
Abstract
Transcriptional networks have been shown to evolve very rapidly, prompting questions as to how such changes arise and are tolerated. Recent comparisons of transcriptional networks across species have implicated variations in the cis-acting DNA sequences near genes as the main cause of divergence. What is less clear is how these changes interact with trans-acting changes occurring elsewhere in the genetic circuit. Here, we report the discovery of a system of compensatory trans and cis mutations in the yeast AP-1 transcriptional network that allows for conserved transcriptional regulation despite continued genetic change. We pinpoint a single species, the fungal pathogen Candida glabrata, in which a trans mutation has occurred very recently in a single AP-1 family member, distinguishing it from its Saccharomyces ortholog. Comparison of chromatin immunoprecipitation profiles between Candida and Saccharomyces shows that, despite their different DNA-binding domains, the AP-1 orthologs regulate a conserved block of genes. This conservation is enabled by concomitant changes in the cis-regulatory motifs upstream of each gene. Thus, both trans and cis mutations have perturbed the yeast AP-1 regulatory system in such a way as to compensate for one another. This demonstrates an example of “coevolution” between a DNA-binding transcription factor and its cis-regulatory site, reminiscent of the coevolution of protein binding partners.
Transcriptional networks are central to understanding both evolution and phenotypic diversity among organisms. Of the many ways in which transcriptional networks can evolve, much attention has been given to changes in the so-called cis-regulatory regions of gene promoters (Wray 2007; Wagner and Lynch 2008). Such changes include gain, loss, or modification of DNA sequence motifs (Cliften et al. 2003; Kellis et al. 2003; Gasch et al. 2004; Stark et al. 2007) as well as alterations in motif spacing relative to the start of transcription, or to other motifs (Ihmels et al. 2005; Tanay et al. 2005). In addition to changes in cis, transcriptional networks can also evolve through alterations to transcription factor (TF) proteins and other trans-acting factors (Wagner and Lynch 2008). Although there have been fewer reports of evolutionary changes in trans, potential mechanisms include mutations to protein structure impacting transcriptional activation or DNA-binding domains (Wagner and Lynch 2008), modulation of TF expression (Sankaran et al. 2009) or post-translational modifications (Holt et al. 2009), or gain and loss of protein–protein interactions among TFs (Tuch et al. 2008; Lavoie et al. 2010).
Recently, a number of genome-scale studies have performed systematic comparisons of TF-binding patterns (Borneman et al. 2007; Tuch et al. 2008; Bradley et al. 2010; Lavoie et al. 2010; Schmidt et al. 2010) or mRNA expression profiles across species (Ihmels et al. 2005; Tanay et al. 2005; Hogues et al. 2008; Field et al. 2009; Wapinski et al. 2010). Almost universally, these studies have identified transcriptional programs that are dramatically rewired over short evolutionary time scales. As with earlier work, many of the observed differences in binding and expression have been linked to changes in cis-regulatory regions. For example, Borneman et al. (2007) found that the TF Tec1 binds only 20% of the same target genes in comparisons between Saccharomyces cerevisiae and the closely related Saccharomyces bayanus and Saccharomyces mikatae, and that this difference is due to gain and loss of canonical Tec1 cis-regulatory motifs. While some recent studies have associated genetic variants in TFs with gene expression changes observed in interspecies hybrids (Wilson et al. 2008; Wittkopp et al. 2008; Tirosh et al. 2009; Bullard et al. 2010; Emerson et al. 2010), in outbred crosses (Brem and Kruglyak 2005; Landry et al. 2005; Gerke et al. 2009; Sung et al. 2009; Zheng et al. 2010), or in human populations (Kasowski et al. 2010), the picture that emerges is that cis-regulatory regions are incredibly plastic over evolutionary time, while TFs (trans) evolve at a comparatively slower rate (Wray 2007).
Given the dramatic changes that appear to be occurring in transcriptional networks, a key question is how such systems retain essential functions over evolutionary time (Wray 2007). One solution is that changes in cis can occur by replacement of one TF cofactor with another, thereby maintaining regulatory control (Tsong et al. 2006). Alternatively, rather than replacing specific cofactors, it is conceivable that the DNA-binding domains of the TFs that bind these cis-regulatory sequences might be altered in lock-step with changes in cis, similarly to the evolution of protein-binding partners (Pazos and Valencia 2008). However, such a mechanism of evolution has yet to be observed. Here, we present a direct example of such “coevolution,” where a specific change to a DNA-binding transcription factor and its cis-regulatory site have occurred in compensatory fashion.
As a model of transcriptional network evolution, we examined the yeast AP-1 (yAP-1) family, which, with a total of eight members, is one of the largest paralogous TF families in S. cerevisiae (Fernandes et al. 1997; Rodrigues-Pousada et al. 2010). Like other paralogous families, AP-1 factors have been born through the process of gene duplication, which gives rise to multiple copies that are free from selective pressure and may functionally diverge from their duplicates by sub- or neofunctionalization (Hittinger and Carroll 2007). AP-1 also provides a classic example of the basic leucine zipper (bZIP) motif, which is widely conserved across eukaryotes (Tan et al. 2008; Rodrigues-Pousada et al. 2010). In humans, AP-1 TFs have been heavily studied due to their crucial role in cell proliferation, death, and differentiation (Shaulian and Karin 2002). In yeast, yAP-1-mediated transcriptional networks carry out overlapping, but distinct biological responses to stress (Tan et al. 2008; Rodrigues-Pousada et al. 2010). In contrast to the widespread divergence in TF binding that has been demonstrated previously (Borneman et al. 2007; Tuch et al. 2008; Lavoie et al. 2010), we show that coupled trans and cis mutations enable conservation of a subset of genes targeted by yAP-1. These results provide an example of compensatory coevolution of a trans and cis regulatory system.
Results
A trans mutation is associated with AP-1 DNA-binding motif specificity
To identify trans mutations that could be associated with AP-1-binding preference, we performed an amino acid sequence alignment of the DNA-binding domains of all eight AP-1-like TFs in S. cerevisiae (Sc). This alignment and its associated phylogenetic tree (Fig. 1A) were searched to identify the key polymorphic amino acids whose patterns of conservation and divergence best explain the phylogeny (Methods, Evolutionary Trace Analysis). Such residues have been shown to frequently play important evolutionary roles (Innis et al. 2000). Using this approach, we identified residue 12 of the DNA-binding domain basic region as the most important evolutionarily divergent position across the yAP-1 family (i.e., the one that was most highly correlated with the phylogeny; Fig. 1A).
A single residue determines yAP-1 DNA-binding motif specificity. (A) Alignment and phylogeny of AP-1 DNA-binding domain basic regions (residues 6 to 20 are shown). Residue 12 (red star) is predictive of preference for overlapping (YRE-O) or adjacent (YRE-A) DNA-binding motifs (left). Note that Yap8 possesses an Asp at residue 12 and binds a 2-bp overlapping YRE-O (Harbison et al. 2004). Positions affecting Gcn4 half-site spacing preference (Kim and Struhl 1995) are shown (gray stars). (B) Recognition of the yAP-1 half-site (Fujii et al. 2000). Residue 12 (red star) is in close proximity to residues conferring AP-1 sequence specificity. (C,D) ScYap1.R79K and ScYap4.K252R mutants have altered half-site spacing preference as evidenced by ChIP-chip (Methods). P-values refer to differences in binding to genes with either YRE-O or YRE-A sites as assessed by Fisher's exact test. (E,F) ScYap1.R79K and ScYap4.K252R mutations cause mRNA expression changes among genes with YRE-O and YRE-A sites among the top 50 most differentially expressed genes. P-values denote the significance of YRE-A and YRE-O motifs among gene promoters compared with the genomic background.
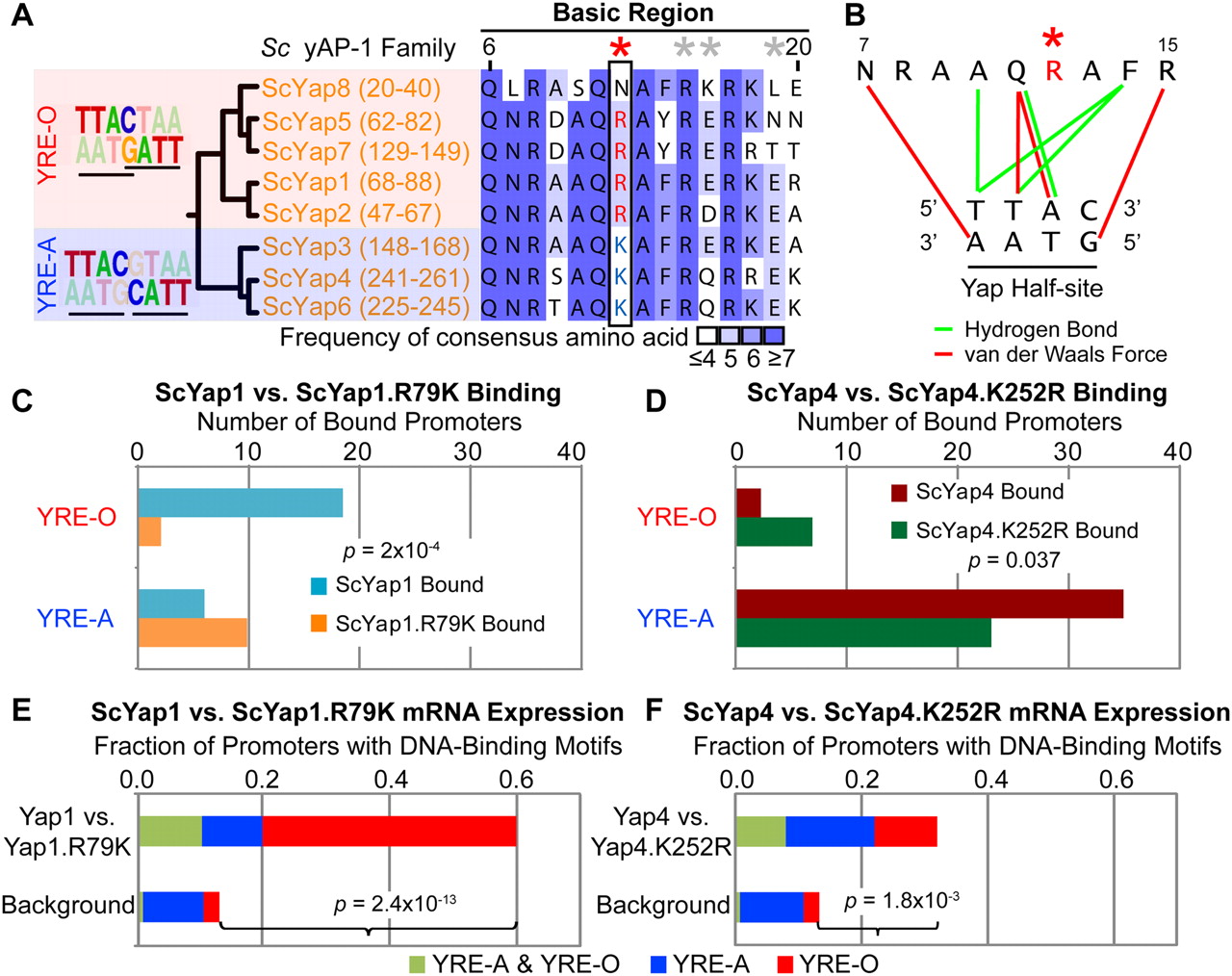
Residue 12 was also predictive of AP-1 family DNA-binding motif preference (Fig. 1A) (MacIsaac et al. 2006; Tan et al. 2008). AP-1 family members bind DNA as homo- or heterodimers, where each constituent monomer recognizes the consensus sequence TTAC (Suckow et al. 1999; Fujii et al. 2000). These “half-sites” are positioned in either adjacent or overlapping fashion (Fig. 1A), which we refer to as yAP-1 response element adjacent (YRE-A) or yAP-1 response element overlapping (YRE-O), respectively. Previous analyses of genome-wide chromatin immunoprecipitation with microarray hybridization (ChIP-chip) data in Sc (Harbison et al. 2004; Tan et al. 2008) have determined that five AP-1 family members (Yap1, Yap2, Yap5, Yap7, Yap8) recognize YRE-O, whereas two family members (Yap4 and Yap6) recognize YRE-A. We examined the binding of the remaining Sc AP-1 member Yap3 by ChIP-chip and determined it preferred YRE-A sites in both complete media and stress conditions (Supplemental Fig. 1). This preference for YRE-O or YRE-A-binding sites in Sc AP-1s correlates precisely with the presence of arginine or lysine at residue 12 (Fig. 1A).
Interestingly, residue 12 is part of an alpha-helical surface that forms multiple contacts to DNA (basic region residues 7–15) (Fig. 1B; Fujii et al. 2000). Previously, this residue was predicted as a likely determinant of DNA half-site spacing preference in Gcn4, another bZIP family TF (Kim and Struhl 1995). Although in vitro testing of Gcn4 mutants was not able to confirm this prediction (Kim and Struhl 1995), it has become apparent that such variations in half-site recognition are best distinguished in vivo (Suckow and Hollenberg 1998; Berger et al. 2008; Maerkl and Quake 2009).
Residue 12 point mutations cause rewiring of AP-1 transcriptional interactions
To further examine the regulatory role of residue 12, we mutated this residue in Yap1, a representative YRE-O-binding factor, and Yap4 (also known as Cin5), a representative YRE-A-binding factor. This process involved generating mutants Yap1.R79K and Yap4.K252R, changing arginine to lysine in Yap1 and lysine to arginine in Yap4 (Methods). Next, Yap1.R79K binding and Yap4.K252R binding were assayed in vivo using ChIP-chip (Methods). Comparison of the top 50 promoters bound by Yap1.R79K with the top 50 promoters bound by wild-type Yap1 (as determined in Tan et al. 2008) showed that mutation of Yap1 significantly altered its preference for YRE-O and YRE-A sites (Fig. 1C; Fisher's exact test P = 0.0002). Comparison of promoters bound by mutant and wild-type Yap4 also showed the predicted shift in binding preference (Fig. 1D; Fisher's exact test P = 0.037). These results were not dependent on the number of promoters examined (Supplemental Fig. 2).
Next, to assess the functional implications of changes in yAP-1 binding, we generated genome-wide mRNA expression profiles for each mutant in comparison to the unmutated parental strain (Methods). Both mutations, Yap1.R79K and Yap4.K252R, altered the expression of genes whose promoters were highly enriched for AP-1-binding sites (YRE-O and YRE-A, Fig. 1E,F; Supplemental Fig. 3). These genes were also enriched for Yap1.R79K and Yap4.K252R binding (P < 10−5), respectively.
An apparent paradox: Candida AP-1 diverges at residue 12, but its targets are conserved
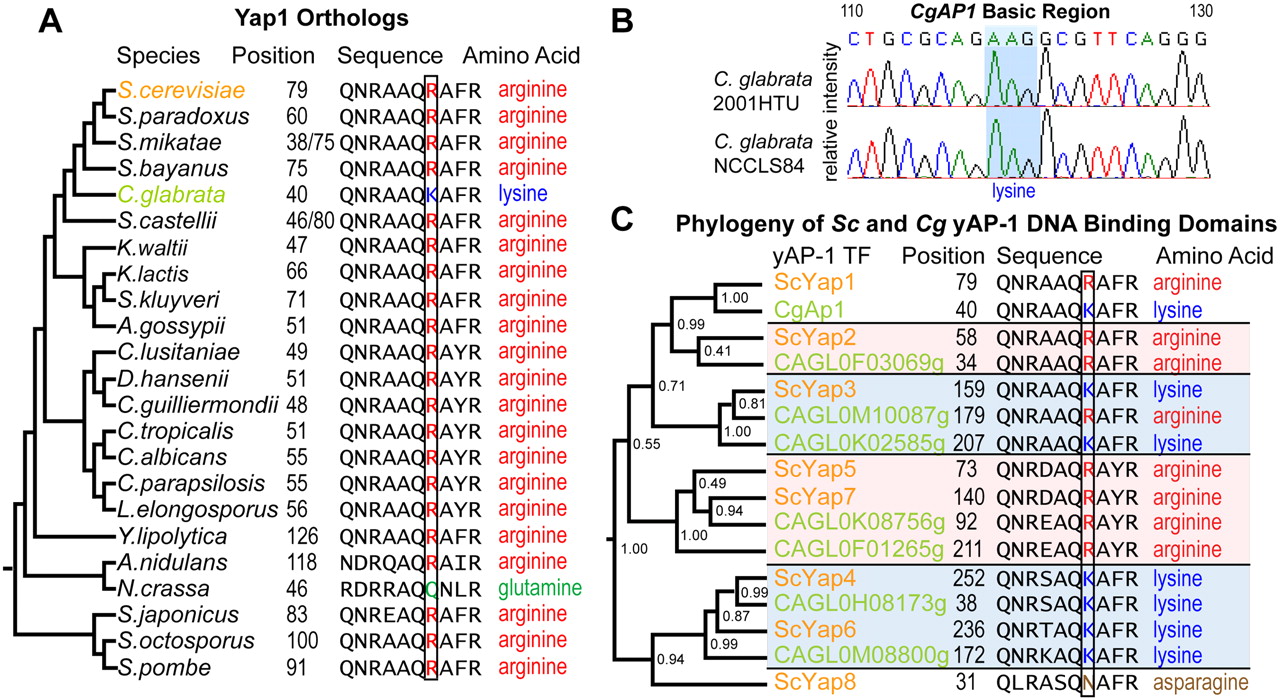
Based on our observation that residue 12 affects binding of AP-1 paralogs in S. cerevisiae, we next asked whether changes in this residue could lead to divergent binding of AP-1 orthologs across species. We searched the yeast phylogeny (Wapinski et al. 2007) for AP-1 orthologs that were anomalous in their use of Arg 12 or Lys 12, suggesting lineage-specific mutation (Supplemental Fig. 4). Among TFs orthologous to Sc YAP1, we found that the Candida glabrata (Cg) ortholog CgAP1 diverges from other yeasts (Fig. 2A–C) due to the presence of lysine at residue 12, in contrast to other yeasts in its clade that possess an arginine. This CgAp1 amino acid substitution was confirmed by sequencing of genomic DNA from two independent Cg isolates, 2001HTU and NCCLS84 (Fig. 2B).
Evolution of the yAP-1 TFs. (A) CgAp1 possesses a lysine at residue 12 (CgAp1.46), while most other species possess an arginine. (B) Sequencing of CgAP1 in two unrelated isolates shows complete identity to the Cg reference genome. (C) Phylogenetic clustering of all Sc and Cg AP-1 DNA-binding domains reveals that CgAp1 and ScYap1 co-cluster. Internal branch point numbers refer to the Bayesian posterior probability, a measure of confidence (Drummond and Rambaut 2007).
We used ChIP-chip to determine whether this Lys 12 substitution had a functional effect on CgAp1 binding (Methods). To facilitate this assay, we tagged CgAp1 with the TAP epitope and designed a custom microarray tiling the Cg genome (Methods). As a control on both the TAP construct and the array design, we used ChIP-qPCR to successfully validate a panel of five randomly chosen Cg gene promoters that were determined to be bound by CgAp1 in the ChIP-chip experiment (Supplemental Fig. 5).
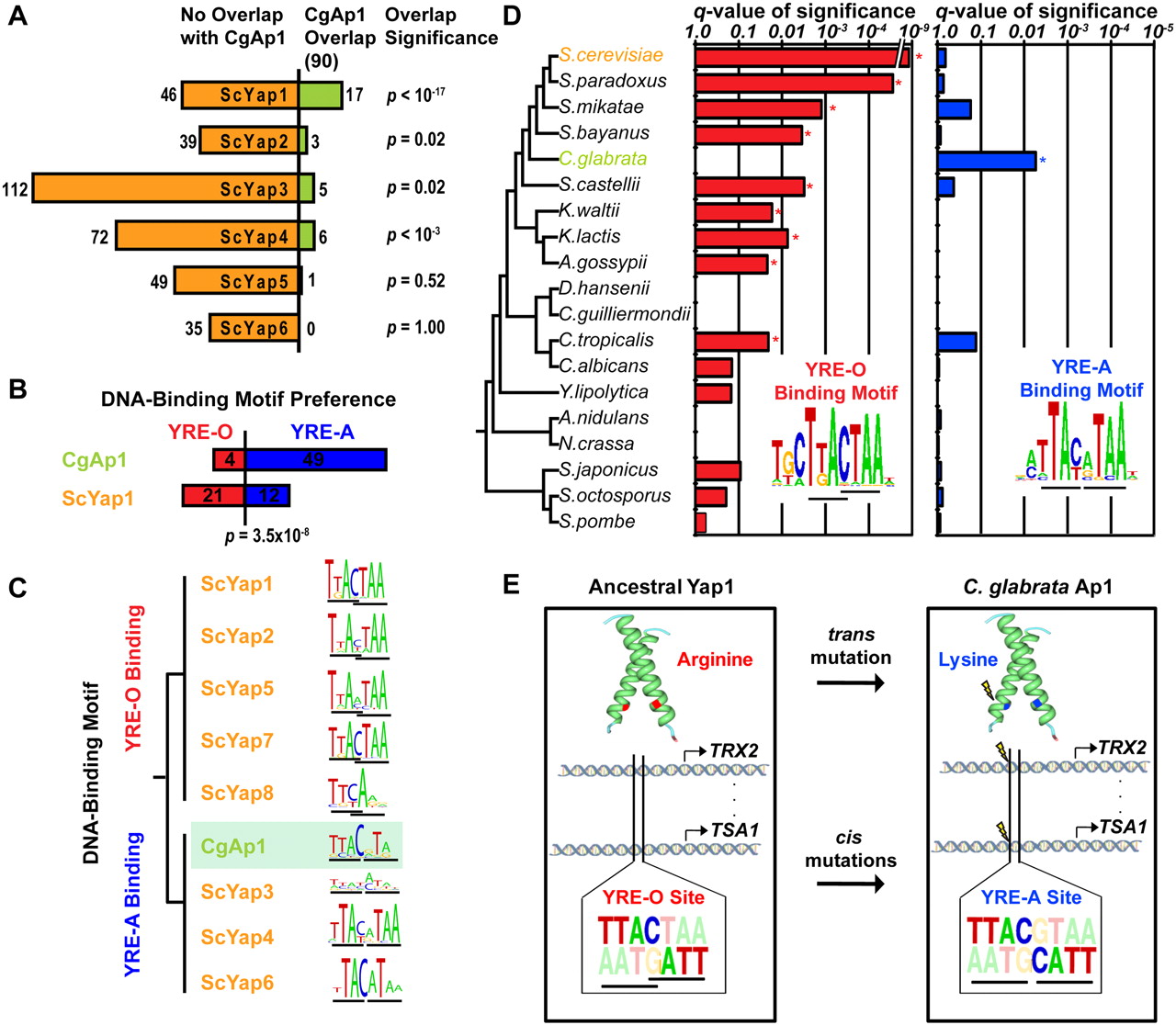
We found that CgAp1 bound the promoters of a total of 114 genes, 90 of which had known orthologs in Sc (Methods). Comparison of these data with ChIP profiles for each of the AP-1 factors in Sc grown under the same treatment conditions (as determined in Tan et al. 2008) showed significant overlap between the targets of CgAp1 and ScYap1 (17 genes, P < 10−17). Overlap with other Sc AP-1 factors was less substantial (Fig. 3A). This pattern of overlap was reinforced by sequence analysis, in which phylogenetic clustering of AP-1 DNA-binding domains places CgAp1 definitively with ScYap1 and not with other Sc AP-1 sequences (Fig. 2C; Methods).
The CgAp1 transcriptional network has been rewired. (A) For the promoters targeted by each yAP-1 transcription factor in Sc, the overlap with CgAp1 targets is shown (of 90 CgAp1 targets total). (B) CgAp1 prefers YRE-A-binding sites compared with ScYap1 (Fisher's exact test). (C) The CgAp1 DNA-binding motif (green) clusters with YRE-A rather than YRE-O motifs. (D) The YRE-O site is enriched (star) among common ScYap1 and CgAp1 targets in other yeasts (hypergeometric test, Q < 0.05), but not Cg (Q ∼ 1.0). The YRE-A site is enriched among these targets in Cg (star) but not other yeasts. (E) Compensatory mutations in both trans and cis maintain AP-1 binding.
We were therefore faced with the following conundrum: On the one hand, the CgAp1 sequence diverges from Yap1 orthologs at residue 12, suggesting a shift in DNA binding. On the other hand, the CgAp1-binding profile is quite specifically conserved with that of Yap1, calling into question the importance of residue 12 for sequence recognition.
CgAp1 prefers YRE-A rather than YRE-O sites
To investigate this apparent contradiction, we next turned to the gene promoters targeted by CgAp1 in the ChIP assay. Promoters targeted by CgAp1 showed a clear preference for YRE-A sites over YRE-O sites (49 vs. four promoters, respectively). This preference significantly differs from ScYap1, which prefers YRE-O over YRE-A (Fisher's exact test P = 3.5 × 10−8; Fig. 3B, 21 vs. 12 promoters, respectively). This preference could not be attributed to threshold effects on binding-site calls, as direct comparison of motif scores confirmed a preference for YRE-A over YRE-O sites (Mann-Whitney U test, P = 0.0072). This preference was also observed via de novo motif search in these promoters (Fig. 3C) and even among the Cg orthologs of all ScYap1 targets (Q = 0.05).
We further analyzed this cis-regulatory preference by examining the orthologs of genes targeted by both CgAp1 and ScYap1 across 20 sequenced yeast genomes (Wapinski et al. 2007). C. glabrata stood out clearly as the only species with enrichment for YRE-A sites (Fig. 3D). In contrast, the YRE-O site was enriched in all neighboring species in the yeast phylogeny, including S. cerevisiae and other sensu stricto species (S. paradoxus, S. mikatae, and S. bayanus) as well as the more diverged Saccharomyces castellii, Kluyveromyces waltii, Kluyveromyces lactis, Ashbya gosspyii, and Candida tropicalis. These results indicate that upstream DNA-binding motifs of CgAp1 targets have evolved from YRE-O to YRE-A (Fig. 3E). Such a switch may have also been accompanied by concordant changes in secondary cis-regulatory DNA motifs (Supplemental material, Yap Cis-Regulatory Motifs Are Coincident with Those of Rtg3 and Aft1; Supplemental Fig. 6) and possible functional divergence (Supplemental material, Divergence and Conservation of Yap1 Function). The most plausible explanation is that these motifs have coevolved with a Lys 12 mutation in CgAp1, with the result that this transcriptional system has retained regulatory control of the same set of target genes over evolutionary time.
Discussion
Which mutation came first: the cis or trans? It is possible to envision two equally plausible scenarios (Fig. 3E): (1) An initial mutation in the Yap1 TF provided selective pressure for subsequent cis-regulatory changes in Yap1 targeted genes; (2) a change from YRE-O to YRE-A-binding site in key Yap1 target(s) provided selective pressure for a mutation in the Yap1 TF. In either scenario, mutations in trans and cis may have been facilitated by other AP-1 family members. The large size and interconnectivity of the AP-1 family may serve as a buffer for accumulation of cis and trans mutations, allowing for highly plastic evolution of the AP-1 regulatory network. In support of this hypothesis, several yAP-1s have been shown to bind each other along with common target genes, which might compensate for some loss in regulation by paralogs (Tan et al. 2008).
Examination of the protein sequences of all AP-1 family members across 20 available yeast genomes (Wapinski et al. 2007) suggests that mutations in residue 12 have occurred frequently during AP-1 family evolution (Supplemental Fig. 4). Interestingly, we found that all yeasts possess at least one AP-1 TF with Arg 12 (Fig. 4). In contrast, several yeasts lack AP-1 TFs with Lys 12, and these species are the most evolutionarily diverged from Sc. These results suggest that the common yeast AP-1 ancestor encoded arginine and that the emergence of TFs using lysine is a more recent evolutionary innovation (Supplemental material, AP-1 Family Ancestry).
All yeasts in the species phylogeny (Wapinski et al. 2007) possess an AP-1 with an arginine. The Candida clade (shaded green) and the whole-genome duplication event (orange star) are noted. Note that the Candida clade does not include Candida glabrata.
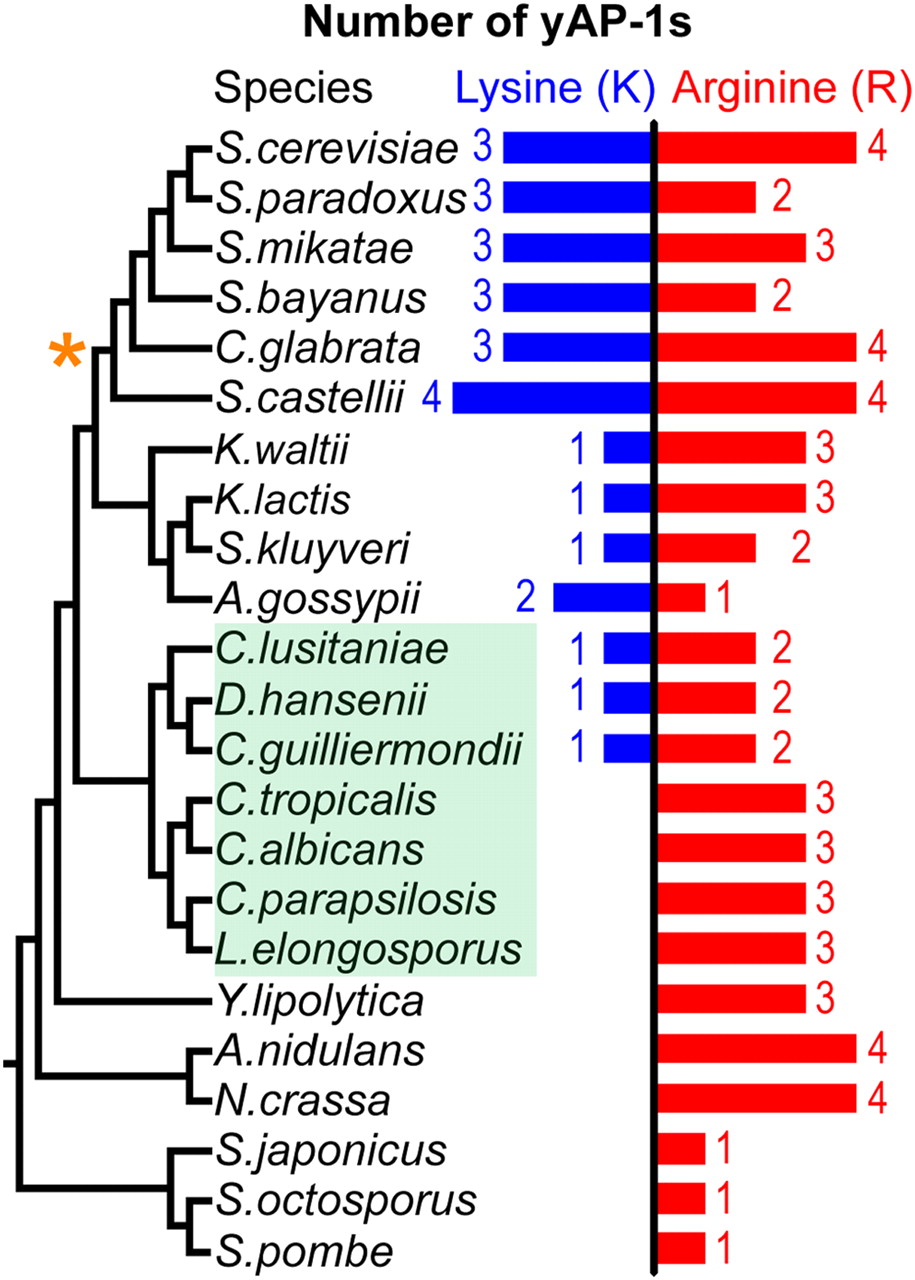
Within the Candida clade, several species (C. tropicalis, C. albicans, C. parapsilosis, and Lodderomyces elongosporus) have AP-1 families based exclusively on Arg 12, while others (C. lusitaniae, Debaryomyces hansenii, and C. guilliermondii) (Fig. 4) represent both Arg 12 and Lys 12 across the AP-1 family. This suggests two equally plausible scenarios for the emergence of Lys 12 in yAP-1 TFs: (1) Lysine emerged following the divergence of Yarrowia lipolytica from other hemi-ascomycetes, followed by a lineage-specific loss within the Candida clade. (2) Lysine emerged following the split of the Candida clade from the rest of the hemi-ascomycetes and emerged again within the Candida clade. In either scenario, a switch from arginine (coded by AGA or AGG) to lysine (coded by AAA or AAG) could be accomplished by a simple single base-pair mutation.
Open questions still remain regarding how arginine and lysine substitutions alter AP-1 DNA-binding motif preference. One hypothesis is that differences in their electrostatic charges alter the space required to accommodate other positively charged residues of the bZIP DNA-binding domain without electrostatic repulsion (Kim and Struhl 1995). This hypothesis suggests that the most positively charged residues such as arginine should be associated with YRE-A rather than YRE-O sites (Kim and Struhl 1995). However, in our findings lysine (pI = 9.59) rather than arginine (pI = 11.15) was associated with YRE-A sites.
An alternative explanation involves a role for AP-1-induced DNA flexibility. Complexes of yAP-1 protein with the YRE-O DNA sequence have been associated with an increase in incorporated water molecules (Dragan et al. 2004a) leading to a decrease in DNA flexibility (Kim and Struhl 1995) compared with yAP-1/YRE-A complexes. A previous report has suggested that changes in DNA flexibility play a key role in determining half-site spacing preference and are responsible for differences between in vivo and in vitro measurements (Suckow and Hollenberg 1998). Since residue 12 is in close proximity to DNA (Fig. 1B) within the protein–DNA complex, residue changes may affect the ability of DNA to incorporate water during binding, thus affecting both yAP-1 DNA motif flexibility and binding (Dragan et al. 2004b). Interestingly, the higher positive charge of arginine induces a stronger dipole than that of lysine, providing a possible mechanism for the increase in the number of incorporated water molecules present at YRE-O sites and associated changes in DNA flexibility and binding preference.
In summary, we have shown that conservation of the AP-1 regulatory program in yeast occurs through coordinated evolution of both the sequence of the TF (trans) and in its DNA-binding motifs (cis). This finding echoes that of previous studies of protein–protein interaction, which have demonstrated cases in which compensatory mutations are required to maintain protein interaction over evolutionary time (Pazos and Valencia 2008). In the context of transcriptional networks, coevolution gives rise to “regulatory homeostasis,” in which both mutations in a TF and its DNA-binding motif occur in compensatory fashion to maintain transcriptional regulation. This series of compensatory mutations, which maintains both the transcriptional circuit and regulatory logic, parallels that of previous work demonstrating evolution of alternative transcriptional circuits producing identical logic (Tsong et al. 2006). Such systems of tightly coupled compensatory mutations might serve to counter the widespread divergence observed in transcriptional networks, and may constitute a general evolutionary mechanism maintaining the regulation of transcriptional networks.
Methods
Yeast strains
All immunoprecipitations were performed on strains where the appropriate gene has been endogenously fused to the TAP epitope (Rigaut et al. 1999). Sc TAP-tagged strains were obtained from Open Biosystems. In Cg, the 2001HTU strain was used for TAP-tagging (see below) and deletions. Cg 2001HTU and NCCLS84 were obtained from ATCC.
ScYap1.R79K and ScYap4.K252R mutants
Endogenously epitope-tagged ScYap1∷TAP and ScYap4∷TAP strains (Open Biosystems) were used to introduce the appropriate mutation (ScYap1.R79K, ScYap4.K252R) via the delitto perfetto method (Storici et al. 2001). In brief, ScYAP1 and ScYAP4 were disrupted with the URA3 selectable marker from pRS306 (Brachmann et al. 1998) using ∼100-bp homology. Complementary 200-bp oligos with a mutation (ScYap1.R79K or ScYap4.K252R) were then transformed by electroporation (Thompson et al. 1998) to remove URA3 by 5-FOA (US Biological) selection and verified by sequencing. This process creates strains possessing endogenous yAP-1 proteins having both the desired mutation and epitope.
CgAp1 TAP-tagged strain
The TAP tag was amplified with ∼100-bp homology (CgAP1) from pFA6a-TAP-HIS3MX6 (Longtine et al. 1998), transformed by electroporation (Thompson et al. 1998), and selected on complete –his media (Amberg et al. 2005). C-terminal integration was verified by PCR and DNA sequencing with protein expression verified by immunoblot (Amberg et al. 2005) with the peroxidase anti-peroxidase antibody (Sigma P1291).
Growth conditions, mRNA expression, and ChIP
Three (CgAp1) or two (ScYap1.R79K, ScYap4.K252R, ScYap3) biological replicates were grown from OD600 0.2 to 0.8 at 30°C in complete media (Amberg et al. 2005) and treated with 0.03% methyl methanesulfonate (Sigma) for 1 h as performed previously (Tan et al. 2008). For mRNA expression analysis, total RNA was isolated by hot phenol/chloroform extraction and labeled with Cy3 or Cy5 dyes (Invitrogen) (Kuo et al. 2010). Samples were hybridized to Agilent expression arrays and washed as recommended by Agilent (Agilent Technologies).
For ChIP, all TAP-tagged strains were treated as previously described (Tan et al. 2008). In brief, cells were fixed with 1% formaldehyde for 20 min, inactivated with glycine and washed with TBS. Cells were lysed for 2 h (Vibrax-VXR 2000) with glass beads and sonicated for four cycles of 20 sec (+100-sec rest) at power setting 2 (Misonex Sonicator 3000) on ice. Lysate was incubated with Dynabeads M-280 conjugated with anti-TAP antibody (Open Biosystems CAB1001) overnight. Cross-link reversal was performed overnight at 65°C with antibody-enriched and unenriched DNA, and amplified (Sigma-Aldrich) and labeled (Invitrogen) with Cy5/Cy3 dyes. Sc and Cg samples were hybridized to commercial or custom (see below) Agilent tiling arrays and washed as recommended by Agilent (Agilent Technologies).
Cg tiling microarray design and validation
We designed a custom microarray tiling the Cg genome at ∼250-bp resolution with ∼44,000 60-mer probes designed to avoid self-dimerization and variability in melting temperatures (Mfold; Zuker 2003), low-complexity and repetitive sequences (RepeatMasker; http://repeatmasker.org), and cross-hybridization (WUBLAST2; Altschul and Gish 1996). Default settings were used for each program. Microarrays were manufactured using Agilent technology (Agilent Technologies). ChIP results were validated by qPCR of five targets compared against CgACT1 (Supplemental Fig. 5).
Microarray data processing
Intensities were background subtracted and normalized by LOESS (Smyth 2005). Expression microarrays were analyzed using the limma package (Smyth 2005) with default parameters. ChIP tiling array errors were estimated by the Rosetta error model (Weng et al. 2006) with resulting P-values of binding for each promoter calculated by combining P-values of adjacent probes as previously described (Tan et al. 2008).
Phylogenetic trees, orthologs, and evolutionary trace analysis
The yAP-1 DNA-binding domain phylogenetic tree was created with BEAST (Drummond and Rambaut 2007) using default settings. Sequences, species trees, and orthologs were obtained from the Fungal Orthogroups Repository (Wapinski et al. 2007). Multiple-sequence alignment was preformed using MUSCLE (Edgar 2004) with default parameters. Evolutionary trace analysis was performed using TraceSuite II (Innis et al. 2000) with default settings.
Motif finding
De novo motifs were identified by SOMBRERO (Mahony et al. 2005) using default parameters and compared with literature (MacIsaac et al. 2006; Tan et al. 2008) using the default settings for STAMP (Mahony et al. 2005). Promoters were scanned with the default settings for Patser (Hertz and Stormo 1999) for motif enrichment by the hypergeometric test with multiple test correction (Storey and Tibshirani 2003). A motif was considered “present” in a promoter for (fraction of maximal information content) ≥0.7.
Acknowledgments
We thank Jonathan Weissman for providing pFA6a-TAP-HIS3MX6, Paul Russell for pFA6a-kanMX6, Nevan Krogan for pFA6a-natMX6, and Richard Kolodner for pRS303 and pRS306. We also thank Ilan Wapinski, Lorraine Pillus, and members of the T.I. laboratory for helpful discussions. D.K. was supported by the National Science and Engineering Research Council of Canada. K.T. and T.I. were supported by the David and Lucille Packard Foundation and NIH grant no. R01 ES014811 to T.I.
Author contributions: D.K., K.T., T.R., and T.I. designed the study. D.K., K.L., S.B., R.C., J.C., and C.L. performed the experimental work. D.K. and K.T. analyzed the data. D.K. and T.I. wrote the manuscript. K.T. and T.I. supervised the work.
References
- ↵SF Altschul, W Gish. 1996. Local alignment statistics. Methods Enzymol 266: 460–480.
- ↵DC Amberg, DJ Burke, JN Strathern. 2005. Methods in yeast genetics: A Cold Spring Harbor Laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- ↵MF Berger, G Badis, AR Gehrke, S Talukder, AA Philippakis, L Pena-Castillo, TM Alleyne, S Mnaimneh, OB Botvinnik, ET Chan, . 2008. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133: 1266–1276.
- ↵AR Borneman, TA Gianoulis, ZD Zhang, H Yu, J Rozowsky, MR Seringhaus, LY Wang, M Gerstein, M Snyder. 2007. Divergence of transcription factor binding sites across related yeast species. Science 317: 815–819.
- ↵CB Brachmann, A Davies, GJ Cost, E Caputo, J Li, P Hieter, JD Boeke. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115–132.
- ↵RK Bradley, X-Y Li, C Trapnell, S Davidson, L Pachter, HC Chu, LA Tonkin, MD Biggin, MB Eisen. 2010. Binding site turnover produces pervasive quantitative changes in transcription factor binding between closely related Drosophila species. PLoS Biol 8: e1000343. doi: 10.1371/journal.pbio.1000343.
- ↵RB Brem, L Kruglyak. 2005. The landscape of genetic complexity across 5,700 gene expression traits in yeast. Proc Natl Acad Sci 102: 1572–1577.
- ↵JH Bullard, Y Mostovoy, S Dudoit, RB Brem. 2010. Polygenic and directional regulatory evolution across pathways in Saccharomyces. Proc Natl Acad Sci 107: 5058–5063.
- ↵P Cliften, P Sudarsanam, A Desikan, L Fulton, B Fulton, J Majors, R Waterston, BA Cohen, M Johnston. 2003. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science 301: 71–76.
- ↵AI Dragan, L Frank, Y Liu, EN Makeyeva, C Crane-Robinson, PL Privalov. 2004a. Thermodynamic signature of GCN4-bZIP binding to DNA indicates the role of water in discriminating between the AP-1 and ATF/CREB sites. J Mol Biol 343: 865–878.
- ↵AI Dragan, Y Liu, EN Makeyeva, PL Privalov. 2004b. DNA-binding domain of GCN4 induces bending of both the ATF/CREB and AP-1 binding sites of DNA. Nucleic Acids Res 32: 5192–5197.
- ↵AJ Drummond, A Rambaut. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7: 214. doi: 10.1186/1471-2148-7-214.
- ↵RC Edgar. 2004. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
- ↵JJ Emerson, LC Hsieh, HM Sung, TY Wang, CJ Huang, HH Lu, MY Lu, SH Wu, WH Li. 2010. Natural selection on cis and trans regulation in yeasts. Genome Res 20: 826–836.
- ↵L Fernandes, C Rodrigues-Pousada, K Struhl. 1997. Yap, a novel family of eight bZIP proteins in Saccharomyces cerevisiae with distinct biological functions. Mol Cell Biol 17: 6982–6993.
- ↵Y Field, Y Fondufe-Mittendorf, IK Moore, P Mieczkowski, N Kaplan, Y Lubling, JD Lieb, J Widom, E Segal. 2009. Gene expression divergence in yeast is coupled to evolution of DNA-encoded nucleosome organization. Nat Genet 41: 438–445.
- ↵Y Fujii, T Shimizu, T Toda, M Yanagida, T Hakoshima. 2000. Structural basis for the diversity of DNA recognition by bZIP transcription factors. Nat Struct Biol 7: 889–893.
- ↵AP Gasch, AM Moses, DY Chiang, HB Fraser, M Berardini, MB Eisen. 2004. Conservation and evolution of cis-regulatory systems in ascomycete fungi. PLoS Biol 2: e398. doi: 10.1371/journal.pbio.0020398.
- ↵J Gerke, K Lorenz, B Cohen. 2009. Genetic interactions between transcription factors cause natural variation in yeast. Science 323: 498–501.
- ↵CT Harbison, DB Gordon, TI Lee, NJ Rinaldi, KD MacIsaac, TW Danford, NM Hannett, JB Tagne, DB Reynolds, J Yoo, . 2004. Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104.
- ↵GZ Hertz, GD Stormo. 1999. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563–577.
- ↵CT Hittinger, SB Carroll. 2007. Gene duplication and the adaptive evolution of a classic genetic switch. Nature 449: 677–681.
- ↵H Hogues, H Lavoie, A Sellam, M Mangos, T Roemer, E Purisima, A Nantel, M Whiteway. 2008. Transcription factor substitution during the evolution of fungal ribosome regulation. Mol Cell 29: 552–562.
- ↵LJ Holt, BB Tuch, J Villen, AD Johnson, SP Gygi, DO Morgan. 2009. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325: 1682–1686.
- ↵J Ihmels, S Bergmann, M Gerami-Nejad, I Yanai, M McClellan, J Berman, N Barkai. 2005. Rewiring of the yeast transcriptional network through the evolution of motif usage. Science 309: 938–940.
- ↵CA Innis, J Shi, TL Blundell. 2000. Evolutionary trace analysis of TGF-beta and related growth factors: Implications for site-directed mutagenesis. Protein Eng 13: 839–847.
- ↵M Kasowski, F Grubert, C Heffelfinger, M Hariharan, A Asabere, SM Waszak, L Habegger, J Rozowsky, M Shi, AE Urban, . 2010. Variation in transcription factor binding among humans. Science 328: 232–235.
- ↵M Kellis, N Patterson, M Endrizzi, B Birren, ES Lander. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423: 241–254.
- ↵J Kim, K Struhl. 1995. Determinants of half-site spacing preferences that distinguish AP-1 and ATF/CREB bZIP domains. Nucleic Acids Res 23: 2531–2537.
- ↵D Kuo, K Tan, G Zinman, T Ravasi, Z Bar-Joseph, T Ideker. 2010. Evolutionary divergence in the fungal response to fluconazole revealed by soft clustering. Genome Biol 11: R77. doi: 10.1186/gb-2010-22-7-r77.
- ↵CR Landry, PJ Wittkopp, CH Taubes, JM Ranz, AG Clark, DL Hartl. 2005. Compensatory cis-trans evolution and the dysregulation of gene expression in interspecific hybrids of Drosophila. Genetics 171: 1813–1822.
- ↵H Lavoie, H Hogues, J Mallick, A Sellam, A Nantel, M Whiteway. 2010. Evolutionary tinkering with conserved components of a transcriptional regulatory network. PLoS Biol 8: e1000329. doi: 10.1371/journal.pbio.1000329.
- ↵MS Longtine, A McKenzie III, DJ Demarini, NG Shah, A Wach, A Brachat, P Philippsen, JR Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961.
- ↵KD MacIsaac, T Wang, DB Gordon, DK Gifford, GD Stormo, E Fraenkel. 2006. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics 7: 113. doi: 10.1186/1471-2105-7-113.
- ↵SJ Maerkl, SR Quake. 2009. Experimental determination of the evolvability of a transcription factor. Proc Natl Acad Sci 106: 18650–18655.
- ↵S Mahony, A Golden, TJ Smith, PV Benos. 2005. Improved detection of DNA motifs using a self-organized clustering of familial binding profiles. Bioinformatics 21: i283–i291.
- ↵F Pazos, A Valencia. 2008. Protein co-evolution, co-adaptation and interactions. EMBO J 27: 2648–2655.
- ↵G Rigaut, A Shevchenko, B Rutz, M Wilm, M Mann, B Seraphin. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032.
- ↵C Rodrigues-Pousada, RA Menezes, C Pimentel. 2010. The Yap family and its role in stress response. Yeast 27: 245–258.
- ↵VG Sankaran, J Xu, T Ragoczy, GC Ippolito, CR Walkley, SD Maika, Y Fujiwara, M Ito, M Groudine, MA Bender, . 2009. Developmental and species-divergent globin switching are driven by BCL11A. Nature 460: 1093–1097.
- ↵D Schmidt, MD Wilson, B Ballester, PC Schwalie, GD Brown, A Marshall, C Kutter, S Watt, CP Martinez-Jimenez, S Mackay, . 2010. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328: 1036–1040.
- ↵E Shaulian, M Karin. 2002. AP-1 as a regulator of cell life and death. Nat Cell Biol 4: E131–E136.
- ↵GK Smyth. 2005. Limma: Linear models for microarray data. In Bioinformatics and computational biology solutions using R and Bioconductor (ed. VCR Gentleman .), pp. 397–420. Springer, New York.
- ↵A Stark, MF Lin, P Kheradpour, JS Pedersen, L Parts, JW Carlson, MA Crosby, MD Rasmussen, S Roy, AN Deoras, . 2007. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature 450: 219–232.
- ↵JD Storey, R Tibshirani. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci 100: 9440–9445.
- ↵F Storici, LK Lewis, MA Resnick. 2001. In vivo site-directed mutagenesis using oligonucleotides. Nat Biotechnol 19: 773–776.
- ↵M Suckow, CP Hollenberg. 1998. The activation specificities of wild-type and mutant Gcn4p in vivo can be different from the DNA binding specificities of the corresponding bZip peptides in vitro. J Mol Biol 276: 887–902.
- ↵M Suckow, B Kisters-Woike, CP Hollenberg. 1999. A novel feature of DNA recognition: A mutant Gcn4p bZip peptide with dual DNA binding specificities dependent of half-site spacing. J Mol Biol 286: 983–987.
- ↵HM Sung, TY Wang, D Wang, YS Huang, JP Wu, HK Tsai, J Tzeng, CJ Huang, YC Lee, P Yang, . 2009. Roles of trans and cis variation in yeast intraspecies evolution of gene expression. Mol Biol Evol 26: 2533–2538.
- ↵K Tan, H Feizi, C Luo, SH Fan, T Ravasi, TG Ideker. 2008. A systems approach to delineate functions of paralogous transcription factors: Role of the Yap family in the DNA damage response. Proc Natl Acad Sci 105: 2934–2939.
- ↵A Tanay, A Regev, R Shamir. 2005. Conservation and evolvability in regulatory networks: The evolution of ribosomal regulation in yeast. Proc Natl Acad Sci 102: 7203–7208.
- ↵JR Thompson, E Register, J Curotto, M Kurtz, R Kelly. 1998. An improved protocol for the preparation of yeast cells for transformation by electroporation. Yeast 14: 565–571.
- ↵I Tirosh, S Reikhav, AA Levy, N Barkai. 2009. A yeast hybrid provides insight into the evolution of gene expression regulation. Science 324: 659–662.
- ↵AE Tsong, BB Tuch, H Li, AD Johnson. 2006. Evolution of alternative transcriptional circuits with identical logic. Nature 443: 415–420.
- ↵BB Tuch, DJ Galgoczy, AD Hernday, H Li, AD Johnson. 2008. The evolution of combinatorial gene regulation in fungi. PLoS Biol 6: e38. doi: 10.1371/journal.pbio.0060038.
- ↵GP Wagner, VJ Lynch. 2008. The gene regulatory logic of transcription factor evolution. Trends Ecol Evol 23: 377–385.
- ↵I Wapinski, A Pfeffer, N Friedman, A Regev. 2007. Natural history and evolutionary principles of gene duplication in fungi. Nature 449: 54–61.
- ↵I Wapinski, J Pfiffner, C French, A Socha, DA Thompson, A Regev. 2010. Gene duplication and the evolution of ribosomal protein gene regulation in yeast. Proc Natl Acad Sci 107: 5505–5510.
- ↵L Weng, H Dai, Y Zhan, Y He, SB Stepaniants, DE Bassett. 2006. Rosetta error model for gene expression analysis. Bioinformatics 22: 1111–1121.
- ↵MD Wilson, NL Barbosa-Morais, D Schmidt, CM Conboy, L Vanes, VL Tybulewicz, EM Fisher, S Tavare, DT Odom. 2008. Species-specific transcription in mice carrying human chromosome 21. Science 322: 434–438.
- ↵PJ Wittkopp, BK Haerum, AG Clark. 2008. Regulatory changes underlying expression differences within and between Drosophila species. Nat Genet 40: 346–350.
- ↵GA Wray. 2007. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8: 206–216.
- ↵W Zheng, H Zhao, E Mancera, LM Steinmetz, M Snyder. 2010. Genetic analysis of variation in transcription factor binding in yeast. Nature 464: 1187–1191.
- ↵M Zuker. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415.
Notes
[1] [Supplemental material is available online at http://www.genome.org. The sequence data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE15818.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.111765.110.