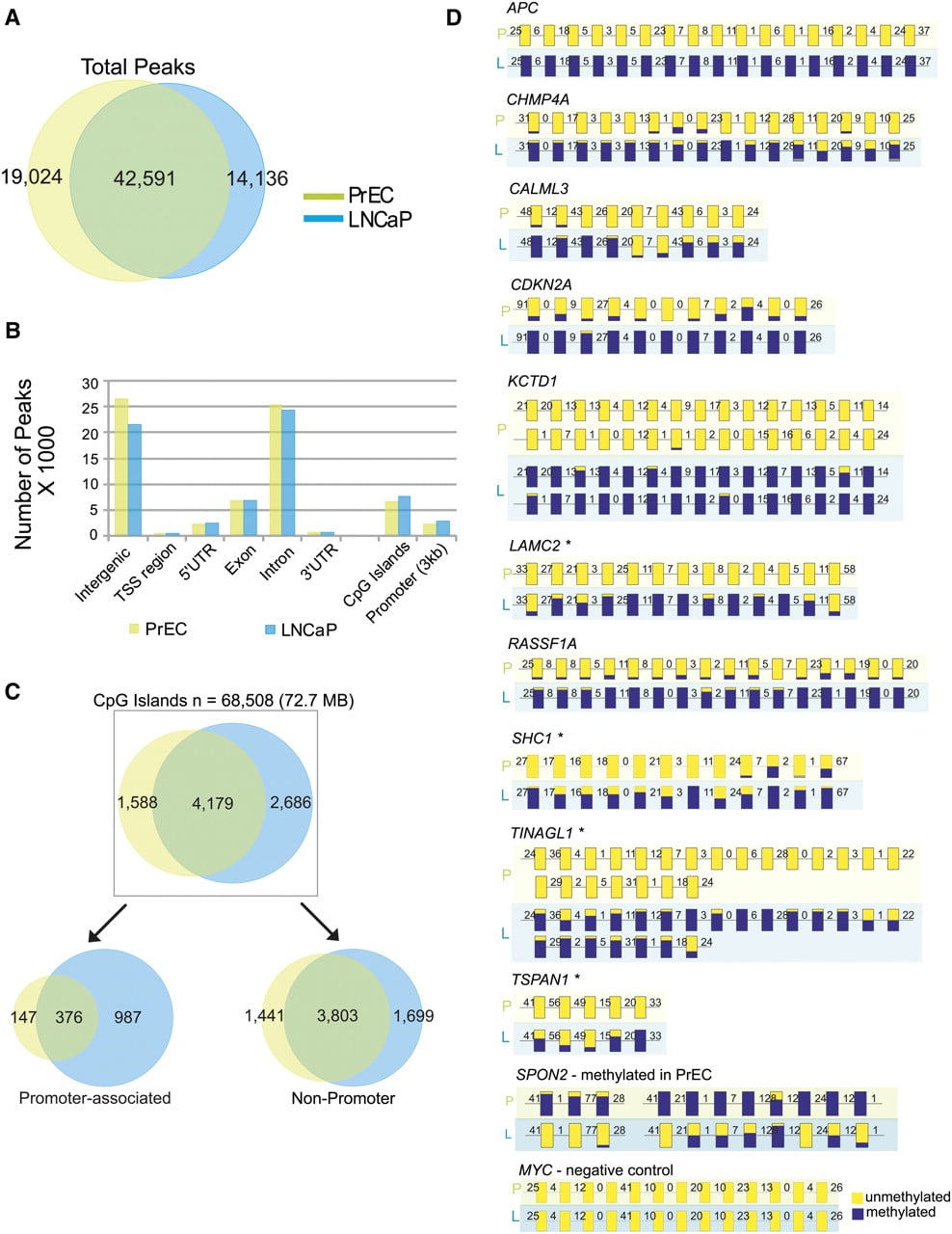
A hidden Markov model (HMM)–based algorithm previously used for ChIP-seq data analysis (Qin et al. 2010) was used to locate peaks from mapped reads obtained in each sequencing run (Supplemental Table 1). We found a 70% overlap in methylated genomic regions between LNCaP (56,727 regions) and PrEC (61,615 regions) cells (Fig. 1A). Methylation located in intergenic and intronic regions of the genomes analyzed had a similar distribution (Fig. 1B); additionally, in LNCaP cells, we also used MeDIP-seq, a methodology that uses 5′-methylcytosine antibody to enrich methylated regions, and we identified approximately 68,000 methylated regions in this cell line, which was comparable to the M-NGS results. Moreover, there was an overall 62% concordance between all the genomic regions (data not shown) and >83% in CGIs identified by M-NGS and MeDIP-seq, thereby validating the two methodologies (Supplemental Fig. 3B).
Characterization of genome-wide methylation patterns in prostate cells by M-NGS. (A) The Venn diagram represents a 70% overlap between the regions methylated in LNCaP (blue) and PrEC (green) cells. (B) In LNCaP (blue) and PrEC (green), the majority of DNA methylation occurred in intergenic and intronic regions, and the genomic distribution of methylation peaks was similar. (C) Promoter-associated CpG islands displayed a sevenfold difference in methylation between LNCaP (blue) and PrEC (green) cells. (D) DNA methylation in APC, CHMP4A, CALML3, CDKN2A, KCTD1, LAMC2, RASSF1, SHC1, TINAGL1, and TSPAN1 gene promoters in LNCaP (L) cells. SPON2 in PrEC (P) cells and a negative control region in MYC were validated by bisulfite sequencing. The methylation status of each CG residue from 10 clones sequenced on both strands was analyzed using the BIQ Analyzer (Bock et al. 2005) program, where the height of the blue bar indicates the percent methylation at a given position, yellow indicates no methylation, and the numbers indicate the distance between analyzed CG dinucleotides. *CpG islands were absent in these promoters. Additional details are provided in Supplemental Figures 5–7 and Supplemental Table 5. Validation of additional candidates, including NAP1L5, C9orf125, AOX1, AMT, NTN4, and PPP1R3C, are presented in Supplemental Figure 8.
The cancer-derived LNCaP cells displayed frequent methylation among the 56 previously reported methylated promoter regions in prostate cancer tissues (36/56 in LNCaP M-NGS and 40/56 in LNCaP MeDIP-seq) compared to PrEC cells (7/56 in PrEC M-NGS) (Supplemental Table 10). However, this difference was absent when we examined the promoters and gene body of known imprinted genes (24/29 in PrEC M-NGS, 23/29 in LNCaP M-NGS, and 26/29 in LNCaP MeDIP-seq) (Supplemental Table 10; Morison et al. 2005).
Global differences in CGI methylation
Because hypermethylation in CpG-rich promoters is a common feature of tumorigenesis (Issa 2004), we compared the extent of CpG island methylation between LNCaP and PrEC cells. Of the 68,508 (72.74 Mb) CpG islands identified using Takai Jones criteria (Takai and Jones 2002) in the human genome, 6865 (7.6 Mb) and 5767 (6.1 Mb) CpG islands were methylated in LNCaP and PrEC, respectively. Globally, we observed a 1.7-fold increase in uniquely methylated CpG islands between LNCaP and PrEC, and this ratio increased to approximately sevenfold specifically in CpG islands associated within gene promoters but not among CGIs located elsewhere (Fig. 1C). In LNCaP cells, methylation in >88% of CpG islands located within promoters and 83% of CGIs in nonpromoters detected by M-NGS were corroborated by the MeDIP-seq data (Supplemental Fig. 3B).
Aberrant promoter methylation is thought to contribute to tumorigenesis by repressing transcription of tumor-suppressor genes (Jones and Baylin 2007). We next looked for methylation on RefSeq gene promoters (±1500 bp flanking the transcription start site) and identified 3496 that were methylated in at least one sample (Supplemental Fig. 4). Visualization of these methylation marks in the context of promoter CGIs revealed the presence of several distinct methylation patterns on gene promoters (Supplemental Fig. 4). Broadly, the promoters fell into two groups based on the presence or absence of a CpG island within this specified region. Interestingly, although 35% of promoters (n = 1232) lacked CpG islands, they exhibited methylation around the transcription start site (TSS) (Supplemental Fig. 4; Supplemental Table 3). The remaining 65% (n = 2264) had CpG islands spanning the TSS, and three distinct methylation patterns were observed in this group: (1) Methylation was mostly confined (39.6%, n = 1383) to the island, and interestingly with much higher frequency (greater than sixfold difference) in LNCaP (n = 952) compared to PrEC (n = 147) cells (Supplemental Fig. 4); (2) methylation was positioned 5′ to the CpG island (11.8%, n = 412); and (3) methylation was positioned 3′ to the CpG island (13.4%, n = 469). In total, methylation flanking 5′ or 3′ of promoter CpG islands accounted for 25.2% of all methylation observed (n = 881). To explore the role of these methylation patterns in prostate cancer pathogenesis, we identified 812 out of 1171 unique gene promoters to be methylated only in LNCaP (Supplemental Table 4) and were considered for further analysis. The remaining 359 promoters were methylated in both LNCaP and PrEC cells.
Validation of DMRs
We next selected 18 regions based on M-NGS data and validated their methylation status using a standard bisulfite sequencing technique in LNCaP and PrEC cells. This included 15 DMRs in LNCaP (RASSF1, KCTD1, CHMP4A, APC, CDKN2A, SHC1, LAMC2, TSPAN1, CALML3, AOX1, AMT, C9orf125, and TINAGL1), one gene in PrEC cells (SPON2), one region methylated in both LNCaP and PrEC cells (NAP1L5), and a control MYC promoter region that was unmethylated in both cell types. The UCSC Genome Browser view of methylation in the two samples by M-NGS and methylation in LNCaP by MeDIP-seq, along with gene schematic, primer sequences, and bisulfite sequence amplicon locations, are presented in Supplemental Figures 5–7 and Supplemental Table 5. Notably, the results for all 18 regions confirmed the data generated by M-NGS (Fig. 1D; Supplemental Fig. 8).
In addition, we observed overexpression of a significant number of LNCaP methylated genes following 5-Aza treatment of cells in a functional validation strategy using gene expression arrays. A total of 973 out of 1171 methylated genes in LNCaP were present in gene expression array data. Significance Analysis of Microarray (SAM) results showed up-regulation of 246 out of 973 methylated genes at a 5% false discovery rate (Supplemental Fig. 9; Supplemental Table 6), supporting epigenetic regulation of these genes.
To identify molecular concepts enriched in our DMRs, we analyzed our data set using the Molecular Concept Map (MCM) analysis derived from the Oncomine database (Rhodes et al. 2007a; Tomlins et al. 2007b). MCM analysis of 789 out of 813 genes methylated only in LNCaP that mapped to the Oncomine database (Supplemental Fig. 10; Supplemental Tables 4, 7) revealed preferential enrichment with underexpressed gene signatures from localized and metastatic PCa samples (lowest P-value < 1.90×10−14) from several independent studies. Furthermore, the signatures, “genes previously known to be methylated in prostate cancer” (P-value < 1.40×10−6) (Ongenaert et al. 2008), and “Gene Ontology-tumor suppressor genes” (P-value < 0.009) were significantly enriched (Supplemental Fig. 10A; Supplemental Table 7). In contrast, PrEC cells did not share this enrichment, and MCM analysis of PrEC-only methylated regions revealed only concepts pertaining to histone modifications and that were common to both PrEC and LNCaP MCM analysis (Supplemental Fig. 10B). Finally, integration with RNA-seq data revealed an association between gene repression and promoter methylation, globally by Gene Set Enrichment Analysis (GSEA) (Supplemental Fig. 11) and upon specific evaluation of select genes (Supplemental Fig. 12). For example, TIG1, GSTP1, CALML3, TASCTD2, and KCTD1 were methylated and repressed specifically in LNCaP, compared to SPON2 and GAGE genes, which were methylated and repressed only in PrEC cells. HIC1 showed basal transcript expression and was methylated in both cell types.
Characterization of DNA methylation in prostate cancer tissues
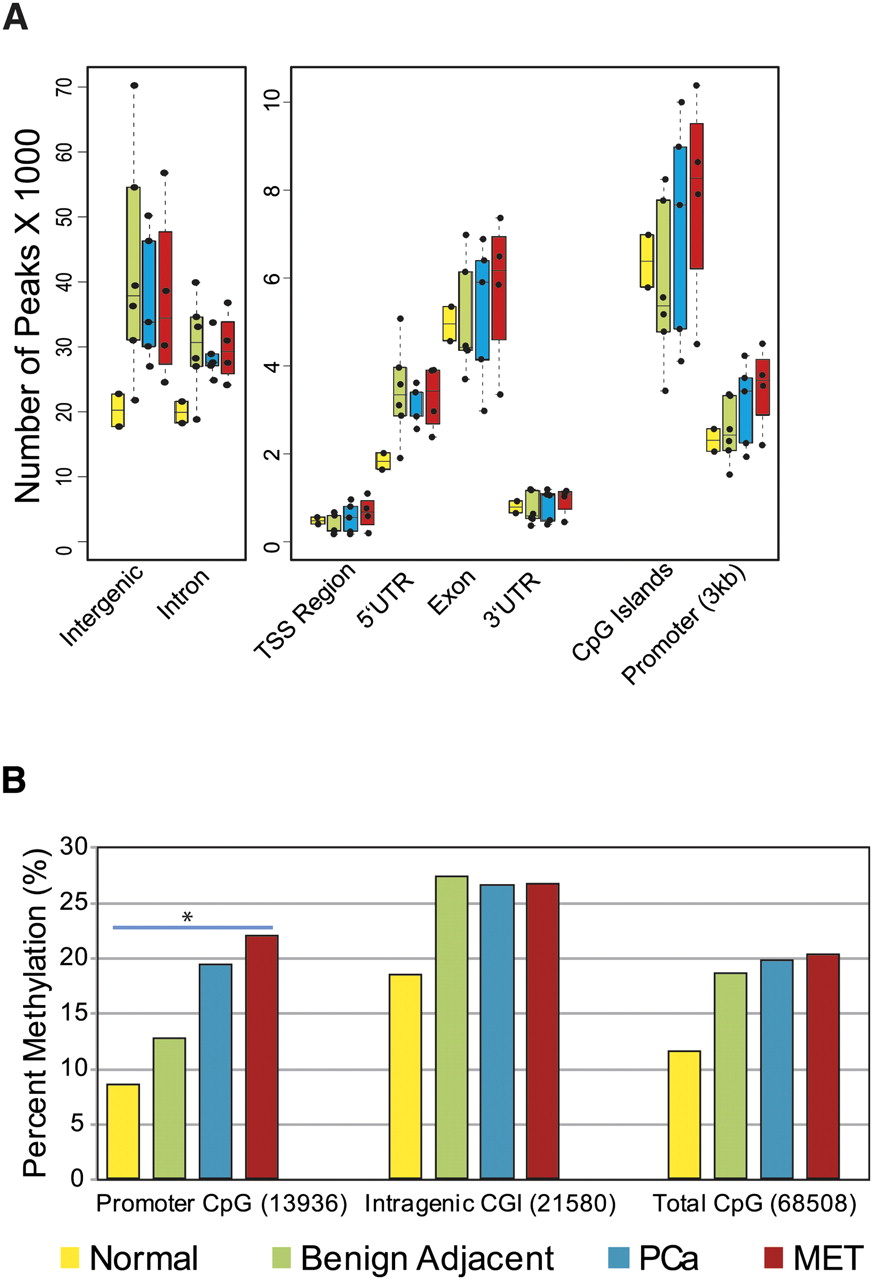
Having established the robustness of M-NGS to identify highly methylated regions in cell line models, we next characterized 17 prostate tissues (six benign adjacent, two normal, five localized prostate cancer, and four metastatic prostate cancer specimens) (Supplemental Table 8). A genome-wide assessment of both benign adjacent and cancer tissues showed a similar number of methylation events within intergenic and intronic regions (Fig. 2A). Of the total 68,508 CGIs present genome-wide, 18.5%, 19.7%, and 20.2% of all CGIs were methylated in benign, localized, and metastatic cancer samples, respectively (Fig. 2B). Importantly, a significant increase in promoter-associated CGI methylation (Pearson's χ2 test, P-value < 2 × 10−16) paralleled prostate cancer progression (benign 12.6%, localized PCa 19.3%, and metastatic PCa 21.8%), whereas methylation of intragenic CGIs remained essentially unchanged (∼26.5%) among the three groups (Fig. 2B).
DNA methylation pattern in prostate tissues. (A) Genome-wide distribution of DNA methylation in various prostate sample groups analyzed. The majority of methylation peaks are confined to intergenic and intronic regions similar to cell lines. (Yellow) Normal prostate; (green) benign adjacent; (blue) localized PCa; (red) metastatic PCa. (B) A gradual increase in percent methylation, with cancer progression among promoter CGIs compared with CGIs located in other genomic regions, was observed. (*) Pearson's χ2 test, P-value < 2 × 10−16.
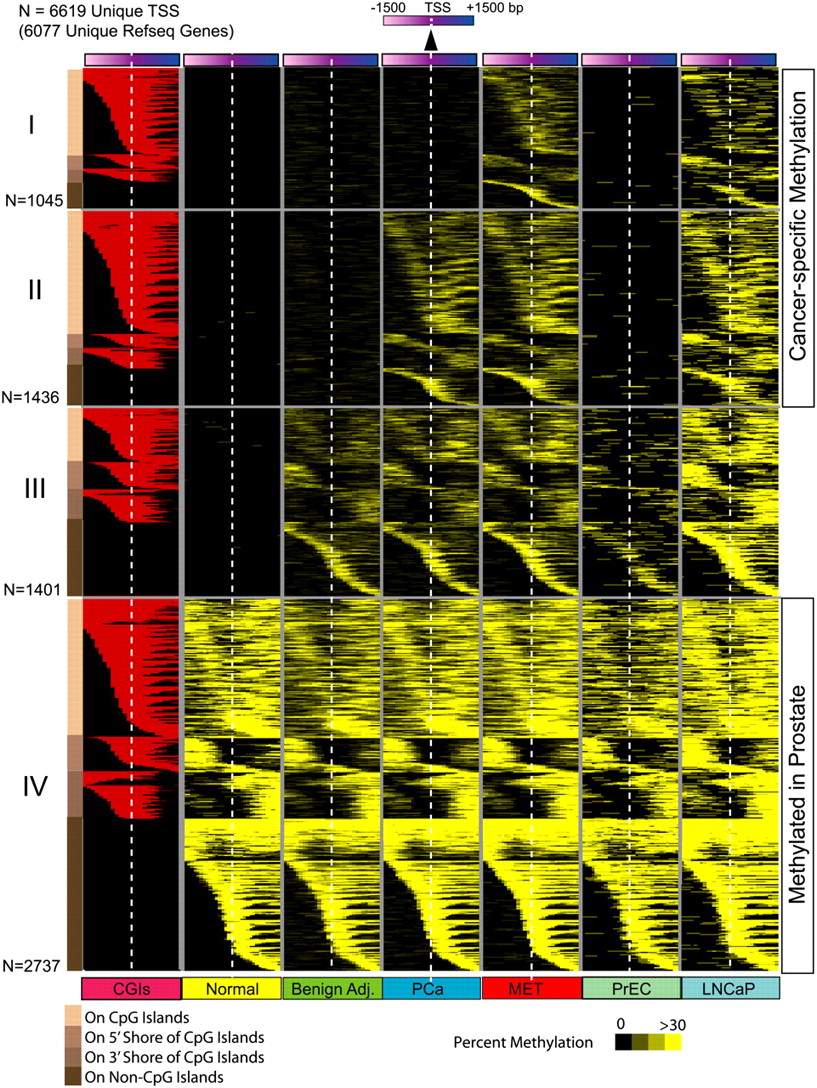
Next, we identified 6619 promoter methylation events (within ±1500 bp flanking the transcriptional start site) present in either normal, benign adjacent, localized, or metastatic prostate cancer samples (Fig. 3; Supplemental Table 9). Of 6619 total methylation events, 2737 were found in all samples, and 1401 of the remaining 3882 were absent in normal prostate samples and PrEC cells but present in benign adjacent prostates. This left 2481 cancer-specific methylation events that may warrant further characterization (Fig. 3). Nearly all of the 56 previously reported prostate cancer methylated regions from pubmeth.org and a recent study (Kron et al. 2009) showed increased methylation in cancer tissues (Supplemental Table 10).
Promoter DNA methylation during prostate cancer progression. A total of 6619 gene promoters from 6077 unique RefSeq genes harbored DNA methylation (yellow) among the various sample groups analyzed (normal, benign adjacent, PCa, or MET). Promoter methylation percentage in sample groups is represented by varying shades of yellow. Each row represents a unique promoter region at 100-bp window size, covering ±1500 bp flanking the transcription start site, indicated by the white dotted line. The location of a CpG island (red) in methylated gene promoters is shown in the first column. Promoters in group IV (n = 2737) are methylated in all sample groups analyzed, promoters in group III (n = 1401) are methylated in all sample groups except normal tissues, while promoters in groups II (n = 1436) and I (n = 1045) are methylated specifically in cancer samples. Promoters are ordered by the location of methylation on a CpG island, adjacent to the island (shores) or on promoters that lacked CpG islands as represented with different shades of brown on the left for groups I to IV. Methylation patterns in prostate cells PrEC and LNCaP are presented alongside for comparison.
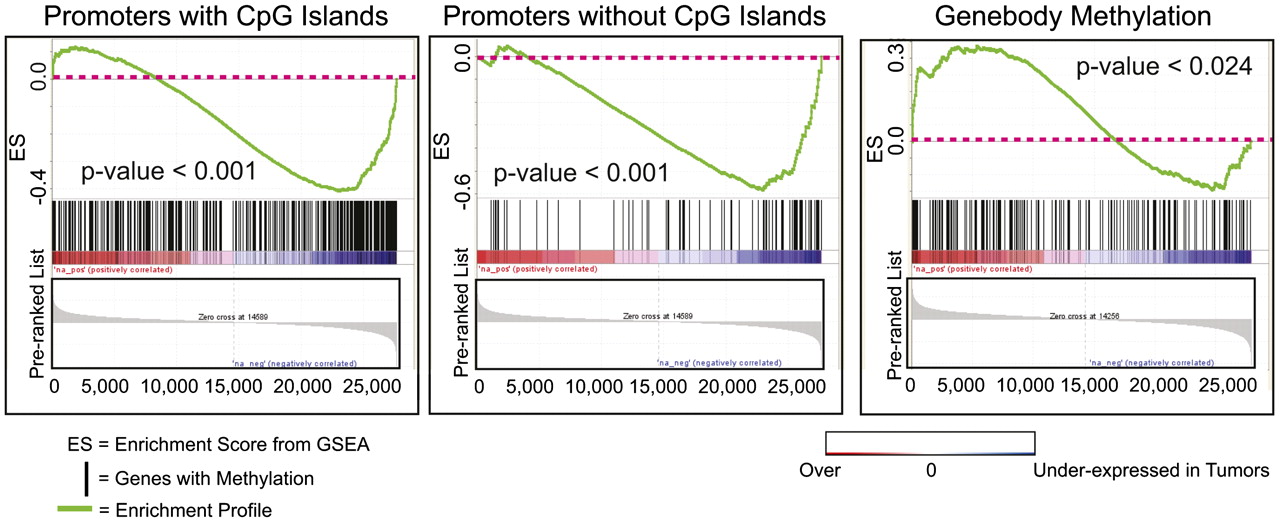
To identify DMRs with functional significance, we next examined promoter methylation events associated with transcriptional changes. Promoters methylated in cancer were significantly associated with gene repression regardless of whether that promoter contained (p < 0.001) or lacked (p < 0.001) a CpG island by GSEA, while genes that displayed coding exon methylation tended to be overexpressed (p < 0.024) (Fig. 4). Oncomine meta-analysis with a data set of 13 different prostate cancer genes' expression further supported methylated candidates' association with gene repression (Supplemental Table 11). Several previously characterized methylation targets (GSTM2, GSTM1, S100A6, PYCARD, and RARRES1) were present among this list, thereby validating the approach.
Promoter methylation and gene repression. Promoter methylation is associated with gene repression. Gene Set Enrichment Analysis (GSEA) of promoters methylated in prostate cancer was performed on microarray expression data from corresponding samples. Significant correlation was observed between gene repression and promoter methylation among both promoters with (P-value < 0.001) or without (P-value < 0.001) CpG islands. Overexpressed transcripts were enriched among genes with gene body methylation (P-value < 0.024).
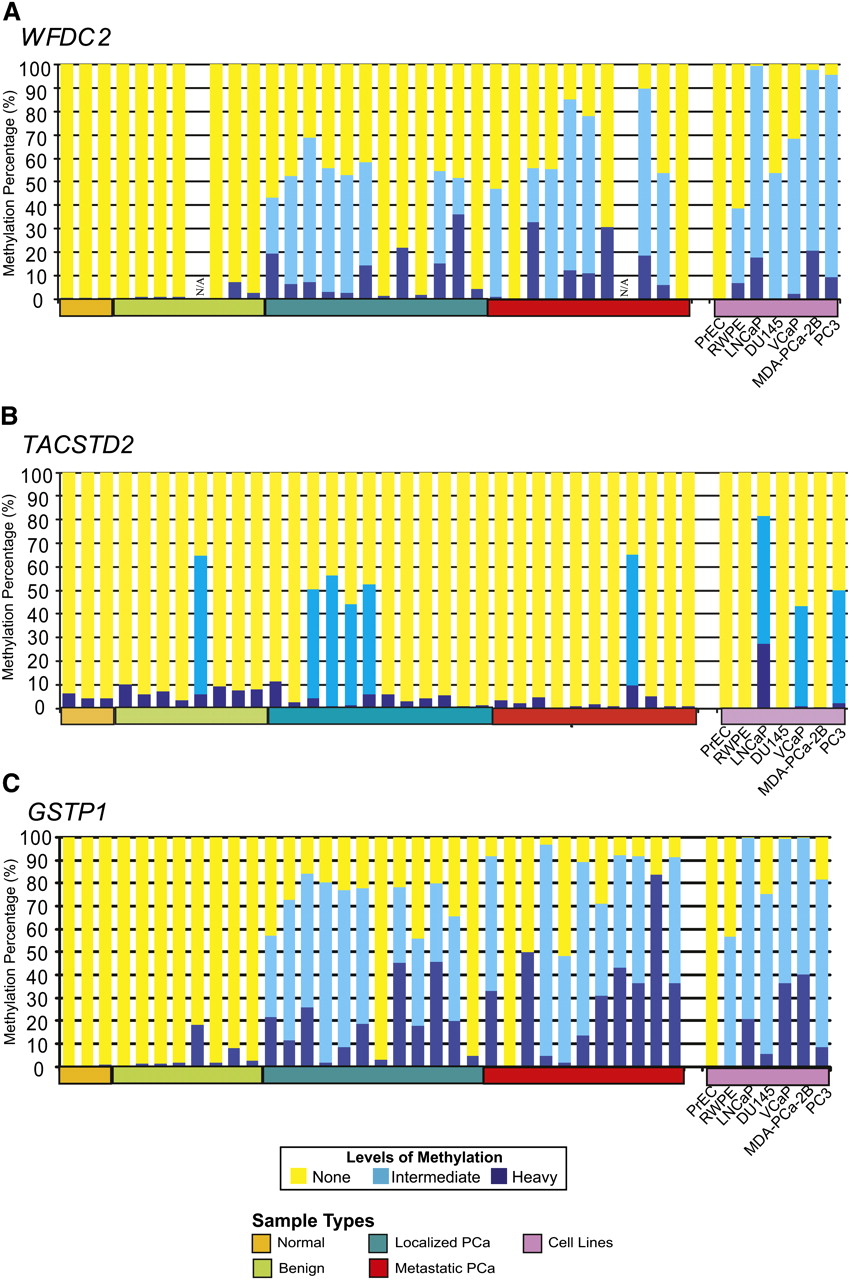
We next used MethylProfiler PCR (Jaspers et al. 2010) as an independent evaluation of the methylation status of a novel target region in WFDC2 (WAP four-disulphide core domain protein 2, previously called HE4), the recently reported prostate methylation target TACSTD2 (Ibragimova et al. 2010), and the well-characterized GSTP1, all identified in this M-NGS study. WFDC2, which ranked 25th in Oncomine meta-analysis, was methylated in 100% (6/6) of transformed prostate cell lines and 77% (17/22) of cancer tissues but not in benign tissues or PrEC (Fig. 5A). In addition, WFDC2 methylation in select samples was independently confirmed by bisulfite sequencing (Supplemental Fig. 13). In comparison, the TACSTD2 promoter was less frequently methylated, with 21% (5/23) of cancer tissues and 9% (1/11) of benign tissues showing hypermethylation, and prostate cell lines similarly exhibited variable levels of methylation (Fig. 5B). In contrast, the well-characterized GSTP1 promoter showed frequent methylation in cancer tissues (86%) and in all transformed cell lines (100%), similar to WFDC2 (Fig. 5C).
WFDC2, TACSTD2 and GSTP1 methylation in prostate tissue panel. MethylProfiler qPCR was used to determine DNA methylation of the WFDC2 (A), TACSTD2 and GSTP1 (B) genes. 17/22 prostate cancer tissues and 6/6 transformed prostate cell lines showed methylation of the WFDC2 promoter, whereas there was no detectable methylation in normal (0/3), benign adjacent tissues (0/7), or the normal PrEC cells. In each sample, the height of the yellow bars indicates no methylation; light blue bars indicate moderate methylation levels; and dark blue bars indicate heavy levels of DNA methylation. Select samples were independently validated by bisulfite sequencing of the corresponding region (Supplemental Fig. 10). (B) Methylation of the TACSTD2 promoter in prostate tissues and cell lines was assessed by MethylProfiler qPCR. Twenty-one percent cancer tissues (5/23) and prostate cancer cell lines, VCaP, LNCaP, and PC3, were methylated. (C) Methylprofiler qPCR analysis of GSTP1. 20/22 prostate cancer tissues, 1/7 benign adjacent tissues, and 6/6 transformed prostate cell lines showed methylation of the GSTP1 promoter, whereas there was no detectable methylation in normal tissues (0/3) or the normal PrEC cells.
Regulation of transcript variant expression by DNA methylation
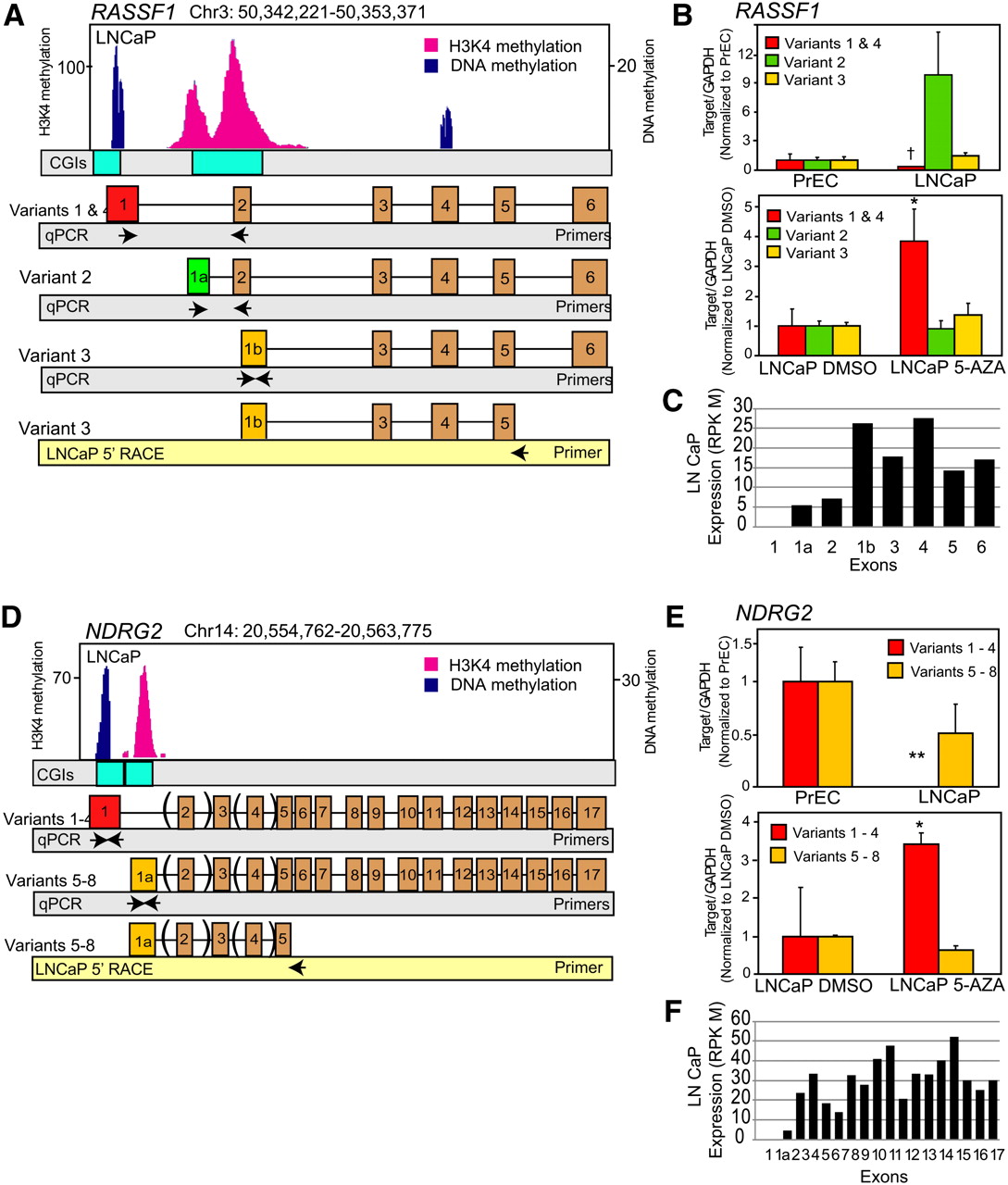
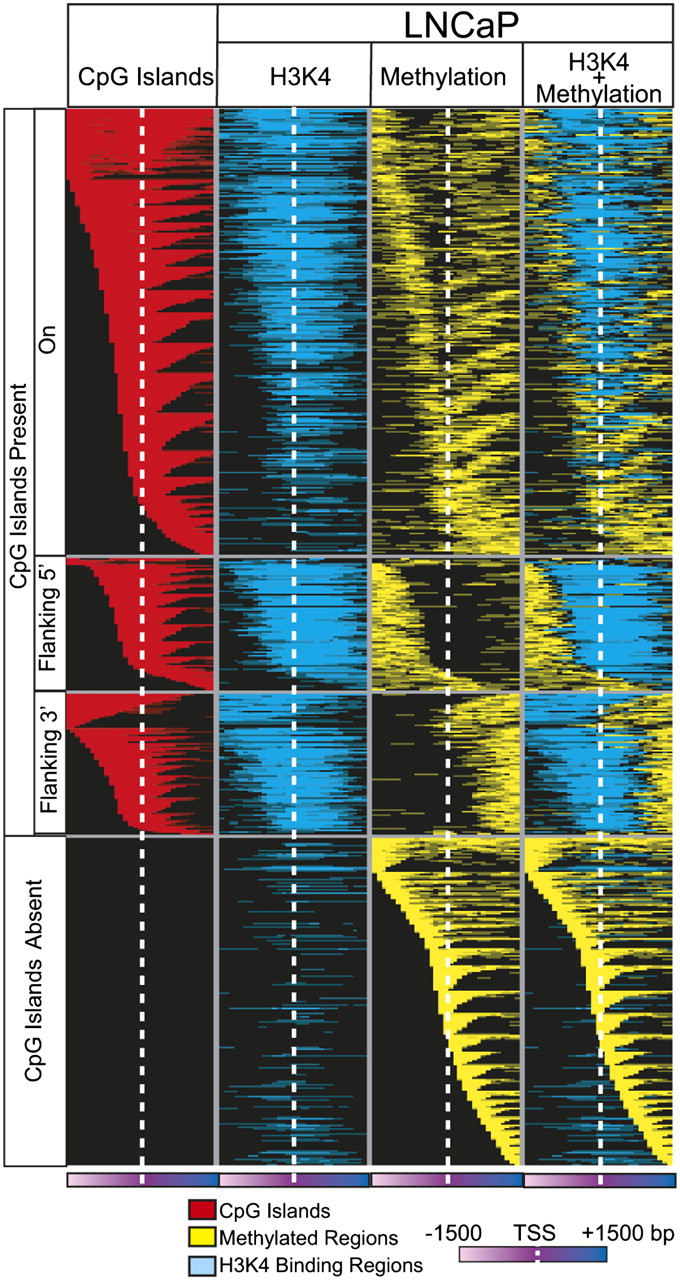
We also observed that a subset of genes displayed selective promoter methylation in a transcript isoform-specific manner, suggesting a mechanism for regulating transcript variant expression in cancer. A well-known example, RASSF1, frequently inactivated by epigenetic alteration in human cancers (Dammann et al. 2005), is composed of three distinct variants. In LNCaP, we observed DNA methylation–mediated silencing of the longer transcript of RASSF1, variant 1, while the smaller isoform, variant 3, that codes for an N-terminal variant protein expressed in multiple cancer cell lines and tissues including PCa, retains high expression (Fig. 6A,B; Dammann et al. 2000; Kuzmin et al. 2002). Active transcription of variant 3 in LNCaP cells is supported by histone 3 lysine 4 trimethylation (H3K4me3) as observed in previously obtained ChIP-seq data (Yu et al. 2010), and 5′ rapid amplification of cDNA ends (5′-RACE) showed the presence of shorter transcripts but not variant 1 in LNCaP (Fig. 6A). Isoform-specific methylation of variant 1 was confirmed by preferential reexpression of this transcript upon 5-Aza treatment of LNCaP cells (Fig. 6B). Interestingly, we found segregation of epigenetic marks into distinct genomic regions in promoters containing CpG islands when we superimposed the promoter methylation and H3K4me3 ChIP-seq data from LNCaP cells (Fig. 7; Supplemental Table 12; Yu et al. 2010). While integration of other epigenetic marks is necessary for a full analysis, these data further suggest that multiple epigenetic modifications may co-occur in distinct patterns to regulate transcript expression in cancer.
Regulation of alternate transcription start site utilization by DNA methylation. (A,D) Cancer-specific DNA methylation enables switching of alternative transcriptional start sites (TSS) leading to transcript isoform regulation. Next-generation sequencing for DNA methylation and histone 3 lysine 4 trimethylation (H3K4me3) in LNCaP cells reveals genome-wide patterning that couples CpG methylation with H3K4 marks to repress or activate specific transcript variants. Independent epigenetic modifications mark specific alternative TSS. In RASSF1 (A) and NDRG2 (D), CpG methylation occurs at the TSS of the longer variants, with H3K4me3 marks positioned on the TSS of the shorter variants. (B,E) Preferential silencing and 5-Aza-induced re-expression of CpG-methylated variants in LNCaP cells. Variants exhibiting CpG methylation on their TSSs show preferential silencing compared to variants with H3K4me3 marks in LNCaP cells. These variants show preferential reexpression upon treatment of LNCaP cells with 6 μM 5-Aza for 48 h. qRT-PCR data are normalized to variant expression levels in PrEC prostate primary epithelial cells or DMSO-treated LNCaP cells in the respective panels. (B,D) 5′-RACE results validated RASSF1 variant-3 and NDRG2 variants 5–8 expression in LNCaP cells. (C,F) Exon expression values from LNCaP RNA-seq data support the corresponding variant transcription of RASSF1 and NDRG2 genes.
Mutually exclusive patterns of promoter DNA methylation and histone H3K4me3 marks in LNCaP cells. Integration of M-NGS DNA methylation data with H3K4me3 ChIP-seq data indicates that DNA methylation and H3K4me3 may be present on the same gene promoter but remain nonoverlapping, adjacent modifications in these promoters. Each row represents a unique promoter region, ±1500 bp flanking the transcription start site (white dotted line) at 100-bp window size. The CpG island location is indicated in red in the first column. The second column represents histone H3K4me3 marks (blue), and the third column (yellow) depicts DNA methylation observed in the corresponding location in LNCaP. Superimposed data are displayed in the fourth column.
Since our M-NGS methodology accurately detected DNA methylation events of RASSF1, we queried our data for differential methylation of transcript variants compared to H3K4me3 marks and identified 34 genes in LNCaP that exhibit isoform-specific promoter methylation (Supplemental Table 13). We validated two genes from this list, namely, NDRG2 and APC (Fig. 6D; Supplemental Fig. 14A). In both of these candidates, the transcript variants (variants 1–4 in NDRG2 and variants 2 and 3 in APC) showing DNA methylation were confirmed to be underexpressed in LNCaP cells compared to PrEC cells by qRT-PCR and 5′-RACE (Fig. 6E; Supplemental Fig. 11A). Furthermore, these variants were preferentially reexpressed upon 5-Aza treatment of LNCaP cells. To determine whether patient tissues demonstrated similar isoform-specific expression patterns, we tested NDRG2 isoforms in two normals, three adjacent normals, five localized PCas, and two metastatic samples by qRT-PCR. Similar to LNCaP cells, variants 1–4 were significantly underexpressed compared to variants 5–8 in localized PCa (P-value = 0.034) and adjacent benign prostate (P-value = 0.012), but not in normal (non–prostate cancer) tissues (Supplemental Fig. 14B). In addition, previously obtained RNA-seq data from LNCaP cells supported the above observation for RASSF1 and NDRG2 genes (Fig. 6C,F).
Methylation differences between ETS-positive and ETS-negative tissues
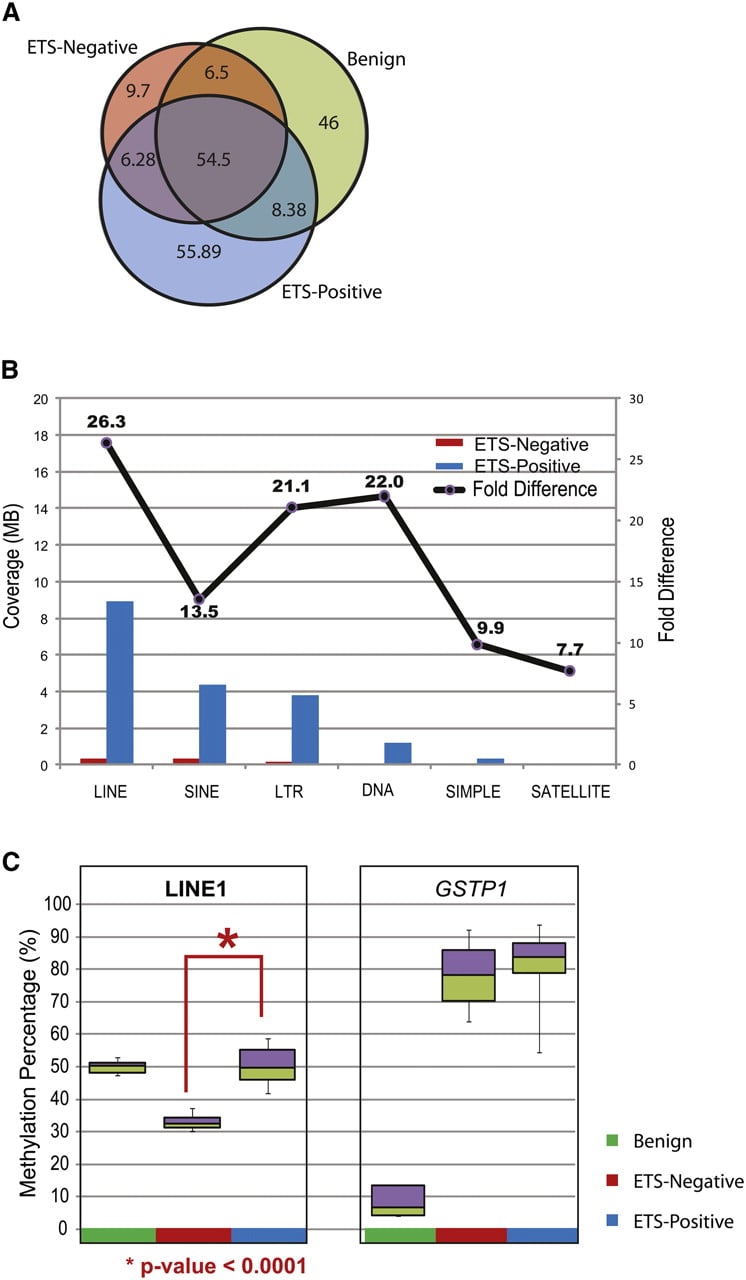
Transcription factor occupancy is suggested to have a protective role in limiting the spread of DNA methylation into affected CpG islands (Gebhard et al. 2010). In prostate cancer, gene fusions involving ETS transcription factors (most commonly ERG and ETV1) occur in ∼40%–50% of patients and serve as the most frequent genetic aberration in this disease (Kumar-Sinha et al. 2008). DNA methylation differences between patients harboring or lacking an ETS gene fusion might therefore provide insights into the transcriptional program of ERG in prostate cancer. We compared the five ERG fusion-positive (ETS-positive) patients and four fusion-negative (ETS-negative) patients in our cohort and observed more than 40 Mb of DMRs specifically associated with ETS-positive samples. Interestingly, the majority of DMRs in ETS-negative samples were also shared with benign samples (Fig. 8A). ETS-positive samples also contained higher repeat-element methylation compared to ETS-negative samples (Fig. 8B). In particular, assessment of global LINE-1 methylation by an independent pyrosequencing analysis on a prostate tissue cohort (n = 20) revealed a significant decrease in LINE-1 element methylation (P-value < 0.0001) in ETS-negative compared to ETS-positive samples (Fig. 8C). These data suggest that previous studies documenting global hypomethylation of LINE-1 elements in prostate cancer may miss subtleties present in different molecular subtypes of this disease.
Differentially methylated regions between ETS-positive and ETS-negative samples. (A) Venn diagram displays the methylation overlap observed between ETS-positive (blue), ETS-negative (red), and benign (green) prostate tissue samples. The inset numbers represent the coverage in each section. (B) The coverage for various repeat elements was higher in ETS-positive compared to ETS-negative samples, indicating higher methylation in the former. The fold difference for methylation in each class of repeat element is indicated by the line plot above. (C) Percent methylation was assessed independently by pyro-sequencing assays for LINE-1 elements and GSTP1 gene promoter methylation in prostate tissue panel (benign n = 5, ETS-positive cancers n = 10, and ETS-negative cancers n = 4). LINE-1 methylation was significantly lower (P-value < 0.0001) in ETS-negative samples compared to ETS-positive tissues, while the GSTP1 gene promoter was highly methylated in both cancer subgroups and not in benign.
Discussion
In this study, we characterized genome-wide methylation patterns in prostate tissues and cell lines using a novel M-NGS methodology. Compared to the bisulfite-based MethylC-seq and enrichment-based MeDIP-seq and MBD-seq, which require microgram quantities of genomic DNA, M-NGS and the reduced representation bisulfite sequencing (Gu et al. 2010) need only nanogram quantities of input DNA and are promising options to characterize clinical samples with limited material availability. Using MeDIP-seq, bisulfite sequencing, and 5-Aza treatments as validation, we demonstrate the accuracy and utility of M-NGS to detect genome-wide methylated regions. A recent study using MeDIP-seq reported methylation in 16% (∼4428/27,679) of all CGIs in the human brain (Maunakea et al. 2010), which is comparable to our MeDIP-seq data (20%) and M-NGS data (up to 20%). The high overlap (>83%) in the methylated CGIs identified by MeDIP-seq and M-NGS in LNCaP cells suggests a comparable performance of these two methodologies. However, a comparative analysis similar to those by Bock et al. (2010) and Harris et al. (2010) may further characterize the advantages and limitations of M-NGS compared to other existing technologies.
This study reveals important DMRs and methylation patterns in both intragenic and intergenic regions in prostate cancer. While globally the total number of genomic regions methylated in all samples was comparable, several thousand DMRs appear to be specific to either the benign or cancer samples. Consistent with prior studies, we found an increasing number of promoter CGIs to accumulate DNA methylation and that this phenomenon correlated with target gene repression (Perry et al. 2010).
We noted patterns of promoter methylation spanning the CGI, but also in the 5′ and 3′ regions flanking the CGI. For these latter categories, Irizarry et al. (2009) have used microarrays to demonstrate ∼70% methylation in regions up to 2 kb away from CpG islands, which were termed “shores” in colon cancer samples. Methylation also occurred on promoters that lacked CGIs, which may also have functional significance. A previous study by Eckhardt et al. (2006) determined that repression of the Oncostatin (OSM) gene occurs by promoter methylation despite the absence of a CGI in the OSM promoter region. Hence the promoter DMRs identified here (including promoter CGI/shores methylation and methylation in promoters that lack CGI) will likely regulate the cancer transcriptome.
Using Gene Set Enrichment Analysis (GSEA) of M-NGS and an expression array data set for promoters methylated in cancer, we revealed enrichment for gene repression regardless of whether the promoter contained a CGI. A similar association between promoter methylation and gene repression was found in multiple public expression data sets using Oncomine meta-analysis. This analysis nominated a novel methylation target, WFDC2, previously shown to be repressed in prostate cancer, starting with the prostatic intraepithelial neoplasia (PIN) stage (Ashida et al. 2004). Methylprofiler PCR analysis showed WFDC2 promoter methylation specifically in >77% cancer but not in benign specimens, indicating that WFDC2 repression is mediated by this epigenetic modification (Fig. 5A). Interestingly, in ovarian cancer, WFDC2 is up-regulated and serves as a biomarker, suggesting that this gene may have different functions in different malignancies (Schummer et al. 1999; Bouchard et al. 2006). In comparison, Ibragimova et al. (2010) recently reported that 17% of prostate cancers contain TACSTD2 gene promoter methylation, while we observed methylation of 26% of cancer specimens in our panel (Fig. 5B). Given the probe limitations of the microarray platforms upon which our analyses are based, we expect that an integrative analysis with NGS transcriptome data will expand our understanding of the role of DNA methylation in cancer further. In addition to WFDC2, several other novel DMRs (MAGI2, MEIS2, NTN4, GPRC5B, C9orf125, FGFR2, AOX1, VAMP5, C14orf159, PPP1R3C, S100A16, and AMT genes) ranked among the top 30 in the meta-analysis (Supplemental Table 11) and merit further examination. Of particular interest, a recent prostate cancer genome sequencing study revealed inactivating deletions in the PTEN-interacting protein MAGI2 (Berger et al. 2011). We observe a DMR in the MAGI2 promoter, thereby suggesting additional regulatory mechanisms to inactivate this gene and indicating that this gene may shave a role in suppressing prostate cancer progression.
While gene silencing mediated by DNA methylation in prostate cancer has been well described (Nelson et al. 2009), a growing body of evidence now supports a role for epigenetic modification in alternate transcription start site utilization. Regulation of alternate transcription by DNA methylation for the PIP5KIA gene in colon cancer (Irizarry et al. 2009) and PARP12 in human B-cells (Rauch et al. 2009) was identified using microarray experiments. More recently, tissue-specific DNA methylation regulating intragenic promoter activity in the SHANK3 locus was demonstrated using MeDIP-seq (Maunakea et al. 2010). Our integrative analysis of DNA methylation and H3K4me3 data nominated candidates for alternate transcription start site utilization as demonstrated in RASSF1, NDRG2, and APC genes. Our analysis further demonstrates that, when present on the same promoter, H3K4me3 modifications and DNA methylation have mutually exclusive boundaries. A similar pattern was observed in mouse neural stem cells, where the GBX2 locus harbors proximal promoter regions containing H3K4me3 marks that are flanked by DNMT3a-bound CpG rich regions containing DNA methylation marks (Wu et al. 2010). Interestingly, H3K4me3, previously considered an active histone mark, is now known to occupy promoters of transcriptionally inactive genes, albeit at threefold lower levels compared to active promoters (Bernstein et al. 2006; Guenther et al. 2007). More recently, binding of CFP1 protein to CpG-rich regions and a 98% overlap between H3K4me3-modified regions and CFP1-binding sites were shown (Thomson et al. 2010). Thus, while the regulation of transcription by DNA methylation and H3K4me3 is well explored, the role for adjacent H3K4me3 and DNA methylation marks in some promoters needs further investigation.
Finally, molecular classification based on ETS gene fusions has enabled subtype-specific analyses of prostate cancer showing distinct copy number aberrations and gene expression patterns in this subtype (Kim et al. 2007; Tomlins et al. 2007b). Here, we define DNA methylation patterns unique to ETS-positive and ETS-negative samples. Specifically, we observed a decrease in repeat-element methylation in ETS-negative compared to ETS-positive samples. Previous work has shown pronounced reduction in both global 5′ methyl cytosine content and LINE-1 hypomethylation in metastatic samples compared to localized PCa (Yegnasubramanian et al. 2008). However, all previous reported assessments of LINE-1 or repeat elements have been global measurements that did not distinguish between specific genomic locations contributing to these measurements. In contrast, next-generation sequencing, including M-NGS, is able to resolve individual repeat elements at certain locations in the genome using uniquely mapped reads. Recent identification of methylation changes in repeat elements at specific genomic locations using MeDIP-seq in malignant nerve sheath tumors demonstrated this potential (Feber et al. 2011). On this basis, we find that ETS-negative prostate cancers show decreased levels of LINE-1 methylation when compared to ETS-positive cancers. Subsequent pyrosequencing validation in a prostate tissue cohort confirmed this difference in LINE-1 methylation, thereby corroborating our M-NGS results. While the mechanism of LINE1 hypomethylation in ETS-negative prostate cancers is unknown, it is interesting to note that previous studies identified TDRD1 (Tudor domain containing protein 1) as a gene that is overexpressed in ERG-positive prostate cancers (Jhavar et al. 2008). Recent data by Reuter et al. (2009) showed derepression of L1 transposons accompanied by a loss of DNA methylation at their 5′ regulatory region in TDRD1 knockout mice, suggesting that TDRD1 may have a role in ETS-specific repeat-element methylation.
Aberrant DNA methylation in prostate cancer is believed to occur in two waves, where epigenetic alteration of some genes, such as GSTP1, can be detected in early disease stages, whereas other genes, such as ESR1, are frequently subject to aberrant DNA methylation in metastatic disease and are considered late events (Nelson et al. 2009). Drug therapies aiming to reverse epigenetic changes, especially those found in castration-resistant prostate cancers, are currently being investigated (Perry et al. 2010). By analyzing both localized and metastatic prostate cancer tissues by M-NGS, we have now identified several hundred differentially methylated regions (DMRs), and because our sample cohort spans the stages of disease progression, we can identify the specific epigenetic alterations associated with early- and late-phase disease (Yegnasubramanian et al. 2004). Ultimately, this information may be used to elucidate epigenetic diagnostic and prognosis markers; a primary example of this is GSTP1, a gene frequently methylated in prostate cancer that may also be detected from clinical samples obtained in a noninvasive fashion (Nelson et al. 2009). The present study thus provides vital information on genomic locations of cancer-specific DMRs that may now facilitate high-throughput screening analyses for prostate cancer disease markers. Moreover, future genome-wide analyses of DNA methylation may improve with larger patient sample sets and with the incorporation of multiple NGS methodologies, such as MeDIP-seq and others, to completely chart an epigenetic landscape (Laird 2010).
In summary, we used a high-throughput M-NGS strategy to characterize the DNA methylome map of prostate cancer tissues and cells using a minimal amount of input DNA. We observe distinct patterns of DNA methylation around TSSs that frequently occur on promoters either containing or lacking a CpG island. This study has uncovered several hundred novel cancer-specific DMRs, similar to the region we characterized in WFDC2, and this information will be used in a future high-throughput screen. We also found additional evidence in prostate that selective regional DNA methylation regulates expression of specific transcript isoforms between normal and cancer cells. Finally, we identified genome-wide differences in DNA methylation between ETS-positive and ETS fusion-negative prostate cancer specimens, along with differences in repeat-element methylation. The comprehensive prostate methylome map generated here provides the precise genomic locations that undergo methylation changes, which will be a highly valuable public resource for investigations aimed at understanding epigenetic regulation of the prostate cancer genome.
Methods
Reagents, cell lines, and prostate tissue samples
Human primary prostate epithelial cells were purchased from Lonza, and the prostate cancer cell line LNCaP was obtained from ATCC. The PrEC and LNCaP cells were grown in PrEGM media (Lonza) and RPMI 1640 containing 10% FBS (Life Technologies), respectively. Human prostate tissue samples were obtained from the University of Michigan SPORE program (Supplemental Table 8). All samples were collected with informed consent of the patients and prior institutional review board approval. CpG island microarrays were purchased from Agilent Technologies. Genomic DNA was isolated from cultured cells and tissue using the DNeasy Blood and Tissue kit (QIAGEN) according to the manufacturer's instructions. 5-Aza-2′-deoxycytidine (5-Aza) was purchased from Sigma-Aldrich and used at 6 μM final concentration dissolved in DMSO.
M-NGS library generation
MethylPlex library synthesis and GC enrichment were obtained through a commercial service at Rubicon Genomics, Inc. (Supplemental Fig. 1). Briefly, 50 ng of gDNA from tissues or cells was digested with methylation-sensitive restriction enzymes 1 and 2 (MSRE1 and MSRE2; Rubicon Genomics) in a 100-μL reaction volume for 12 h at 37°C followed by incubation for 2 h at 60°C. The samples were precipitated with two volumes of ethanol in the presence of sodium acetate (pH 5.2) and pellet paint (VWR). DNA pellets were washed with 70% ethanol, air dried, and suspended in 20 μL of TE buffer (pH 8.0). To prepare MethylPlex libraries, 10 μL of the samples from the previous step was denatured for 4 min at 95°C, cooled to 4°C, and mixed with 4 μL of library synthesis mix (Rubicon Genomics). The tubes were incubated for 2 min at 95°C and returned to 4°C before adding 1 μL of library synthesis enzyme (Rubicon Genomics). The reaction was carried in a thermocycler under the following conditions: 20 min at 16°C, 20 min at 24°C, 20 min at 37°C, and 10 min at 75°C, then returned to 4°C. Subsequently, 15 μL of the MethylPlex library was amplified in a Bio-Rad iCycler real-time PCR machine after mixing with 60 μL of library amplification mix (Rubicon Genomics), under the following cycle conditions, 2 min at 95°C (1 cycle), followed by 9 to 13 cycles of 20 sec at 96°C, 2 min at 65°C, and 1 min at 75°C. The amplified DNA was purified using the QIAquick PCR purification kit (QIAGEN), eluted in a 50-μL volume and subjected to GC enrichment following the manufacturer's protocol (Rubicon Genomics). The GC-enriched DNA was purified using the DNA Clean and Concentrator kit (Zymo Research) and eluted in 35 μL of Tris-EDTA buffer (pH 8.0). One and five micrograms of the purified products from each cell line were directly incorporated into the genomic DNA sequencing sample preparation kit procedure from Illumina at the end repair step, skipping the nebulization process. An adenine base was then added to the purified end repaired products using Klenow exo (3′-to-5′ exo minus) enzyme. The reaction product was purified, ligated to Illumina adaptors with DNA ligase, and resolved on an agarose gel. For LNCaP and PrEC libraries, gel pieces were excised at 200-bp and 400-bp positions, and the DNA was extracted using a gel extraction kit (QIAGEN). Subsequently for all tissue samples, a 350–450-bp gel cut was used. One microliter of this eluate was used as a template in a PCR amplification reaction with Phusion DNA polymerase (Finnzymes) to enrich for the adapter-modified DNA fragments. The PCR product was purified and analyzed by Bioanalyzer (Agilent Technologies) before using it for flow cell generation, where 10 nM library was used to prepare flowcells with approximately 30,000 clusters per lane. The raw sequencing image data were analyzed by the Illumina analysis pipeline and aligned to the unmasked human reference genome (NCBI v36, hg18) using the ELAND software (Illumina) to generate sequence reads of 25–32 bp. Additional information on sequencing runs for all cells and tissue sample runs can be found in Supplemental Tables 1 and 8. The M-NGS data have been deposited under accession number GSE27619 in the GEO database.
Total RNA isolation and quantitative real-time PCR (QPCR)
Total RNA was isolated from cells using the RNeasy mini kit (QIAGEN) according to the manufacturer's instructions. A DNase I treatment step was included during the total RNA isolation procedure to remove genomic DNA from the samples. One microgram of total RNA was used in cDNA synthesis using Superscript III reverse transcriptase (Invitrogen). Quantitative real-time PCR (QPCR) was performed on prostate-cell-line cDNA samples using SYBR Green Mastermix (Applied Biosystems) on an Applied Biosystems 7900 Real Time PCR system as described (Tomlins et al. 2007b). All oligonucleotide primers were synthesized by Integrated DNA Technologies and are listed in Supplemental Table 5. GAPDH primer sequences were as described (Vandesompele et al. 2002). The amount of target transcript and GAPDH in each sample was normalized by standard ddCt methodology, and then to the reference PrEC or DMSO-treated LNCaP samples accordingly.
CpG island annotation
The genomic coordinates for human CGIs were downloaded from ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/ARCHIVE/BUILD.36.1/mapview/seq_cpg_islands.md.gz. Only islands annotated as strict CpGs were used in this study.
RNA-seq library preparation
Poly(A) RNA from LNCaP and PrEC cells (200 ng) was isolated from total RNA using SeraMag Magnetic Oligo(dT) Beads (Thermo Fisher Scientific). RNA was fragmented for 5 min at 70°C in a fragmentation buffer (Ambion) and converted to first-strand cDNA using SuperscriptII (Life Technologies). Second-strand cDNA synthesis was performed with Escherichia coli DNA Pol I (Life Technologies). The double-stranded cDNA library was further processed following the Illumina Genomic DNA sample preparation protocol, which involved end repair using T4 DNA polymerase, Klenow DNA polymerase, and T4 Polynucleotide kinase followed by a single “A” base addition using Klenow 3′-to-5′ exo− polymerase. Illumina's adaptor oligo was ligated using T4 DNA ligase. The adaptor-ligated library was size-selected by separating on a 4% agarose gel and cutting out the library smear at 200 bp. The library was PCR-amplified by Phusion polymerase (Finnzymes) and purified by a PCR purification kit (QIAGEN). The library was quantified with a Bio-Analyzer (Agilent Technologies), and 10 nM each library was used to prepare flowcells with approximately 30,000 clusters per lane. The GEO accession number for the LNCaP and PrEC RNA-seq libraries is GSE29155.
Statistical analysis
HMM analysis of M-NGS data
Hidden Markov model (HMM)–based next-generation sequencing analysis is conducted in a two-step process that takes in raw reads and outputs refined boundaries of enriched chromosomal regions (Qin et al. 2010). The first step includes the formation of hypothetical DNA fragments (HDFs) from uniquely mapped reads, where the coverage of HDFs is determined by the specified DNA fragment size, and overlapped HDFs are merged to represent one consecutive genomic region. The second step is designed to refine the boundaries of the enriched region using HMM with a bin size of 25 bp (by default). Under the null hypothesis, raw reads are assumed to land on the genome following a Poisson distribution with the background rate of r0, and enriched regions are expected to have more HDFs with statistical significance. The rate of the Poisson distributions in a given sample is assumed to be r1, and the transition probabilities are estimated empirically, based on the inferred enriched regions defined in the first step. The output from HMM is selected based on the posterior probability of being in the enriched regions and then further filtered using maximum read counts. The threshold for maximum read counts is determined from a Bonferroni-corrected P-value of 0.001 calculated using a Poisson distribution with background rate r0. The output is provided in BED format as well as Wiggle format for UCSC Genome Browser visualization. The output file annotation field contains information such as enriched genomic position and length, maximum height, GC content, repeated sequencing genomic position and length, mean and standard deviation of conservative scores for the enriched region, relationship with nearest genes including whether the enriched region is located within the gene or between genes, gene name, GB accession number, strand, and distance to the gene transcription start site.
Calculating gene expression from RNA-seq data
Gene expression levels of passing filter reads from RNA-seq data that mapped by ELAND to exons (March 2006 assembly of UCSC KnownGene table) in LNCaP and PrEC cell lines are quantified as described (Maher et al. 2009).
One-class SAM analysis
Significance analysis of microarray (SAM) (Tusher et al. 2001) (http://www-stat.stanford.edu/∼tibs/SAM/) was performed on the gene expression data set obtained from 5-Aza and DMSO-treated LNCaP cells by selecting genes that were methylated in LNCaP. From 1171 methylated genes from LNCaP M-NGS (Supplemental Table 4), a total of 973 genes was mapped to Agilent expression profiling data. One-class SAM analysis was done using default settings, and significant genes were calculated with a false discovery rate (FDR) of 0.05.
Gene Set Enrichment Analysis
Gene Set Enrichment Analysis (GSEA) is a computational method that assesses whether a defined set of genes shows statistically significant, concordant differences between any two given conditions. The fold change between the raw counts from RNA-seq NGS data on LNCaP and PrEC (representing 24,167 unique genes) was calculated, and genes were ranked by the order of expression in LNCaP. This list was uploaded as a pre-ranked gene list to GSEA v2.04 (Broad Institute, Cambridge, MA), and using respective gene lists of methylated targets in LNCaP and PrEC cell lines, GSEA was performed using a weighted enrichment statistic and default normalization mode. Similarly, the fold change between the average expression value from normal/benign (n = 4) and cancer samples (n = 9) profiled on the Agilent Human GE 44K microarray was calculated and pre-ranked (representing 27,928 unique probes). This list was uploaded to GSEA, and enrichment analysis was performed using methylation target gene lists (the methylation present in promoters with CGIs and without CGIs, and in the gene body) in tumor samples.
Oncomine meta-analysis
A complete description of meta-analysis performed in Oncomine is available (Rhodes et al. 2007a). In brief, a genelist of interest is uploaded to the Oncomine database, and the built-in meta-analysis tool rank-orders the genelist by the P-value, which is determined by a Student's t-test for comparisons made within each available data set (e.g., cancer vs. normal). The ranked genes were visualized with pink and green shades (top-ranked ones with darker shades, pink for overexpression, and green for repression) in heatmap format, with each row representing genes and each column representing the data set. The final order of the genes is determined by averaging ranks across the data sets.
Molecular Concepts Map analysis
A complete description of the methods used to identify biological concept signatures in Molecular Concepts Map (MCM) is available (Rhodes et al. 2007b). In addition to more than 15,000 biological concepts from Oncomine, which include manual curation of the literature, target gene sets from genome-scale regulatory motif analyses, and reference gene sets from several gene and protein annotation databases, we have uploaded a gene list from differentially methylated regions identified from an independent Differential Methylation Hybridization profiling (concept named “DMH-Tissue Methylated in PCa”) (data not shown), as well as known methylated genes in cancers provided from the Pubmeth database. In brief, MCM analysis uses a Fisher's exact test to find various significantly enriched concepts in an uploaded gene list and provides visual interaction networks.
Repeat-element methylation analysis
The list of repeat elements predicted by the RepeatMasker (RepeatMasker Open-3.0; http://www.repeatmasker.org) program was downloaded from the UCSC Genome Browser. The MethylPlex-NGS data from localized and metastatic prostate tissue samples were divided into two groups based on their ETS gene fusion status (ETS-positive and ETS-negative). The samples in each group were pooled together for HMM analysis, and the regions identified were mapped to repeat-element location.
ChIP-sequencing
LNCaP cells' ChIP-seq data obtained using the H3K4me3 antibody (Abcam) and PanH3 (Abcam) were reported previously by Yu et al. (2010); the GEO accession number for the data set is GSE14097. ChIP samples were prepared for sequencing using the Genomic DNA sample prep kit (Illumina) following the manufacturer's protocols. To facilitate ChIP-seq data analysis, a hidden Markov model (HMM)–based enriched region identifying algorithm (described in the Methods section under “Statistical Analysis”) was used.
MeDIP-sequencing
Six micrograms of genomic DNA isolated from LNCaP cells was sonicated to an ∼100–500-bp range and purified using the QIAGEN PCR purification kit. Using standard Illumina protocol/reagents, we end-repaired, A-tailed, and added adaptors to the fragmented DNA. The DNA was then heat-denatured for 10 min at 95°C and snap-cooled on ice. The DNA was incubated with 6 μg of anti-methyl cytosine antibody in IP buffer (10 mM sodium phosphate buffer containing 140 mM sodium chloride and 0.05% Triton X-100) overnight at 4°C in a shaker. The methylated fragments were collected by incubating with 100 μL of protein A beads (Invitrogen) for 2 h at 4°C. The beads were washed four times at 4°C in IP buffer and resuspended in 200 μL of TE buffer containing 0.25% SDS and 5 μg of proteinase K and incubated for 2 h at 55°C. The samples were purified using a DNA Clean and Concentrator-5 kit (Zymo Research), and the libraries were prepared following Illumina ChIP-seq protocol. The library was quantified with a Bio-Analyzer (Agilent Technologies), and 10 nM each library was used to prepare flowcells with approximately 30,000 clusters per lane.
Methyl-Profiler
Methyl-Profiler (SABiosciences) is a restriction enzyme digestion–based novel technology for CGI methylation profiling, requiring <500 ng of input genomic DNA (Jaspers et al. 2010). The samples were first digested with methylation-sensitive (Ms) and/or methylation-dependent (Md) restriction enzymes along with mock digestion according to the manufacturer's instruction. PCR reactions were performed with an ABI StepOne qPCR machine (Applied Biosystems) with RT2 SYBR Green/ROX qPCR Master Mix (SABiosciences) and primers targeting the region of interest. The PCR reactions were carried out with the following conditions: 10 min at 95°C, followed by 40 cycles of 15 min at 97°C and 1 min at 72°C, as described in the manufacturer's protocol. Using delta-Ct values, the relative amounts of methylation are calculated using an automated Excel-based data analysis template provided by the manufacturer. The mock-digested template is used for initial DNA input quantification, the Ms enzyme is used for hypermethylation quantification, and the Md enzyme is used for quantifying unmethylated DNA. A mixture of these two enzymes (Msd) is used to quantify the undigested amount of DNA. A methylation rate below 5% is considered not significant. While the calculated methylation percentage between 10% and 60% is considered intermediate, the values above 60% are taken as heavy methylation.
Bisulfite sequencing
Bisulfite conversion was carried out using an EZ DNA methylation gold kit (Zymo Research) according to the manufacturer's instructions. Briefly, 500 ng of genomic DNA from either LNCaP or PrEC cells in a 20-μL volume was mixed with 130 μL of CT conversion reagent and was initially incubated for 10 min at 98°C followed by incubation for 2.5 h at 64°C. M-biding buffer (600 μL) was added to the above reaction and DNA was purified using a Zymo spin column. Sequential washes were performed with 100 μL of M-Wash buffer, 200 μL of M-sulphonation buffer, and 200 μL of M-wash buffer was carried out before eluting the DNA in 30 μL of M-elution buffer. Purified DNA (2 μL) was used as template for PCR reactions with primers (Integrated DNA Technologies) and synthesized according to bisulfite-converted DNA sequences for the regions of interest using the Methprimer software (Li and Dahiya 2002). The PCR product was gel-purified and cloned into the pCR4 TOPO TA sequencing vector (Life Technologies). Plasmid DNA isolated from 10 colonies from each sample was sequenced by conventional Sanger Sequencing (University of Michigan DNA Sequencing Core). The “BIQ Analyzer” (Bock et al. 2005) online tool was used to calculate the methylation percentage and to generate the bar graphs.
Microarray profiling
Expression profiling of 5-Aza-treated LNCaP cells
For 5-Aza stimulation experiments, LNCaP cells cultured in RPMI 1640 were treated with vehicle, dimethyl sulfoxide (DMSO), or 6 μM 5-Aza for 4 or 6 d in duplicates. Total RNA was isolated with TRIzol (Life Technologies) and further purified using the RNAeasy Micro Kit (QIAGEN) according to the manufacturer's instructions. Expression profiling was performed using the Agilent 44K expression array. One microgram of total RNA was converted to cRNA and then labeled according to the manufacturer's protocol (Agilent). Hybridizations were performed for 16 h at 65°C. Scanned images from an Agilent microarray scanner were analyzed and extracted using Agilent Feature Extraction Software 9.1.3.1 with linear and lowess normalization performed for each array. A total of four hybridizations were performed including two 4-d and two 6-d 5-Aza-treated samples (Cy5) against control DMSO-treated samples (Cy3). The accession number for the gene expression data set in the GEO database is GSE27619.
Expression profiling of prostate tissues
Prostate tissues characterized by M-NGS, normal/benign (n = 4) and cancer (n = 9), were profiled on an Agilent Human GE 44K microarray as described for LNCaP cells above. Total RNA from pooled normal prostate tissues obtained from a commercial source (Clontech Laboratories) was used as the reference. This microarray data set was used in GSEA analysis to study the association between DNA methylation and gene expression. The data set has been deposited in the GEO database.
MethylPlex library Agilent CpG array hybridization
Two micrograms of the purified products from each PrEC and LNCaP MethylPlex DNA were labeled following the mammalian ChIP-on-chip protocol (Agilent Technologies) starting at the sample labeling stage, which uses a random primed, Klenow-based extension protocol. The samples were hybridized to an Agilent Human CpG 244K array (Cat# G4492A; Agilent Technologies), where LNCaP sample was coupled with Cy5 and PrEC to Cy3. The slides were washed according to the manufacturer's instructions. A dye-flip experiment was also performed. The scanned images were analyzed and extracted using Agilent Feature Extraction Software 9.1.3.1. Methylated regions identified by the array data were compared to M-NGS targets; their overlap is presented in Supplemental Figure 3, and the data are provided in Supplemental Table 2. This data set has been deposited in GEO under accession number GSE27619.
5′ rapid amplification of cDNA ends (5′-RACE)
5′-RACE was performed as previously described (Han et al. 2008). First-strand cDNA was amplified with gene-specific reverse primers RASSF1, APC, and NDRG2 (Supplemental Table 5) and 5′ GeneRacer primers (Life Technologies) using Platinum Taq High Fidelity enzyme (Life Technologies) after the touchdown PCR protocol according to the manufacturer's instructions. PCR amplification products were cloned into a pCR4-TOPO TA vector (Life Technologies) and sequenced bidirectionally using vector primers as described (Tomlins et al. 2007a).
Pyrosequencing
LINE-1 element methylation was estimated using the PyroMark Q24 LINE-1 methylation assay (QIAGEN) according to the manufacturer's instructions. Briefly, bisulfite-converted gDNA (described above), LINE-1 primers, and components of Hotstart Master Mix (QIAGEN) were used in a PCR reaction to amplify LINE regions from the sample. The amplification was obtained from 45 cycles of 20 sec at 95°C, 20 sec at 50°C, and 20 sec at 72°C, after an initial denaturation/enzyme activation for 15 min at 95°C, and final elongation of 5 min at 72°C. The PCR products were captured on Streptavidin Sepharose beads (GE Healthcare), denatured to produce single strands, washed, and annealed to sequencing primer, and the sequence was determined using the PyroMark Q24 system (QIAGEN). The mean methylation of three individual positions within the PCR product is considered in this assay.
Competing interest statement
The University of Michigan has filed a patent on the findings discussed in the manuscript. A.M.C., S.M.D. and J.H.K. are named as co-inventors. A.M.C. has served on the scientific advisory board of Rubicon Genomics in the past.
Acknowledgments
We thank Emmanuel Kamberov, Takao Kurihara, Tim Tesmer, Vladimir Marakarov, and John Langmore from Rubicon Genomics for early access to the MethylPlex technology. We thank Terrence Barrette, Mike Quist, Bharathi Laxman, Chandan Kumar, Jianjun Yu, Hao Wu, Stephanie Huang, and Yi-Mi Wu for help with data analysis; Roger Morey for help with Illumina sequencing; Bo Han for providing prostate tissue samples; and Jyoti Athanikar for editing the manuscript. C.A.M. was supported by the American Association of Cancer Research Amgen Fellowship in Clinical/Translational Research, Canary Foundation and American Cancer Society Early Detection Postdoctoral Fellowship, and a Prostate Cancer Foundation Young Investigator Award. J.Y. was supported by NIH Grant 1K99CA129565-01A1 and Department of Defense (DOD) Grant PC080665. Z.S.Q. and M.H. were supported by NIH Grant R01HG005119. J.R.P. was supported by NIH Cancer Biology Training Grant CA009676-18. J.R.P. is a Fellow of the University of Michigan Medical Scientist Training Program. A.M.C. was supported in part by the NIH (Prostate SPORE P50CA69568, R01 R01CA132874), the DOD (BC075023, W81XWH-08-0110), the Early Detection Research Network (U01 CA111275), a Burroughs Welcome Foundation Award in Clinical Translational Research, and a Doris Duke Charitable Foundation Distinguished Clinical Investigator Award. This work was also supported by National Center for Integrative Biomedical Informatics Grant U54 DA021519.
References
- ↵S Ashida, H Nakagawa, T Katagiri, M Furihata, M Iiizumi, Y Anazawa, T Tsunoda, R Takata, K Kasahara, T Miki, . 2004. Molecular features of the transition from prostatic intraepithelial neoplasia (PIN) to prostate cancer: Genome-wide gene-expression profiles of prostate cancers and PINs. Cancer Res 64: 5963–5972.
- ↵MF Berger, MS Lawrence, F Demichelis, Y Drier, K Cibulskis, AY Sivachenko, A Sboner, R Esgueva, D Pflueger, C Sougnez, . 2011. The genomic complexity of primary human prostate cancer. Nature 470: 214–220.
- ↵BE Bernstein, TS Mikkelsen, X Xie, M Kamal, DJ Huebert, J Cuff, B Fry, A Meissner, M Wernig, K Plath, . 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326.
- ↵A Bird. 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21.
- ↵C Bock, S Reither, T Mikeska, M Paulsen, J Walter, T Lengauer. 2005. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 21: 4067–4068.
- ↵C Bock, EM Tomazou, AB Brinkman, F Muller, F Simmer, H Gu, N Jager, A Gnirke, HG Stunnenberg, A Meissner. 2010. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 28: 1106–1114.
- ↵D Bouchard, D Morisset, Y Bourbonnais, GM Tremblay. 2006. Proteins with whey-acidic-protein motifs and cancer. Lancet Oncol 7: 167–174.
- ↵AL Brunner, DS Johnson, SW Kim, A Valouev, TE Reddy, NF Neff, E Anton, C Medina, L Nguyen, E Chiao, . 2009. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res 19: 1044–1056.
- ↵SJ Cokus, S Feng, X Zhang, Z Chen, B Merriman, CD Haudenschild, S Pradhan, SF Nelson, M Pellegrini, SE Jacobsen. 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215–219.
- ↵R Dammann, C Li, JH Yoon, PL Chin, S Bates, GP Pfeifer. 2000. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 25: 315–319.
- ↵R Dammann, U Schagdarsurengin, C Seidel, M Strunnikova, M Rastetter, K Baier, GP Pfeifer. 2005. The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol Histopathol 20: 645–663.
- ↵TA Down, VK Rakyan, DJ Turner, P Flicek, H Li, E Kulesha, S Graf, N Johnson, J Herrero, EM Tomazou, . 2008. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 26: 779–785.
- ↵F Eckhardt, J Lewin, R Cortese, VK Rakyan, J Attwood, M Burger, J Burton, TV Cox, R Davies, TA Down, . 2006. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 38: 1378–1385.
- ↵A Feber, GA Wilson, L Zhang, N Presneau, B Idowu, TA Down, VK Rakyan, LA Noon, AC Lloyd, E Stupka, . 2011. Comparative methylome analysis of benign and malignant peripheral nerve sheath tumours. Genome Res 21: 515–524.
- ↵C Gebhard, C Benner, M Ehrich, L Schwarzfischer, E Schilling, M Klug, W Dietmaier, C Thiede, E Holler, R Andreesen, . 2010. General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res 70: 1398–1407.
- ↵H Gu, C Bock, TS Mikkelsen, N Jager, ZD Smith, E Tomazou, A Gnirke, ES Lander, A Meissner. 2010. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat Methods 7: 133–136.
- ↵MG Guenther, SS Levine, LA Boyer, R Jaenisch, RA Young. 2007. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130: 77–88.
- ↵B Han, R Mehra, SM Dhanasekaran, J Yu, A Menon, RJ Lonigro, X Wang, Y Gong, L Wang, S Shankar, . 2008. A fluorescence in situ hybridization screen for E26 transformation-specific aberrations: Identification of DDX5-ETV4 fusion protein in prostate cancer. Cancer Res 68: 7629–7637.
- ↵RA Harris, T Wang, C Coarfa, RP Nagarajan, C Hong, SL Downey, BE Johnson, SD Fouse, A Delaney, Y Zhao, . 2010. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 28: 1097–1105.
- ↵E Hodges, A Smith, J Kendall, Z Xuan, K Ravi, M Rooks, M Zhang, K Ye, A Battacharjee, L Brizuela, . 2009. High definition profiling of mammalian DNA methylation by array capture and single molecule bisulfite sequencing. Genome Res 19: 1593–1605.
- ↵I Ibragimova, I Ibanez de Caceres, AM Hoffman, A Potapova, E Dulaimi, T Al-Saleem, GR Hudes, MF Ochs, P Cairns. 2010. Global reactivation of epigenetically silenced genes in prostate cancer. Cancer Prev Res (Phila) 3: 1084–1092.
- ↵RS Illingworth, AP Bird. 2009. CpG islands–‘a rough guide.' FEBS Lett 583: 1713–1720.
- ↵RA Irizarry, C Ladd-Acosta, B Wen, Z Wu, C Montano, P Onyango, H Cui, K Gabo, M Rongione, M Webster, . 2009. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41: 178–186.
- ↵JP Issa. 2004. CpG island methylator phenotype in cancer. Nat Rev Cancer 4: 988–993.
- ↵I Jaspers, KM Horvath, W Zhang, LE Brighton, JL Carson, TL Noah. 2010. Reduced expression of IRF7 in nasal epithelial cells from smokers after infection with influenza. Am J Respir Cell Mol Biol 43: 368–375.
- ↵S Jhavar, A Reid, J Clark, Z Kote-Jarai, T Christmas, A Thompson, C Woodhouse, C Ogden, C Fisher, C Corbishley, . 2008. Detection of TMPRSS2-ERG translocations in human prostate cancer by expression profiling using GeneChip Human Exon 1.0 ST arrays. J Mol Diagn 10: 50–57.
- ↵PA Jones, SB Baylin. 2007. The epigenomics of cancer. Cell 128: 683–692.
- ↵JH Kim, SM Dhanasekaran, R Mehra, SA Tomlins, W Gu, J Yu, C Kumar-Sinha, X Cao, A Dash, L Wang, . 2007. Integrative analysis of genomic aberrations associated with prostate cancer progression. Cancer Res 67: 8229–8239.
- ↵K Kron, V Pethe, L Briollais, B Sadikovic, H Ozcelik, A Sunderji, V Venkateswaran, J Pinthus, N Fleshner, T van der Kwast, . 2009. Discovery of novel hypermethylated genes in prostate cancer using genomic CpG island microarrays. PLoS ONE 4: e4830. doi: 10.1371/journal.pone.0004830.
- ↵C Kumar-Sinha, SA Tomlins, AM Chinnaiyan. 2008. Recurrent gene fusions in prostate cancer. Nat Rev Cancer 8: 497–511.
- ↵I Kuzmin, JW Gillespie, A Protopopov, L Geil, K Dreijerink, Y Yang, CD Vocke, FM Duh, E Zabarovsky, JD Minna, . 2002. The RASSF1A tumor suppressor gene is inactivated in prostate tumors and suppresses growth of prostate carcinoma cells. Cancer Res 62: 3498–3502.
- ↵PW Laird. 2010. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet 11: 191–203.
- ↵LC Li, R Dahiya. 2002. MethPrimer: designing primers for methylation PCRs. Bioinformatics 18: 1427–1431.
- ↵LC Li, PR Carroll, R Dahiya. 2005. Epigenetic changes in prostate cancer: Implication for diagnosis and treatment. J Natl Cancer Inst 97: 103–115.
- ↵CA Maher, C Kumar-Sinha, X Cao, S Kalyana-Sundaram, B Han, X Jing, L Sam, T Barrette, N Palanisamy, AM Chinnaiyan. 2009. Transcriptome sequencing to detect gene fusions in cancer. Nature 458: 97–101.
- ↵AK Maunakea, RP Nagarajan, M Bilenky, TJ Ballinger, C D'Souza, SD Fouse, BE Johnson, C Hong, C Nielsen, Y Zhao, . 2010. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466: 253–257.
- ↵A Meissner, TS Mikkelsen, H Gu, M Wernig, J Hanna, A Sivachenko, X Zhang, BE Bernstein, C Nusbaum, DB Jaffe, . 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770.
- ↵IM Morison, JP Ramsay, HG Spencer. 2005. A census of mammalian imprinting. Trends Genet 21: 457–465.
- ↵WG Nelson, AM De Marzo, S Yegnasubramanian. 2009. Epigenetic alterations in human prostate cancers. Endocrinology 150: 3991–4002.
- ↵M Ongenaert, L Van Neste, T De Meyer, G Menschaert, S Bekaert, W Van Criekinge. 2008. PubMeth: a cancer methylation database combining text-mining and expert annotation. Nucleic Acids Res 36: D842–D846.
- ↵AS Perry, RW Watson, M Lawler, D Hollywood. 2010. The epigenome as a therapeutic target in prostate cancer. Nat Rev Urol 7: 668–680.
- ↵ZS Qin, J Yu, J Shen, CA Maher, M Hu, S Kalyana-Sundaram, AM Chinnaiyan. 2010. HPeak: an HMM-based algorithm for defining read-enriched regions in ChIP-Seq data. BMC Bioinformatics 11: 369.
- ↵TA Rauch, GP Pfeifer. 2009. The MIRA method for DNA methylation analysis. Methods Mol Biol 507: 65–75.
- ↵TA Rauch, X Wu, X Zhong, AD Riggs, GP Pfeifer. 2009. A human B cell methylome at 100-base pair resolution. Proc Natl Acad Sci 106: 671–678.
- ↵M Reuter, S Chuma, T Tanaka, T Franz, A Stark, RS Pillai. 2009. Loss of the Mili-interacting Tudor domain-containing protein-1 activates transposons and alters the Mili-associated small RNA profile. Nat Struct Mol Biol 16: 639–646.
- ↵DR Rhodes, S Kalyana-Sundaram, V Mahavisno, R Varambally, J Yu, BB Briggs, TR Barrette, MJ Anstet, C Kincead-Beal, P Kulkarni, . 2007a. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 9: 166–180.
- ↵DR Rhodes, S Kalyana-Sundaram, SA Tomlins, V Mahavisno, N Kasper, R Varambally, TR Barrette, D Ghosh, S Varambally, AM Chinnaiyan. 2007b. Molecular concepts analysis links tumors, pathways, mechanisms, and drugs. Neoplasia 9: 443–454.
- ↵M Schummer, WV Ng, RE Bumgarner, PS Nelson, B Schummer, DW Bednarski, L Hassell, RL Baldwin, BY Karlan, L Hood. 1999. Comparative hybridization of an array of 21,500 ovarian cDNAs for the discovery of genes overexpressed in ovarian carcinomas. Gene 238: 375–385.
- ↵D Takai, PA Jones. 2002. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci 99: 3740–3745.
- ↵JP Thomson, PJ Skene, J Selfridge, T Clouaire, J Guy, S Webb, AR Kerr, A Deaton, R Andrews, KD James, . 2010. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 464: 1082–1086.
- ↵SA Tomlins, B Laxman, SM Dhanasekaran, BE Helgeson, X Cao, DS Morris, A Menon, X Jing, Q Cao, B Han, . 2007a. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 448: 595–599.
- ↵SA Tomlins, R Mehra, DR Rhodes, X Cao, L Wang, SM Dhanasekaran, S Kalyana-Sundaram, JT Wei, MA Rubin, KJ Pienta, . 2007b. Integrative molecular concept modeling of prostate cancer progression. Nat Genet 39: 41–51.
- ↵VG Tusher, R Tibshirani, G Chu. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci 98: 5116–5121.
- ↵J Vandesompele, K De Preter, F Pattyn, B Poppe, N Van Roy, A De Paepe, F Speleman. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034.
- ↵M Weber, JJ Davies, D Wittig, EJ Oakeley, M Haase, WL Lam, D Schubeler. 2005. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37: 853–862.
- ↵H Wu, V Coskun, J Tao, W Xie, W Ge, K Yoshikawa, E Li, Y Zhang, YE Sun. 2010. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 329: 444–448.
- ↵S Yegnasubramanian, J Kowalski, ML Gonzalgo, M Zahurak, S Piantadosi, PC Walsh, GS Bova, AM De Marzo, WB Isaacs, WG Nelson. 2004. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res 64: 1975–1986.
- ↵S Yegnasubramanian, MC Haffner, Y Zhang, B Gurel, TC Cornish, Z Wu, RA Irizarry, J Morgan, J Hicks, TL DeWeese, . 2008. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res 68: 8954–8967.
- ↵J Yu, RS Mani, Q Cao, CJ Brenner, X Cao, X Wang, L Wu, J Li, M Hu, Y Gong, . 2010. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 17: 443–454.
Footnotes
[1] [Supplemental material is available for this article. The next-generation sequencing and microarray data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession nos. GSE29155 and GSE27619, respectively.]
[2] Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.119347.110.