
Abstract
Promising new sequencing technologies, based on sequencing-by-synthesis (SBS), are starting to deliver large amounts of DNA sequence at very low cost. Polymorphism detection is a key application. We describe general methods for improved quality scores and accurate automated polymorphism detection, and apply them to data from the Roche (454) Genome Sequencer 20. We assess our methods using known-truth data sets, which is critical to the validity of the assessments. We developed informative, base-by-base error predictors for this sequencer and used a variant of the phred binning algorithm to combine them into a single empirically derived quality score. These quality scores are more useful than those produced by the system software: They both better predict actual error rates and identify many more high-quality bases. We developed a SNP detection method, with variants for low coverage, high coverage, and PCR amplicon applications, and evaluated it on known-truth data sets. We demonstrate good specificity in single reads, and excellent specificity (no false positives in 215 kb of genome) in high-coverage data.
Although the cost of DNA sequencing by Sanger chemistry has dropped dramatically over the past decade, this cost is still too high for many important research projects to be practical. Several new methods based on sequencing-by-synthesis (SBS) promise to yield large amounts of DNA sequence at dramatically lower cost and thus open new areas to research. However, current SBS reads are much shorter than Sanger chemistry reads (e.g., from the ABI 3730) and are of lower quality per base.
Indeed, SBS data differ fundamentally from the now-familiar data produced using Sanger sequencing chemistry, and these differences significantly impact many applications of the data. For example, the 454 system (Margulies et al. 2005) does not read individual bases directly. Rather, it reads the lengths of homopolymer runs: the number of As, Cs, Gs, or Ts at the current position. As a consequence, the typical read errors are overcalls and undercalls (insertions or deletions of bases from the sequence), in contrast to the miscall errors typical of Sanger chemistry sequencing. As another example, the Illumina/Solexa system produces data in four color channels, one for each base. The intensity of each color reflects the proportion of molecules incorporating that base. Miscalls are the most common errors. The very short reads it currently produces (25–50 b) require special tools. In general, new SBS systems are unlikely to produce data that can function as drop-in replacements for Sanger chemistry reads.
A common language is essential to compare results from different systems and to make sensible decisions about which sequencing method is suitable to each application. This problem has been solved for Sanger chemistry sequencing: Reliable, validated base quality scores (also known as Q scores) provide a standard with which to compare data. Originating with the phred program (Ewing and Green 1998; Ewing et al. 1998), they are virtually universally used and have become a critical tool for comparing results. Other basecallers such as LifeTrace (Walther et al. 2001) and KB (Applied Biosystems, Inc. 2004) also produce phred-like quality scores. The quality scores compress a variety of types of information about the quality of basecalls into a readily usable probability-of-error value. Many analysis tools and virtually all assemblers require quality score input to deliver accurate results. To date, the vendor-generated quality scores for available SBS systems have fallen short as substitutes for phred quality scores; for instance, 454 quality scores by design do not address the probability of undercall errors (Margulies et al. 2005). Accurate quality scores with properties similar to those generated by phred are essential to provide a simple uniform foundation on which to build all applications that are concerned with read quality. In particular, they will: (1) facilitate the creation of meaningful quality scores for assemblies; (2) support sensitive and specific polymorphism detection; (3) enable accurate statistical modeling of the significance of read alignments; (4) provide a quantitative basis for comparison of sequencing results from different technologies or laboratories; and (5) provide a quantitative foundation for the joint use of sequence from SBS technologies and Sanger chemistry methods.
Polymorphism detection will be a key application of the new SBS technologies but will present new problems and require new solutions. Because of the high volume of data, the data analysis steps must be fully automated. There are several key considerations in the design of polymorphism detection algorithms and experiments. First, individual bases are currently of lower quality than are typical Sanger chemistry bases, which means it is difficult to obtain high specificity in detecting polymorphisms. Second, since these technologies sequence hundreds of thousands to millions of reads from one sample, in many cases samples must be pooled for efficiency. Such pooling inevitably leads to concentration differences between the different samples, affecting the sensitivity of the assay. Finally, PCR is typically used to select regions of interest for targeted sequencing, but this can lead both to bias (because some regions are amplified more readily than others) and also to miscalls due to polymerase errors. This last problem is more serious for SBS technologies: consider a PCR process that yields 99% perfect copies of the original template. In sequencing of a PCR product by Sanger chemistry, all molecules are sequenced together, so the result is an average of the signal for each molecule: that is, the 1% error merely adds a small amount of noise. On the other hand, each SBS read is derived from a single DNA molecule, so that the polymerase error rate has a direct impact on the overall error rate of the system: Even absent any sequencing error, 1% of the molecules will be read incorrectly.
In this report, we address these issues and give examples of successful applications of these new technologies in automated polymorphism detection. We define more accurate quality scores and apply them to help overcome the challenges described above. The software described here is publicly available (Supplemental materials). Because the 454 platform was the first SBS technology available, our data are from that source.
Quality score generation
In order to develop quality scores for SBS data, it is essential to understand the common error patterns in the data, which arise from the idiosyncrasies of each individual SBS platform. Once these are understood, measures can be developed that capture predictors of possible errors. Then the predictors can be combined to yield a single accurate probability of error that can be expressed as a quality score.
To combine the error predictors that we developed, we used the same algorithm first used in phred for Sanger chemistry data (Ewing and Green 1998). Applying this algorithm to large training data sets (see Supplemental Table 1), for which the true sequence of the DNA is known, yields a table that summarizes the expected quality for bases with different combinations of the error predictors. This table can then be readily applied to new sequencing runs to produce quality scores.
Error predictors for 454 data
Processed data from the 454 platform present as a flowgram: a series of intensity values for successive reagent “flows.” During each flow, the incorporation of zero, one, or more instances of a single base is possible. Each flow corresponds to one of the four bases, and they repeat in a predetermined order called a flow-cycle. The signal intensity for a flow is rounded to an integer to give the number of monomers of the corresponding base that were incorporated. For example, the flowgram (T:1.1, A:0.1, C:0.9, G:0.1, T:1.6, A:0.0, C:0.4, G:1.0) would correspond after rounding to the sequence TCTTG. A read error occurs whenever the signal intensity is more than 0.5 from the true value: for example, interpreting 1.6 as a 2 when there was only one base, or 0.4 as a 0 when there really was a base at that position. Therefore most errors are overcalls (65%–75% of read errors) or undercalls (20%–30%). Miscalls are much rarer (∼5% of errors) and are typically due to undercall/overcall pairs (e.g., the flowgram above might be a miscall of the true sequence TCTCG due to an overcalled T and undercalled C).
The quality score assigned to a base by the 454 software represents the probability that the base is an overcall, given the observed signal intensity for the corresponding flow; it is computed from the signal distributions observed in the run (Margulies et al. 2005). However, noise in this system comes from a variety of sources, many of which vary within the run: optical and chemical noise, multiple templates on a bead, signal contamination from nearby wells, and loss of synchrony between the ∼107 copies of the template that are on each bead (Margulies et al. 2005). To explain this last point: In any flow, some small fraction of DNA strands will fail to incorporate the appropriate base (e.g., in an A flow, incorporating two As instead of three). Others may incorporate too many bases (e.g., in an A flow incorporating a stray T nucleotide). As a result, signal intensity is transferred from one flow to another, typically to the previous or the next flow of the same base; loss of synchrony accumulates throughout the read.
To capture these diverse sources of sequencing error, we devised six noise predictors as input to our quality-scoring algorithm. While the algorithm relies on multidimensional combinations of predictor thresholds to achieve its accuracy, it is possible to roughly rank the predictors by importance. The following list is from most to least important:
Observed noise in the neighborhood of a given flow
Observed noise in the whole read
Observed noise at a given flow
Homopolymer count: having more bases to incorporate in a flow yields more errors
Homopolymer count for the same flow in the previous flow-cycle; a long base run induces errors via synchrony loss and partial transfer of its strong signal to subsequent flows
Position on read: later in read yields more errors
In contrast, the 454 quality-score algorithm has less power because it uses only the homopolymer count and noise level at a given flow. In addition, it assigns scores that decline steadily across a homopolymer run, whereas to maintain consistency with prior scoring methods, our algorithm generally assigns the same score to all bases in a run.
We used extensive known-truth 454 GS20 sequence data to assess the effectiveness of our algorithms for SNP calling and quality scoring.
Results
Accuracy, usefulness, and consistency of quality scores
We produced a quality score table as described above by training on four data sets of 454 data from four genomes, with a total of 58 Mb (Supplemental Table 1). We then used the table to generate quality scores for 13 additional test data sets from five genomes, with a total of 200 Mb (Supplemental Table 2), and compared the resulting quality scores to those produced by the 1.0.52 version of the 454 software. For simplicity, we will refer to the former as “new quality scores” and the latter as “old quality scores.” The genomes used all had pre-existing high-quality references (either finished genomes or the high-quality portions of draft genomes), which is critical for comparing quality scores.
To measure quality, we selected reads with a unique alignment having at least 80% identity. For each predicted quality score, we tabulated the total number of bases and the number of errors, charging undercall errors to the neighboring base with the lower quality score. This computation gave us an error rate for each predicted quality score (5 through 35), or equivalently an observed quality score (Q = −10 log10[error rate]). Figure 1A shows the relationship between the predicted and observed quality scores aggregated across the 13 data sets. The new scores clearly better predict actual quality than do the old scores.
New quality scores for 454 reads. Old: quality scores from 454 software v.1.0.52. New: quality scores developed for this work. Data for panels A, B, and C come from 13 different runs on DNA from five different species. (A) Predicted vs. observed quality for old and new quality scores. New quality scores are much closer to the ideal, 1:1 line. (B) Proportion of bases greater than a given actual quality. The new quality scores accurately identify many more bases at quality ≥30 (63% vs. 23%). (C) Error prediction by error type. New quality scores accurately predict different types of errors: predicted vs. actual quality when errors are separated into overcalls, undercalls, and miscalls. (D) New quality scores are stable across machines. Predicted vs. actual quality for three runs of the same human BAC library on three different machines. (E) New quality scores are stable across genomes. Predicted vs. actual quality for five genomes varying in GC content and proportion of bases in homopolymers.
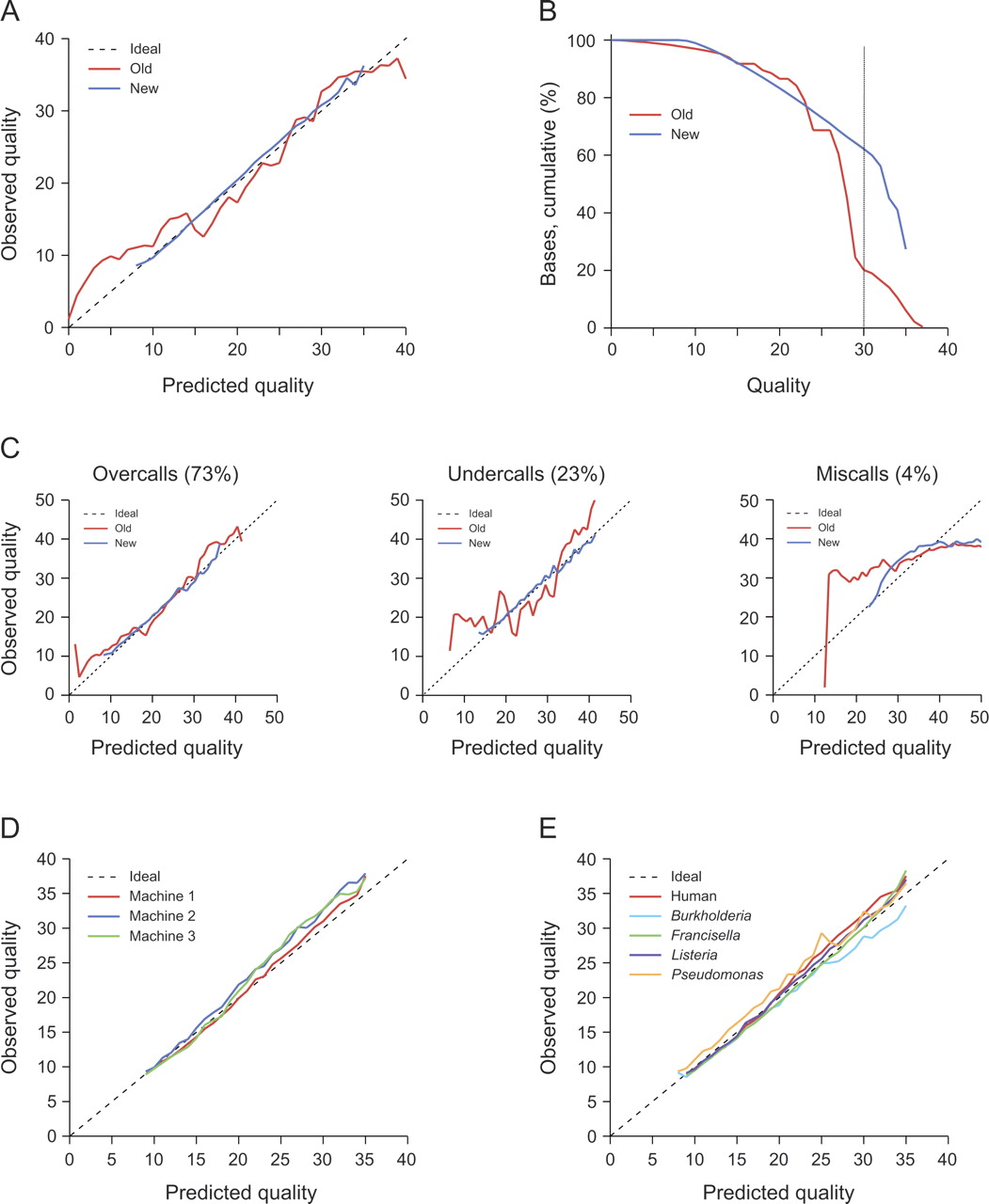
An effective measure of the predictive ability of quality scores is the R2 with respect to the ideal relationship of identity (y = x). This measure, R2i, shows a strong difference between the new quality scores (R2i = 0.99) and the old ones (R2i = 0.94).
An effective measure of the usefulness of predictive quality scores is their success at identifying high-quality bases (Ewing and Green 1998). The new quality scores correctly identify many more bases at Q30 or higher than do the old ones, 61% versus 23% (Fig. 1B).
The new quality scores are designed to treat overcalls, undercalls, and miscalls evenhandedly. As Figure 1C shows, they provide similarly accurate information about each, in contrast to the old scores. As expected, the improvement is particularly striking for undercalls and miscalls, because the old quality scores were only designed to reflect the probability of an overcall (Margulies et al. 2005).
It is also important to evaluate to what extent the new quality scores are stable across machines. This stability would minimize training requirements by allowing a single quality table to serve for many machines. For three runs produced on three different machines at the Broad Institute, with the same libraries of human BACs, the actual quality was consistently slightly better than prediction: R2i = 0.99, 0.95, 0.96 (Fig. 1D).
Finally, a key question is whether the quality scores provide similarly reliable information for genomes with different characteristics. We evaluated the quality scores on five genomes having diverse GC and homopolymer content, finding similar levels of predictive power: R2i = 0.96–0.99 (Table 1; Fig. 1E).
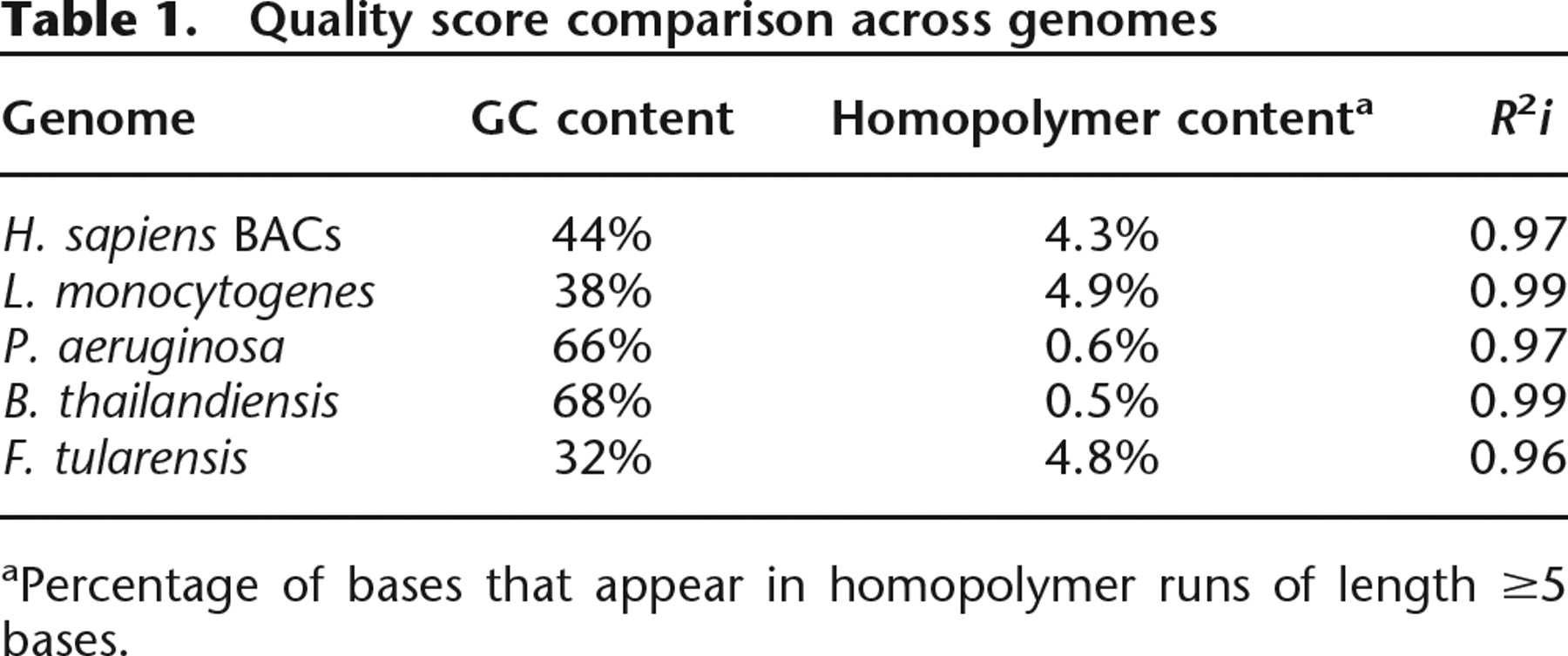
aPercentage of bases that appear in homopolymer runs of length ≥5 bases.
Detection of single-nucleotide polymorphisms
We developed a method for automated single-nucleotide polymorphism (SNP) detection making use of our new quality scores. In outline, the method is straightforward:
Align reads to reference
Accept unambiguous reads with adequate identity
Select Neighborhood Quality Standard (NQS) windows (Altshuler et al. 2000; International SNP Map Working Group 2001) within accepted reads
Call SNPs when sufficient evidence is found
We evaluated the method for three SNP discovery contexts: low-coverage genomic data, high-coverage genomic data, and PCR amplicon data. In these different contexts, we used different standards (described below) for the SNP evidence, but otherwise we used a consistent method. In more detail: (1) Alignment was done with QueryLookupTable (Methods). (2) We ignored alignments having <80% identity. We scored alignments by adding indels plus mismatches, and declared a read ambiguous if the score for its best alignment exceeded one-fourth of the score of the second-best. (3) We required 11-base NQS 20/15, i.e., quality 20 at the central base, and a window of five bases on each side with quality 15. We allowed at most two mismatches and zero indels in the window.
In each context, we evaluated the method using reads from genomes of known sequence, which is vital for an unambiguous determination of specificity.
Low coverage SNP discovery with single reads
One important way to generate a low-cost sample of SNPs from an entire genome is to call SNPs from very low coverage reads (e.g., 0.01×). In this context our method calls all SNPs found in an NQS window in an accepted read. To assess SNP calling using single 454 reads, we chose a set of six finished human BACs as a practically sized, thoroughly known target. We generated three mutated references for each BAC by applying sets of synthetic SNPs randomly selected from SNPs found in the human reference regions corresponding to these BACs (Methods). We separately analyzed five data sets (Methods; Supplemental Table 3), calling SNPs against each of the three references. Thus we carried out 15 experiments, each of which had ∼70,000 reads. On average, for each read landing on a SNP, the sensitivity (probability of reporting the SNP) was 60%–73%. There were 55–72 false positives per million bases (Mb) accepted for SNP calling. For context, the SNP rate in humans is estimated at ∼1000 per Mb of genome (International HapMap Consortium 2003), and the false-positive rate of an analogous SNP-discovery method using Sanger chemistry-based whole-genome shotgun reads is ∼30 per accepted Mb (International SNP Map Working Group 2001).
To estimate the benefit from using quality information, we compared SNP calling without quality filtering (NQS 0/0). This gave sensitivity of 85%–93% but a false-positive rate of 97–233/Mb called, showing that using quality information both improves specificity and makes it more consistent.
In contrast, higher-quality bases (NQS 30/30) did not significantly improve on the false-positive rate of ∼65/Mb. This result is consistent with Figure 1C, which shows in a different data set that the highest quality bases (measured by either old or new quality scores) show miscall errors at ∼50/Mb.
High coverage genomic SNP discovery
We set out to do comprehensive SNP calling with very high specificity (<10 false positives/Mb) using high coverage sequence reads. In this context, we required the evidence for a SNP to (1) include reads in both directions and (2) display frequency above a certain threshold among all SNP-calling-accepted bases at that position. The thresholds were arbitrarily set at 66% for haploid and 10% for diploid genomes, but the results are reasonably robust to the choice of threshold: between 60% and 70% for haploid and between 5% and 15% for diploid produce virtually the same results.
To test haploid SNP discovery, we used the finished human BAC reads described above, which are a high coverage shotgun data set. In this case we divided the accepted reads into groups for each of the six BAC targets, using the same criteria for read acceptance as before.
To test diploid SNP discovery, we used the fact that the BACs were chosen in overlapping pairs. Thus, reads from the overlap regions can be used to simulate shotgun reads from a diploid genome containing natural heterozygous SNPs (Methods).
In both cases, we simulated various levels of coverage (Methods) by randomly sampling the reads. For each target, the coverage was measured as the ratio of the number of SNP-calling-accepted bases for that target to the number of bases in the target.
Haploid genomic results
We assessed SNP calling in this case by calling SNPs against the same three mutated references (total size, 544 kb). At all tested coverage levels (Methods), this algorithm produced no false positives. (A 95% confidence interval is at most eight false positives per Mb; i.e., for false-positive rates above 8/Mb, the likelihood of observing zero false positives is <5%.)
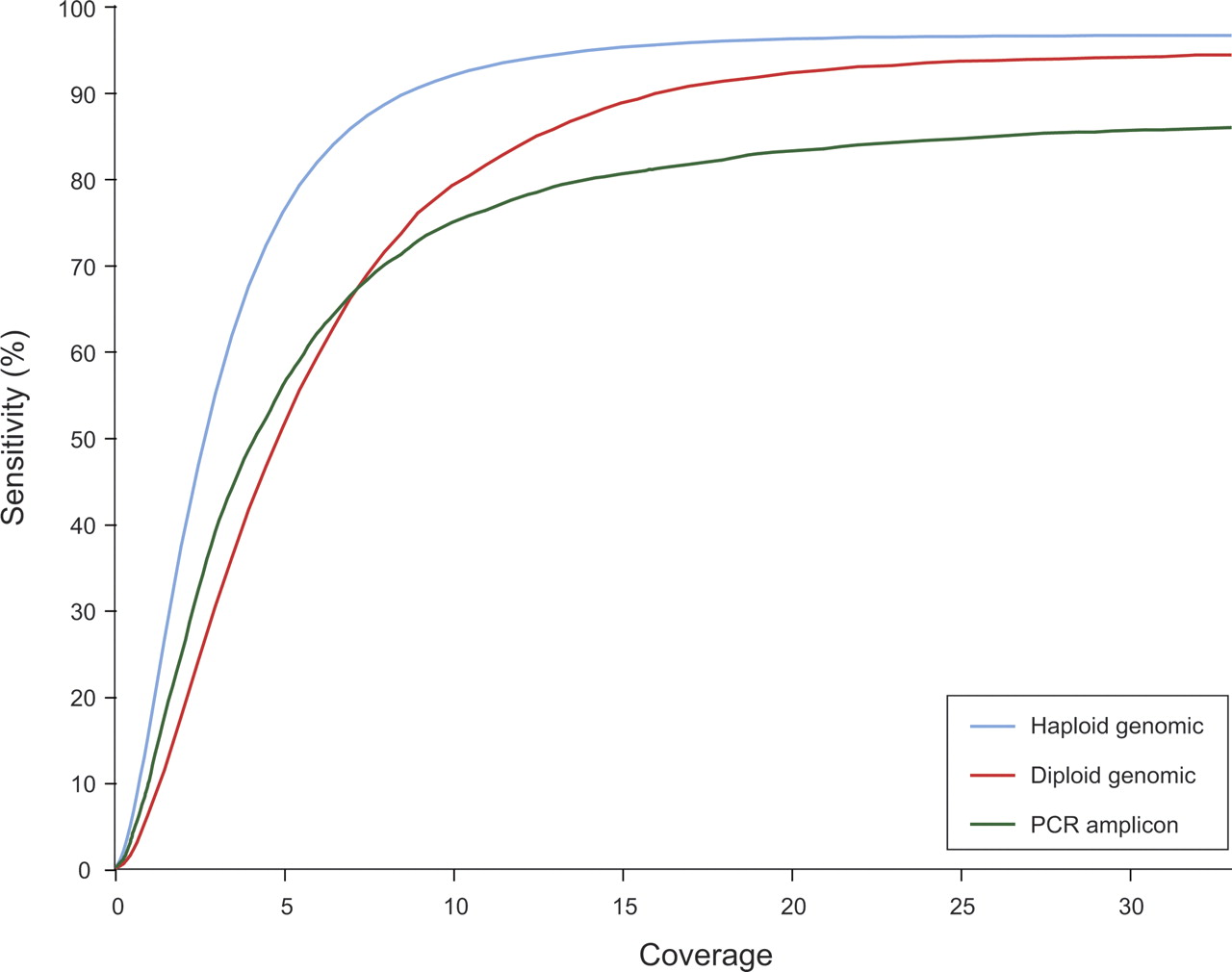
As shown in Figure 2, at 20× coverage, we observe 94% sensitivity with little gain at higher coverage. Of the missing 6% of SNPs, ∼75% are lost due to systematic sequence-dependent variation in the read quality. For example, the predicted quality is lower near long homopolymers. Repeat regions and coverage variation account for the remaining ∼25% of losses. It is important to note that the loss-of-sensitivity effects of these three factors depend on sequence context, so the impact of each will vary greatly between different genomes. We observed a sensitivity range (at 20×) from 90% to 97% for these BACs.
Sensitivity of SNP calling as a function of coverage. Coverage is counted by bases accepted for SNP calling. At all coverages, the fraction of the reference that could be correctly called in haploid DNA (a BAC) exceeds the sensitivity for heterozygous SNPs that can be called in diploid DNA (a mixture of two different, overlapping BACs). Sensitivity is generally lower for heterozygous SNPs in PCR amplicons, due to pooling variation. No false positives were found at any coverage level in genomic data: ∼545 kb haploid, ∼220 kb diploid. For PCR amplicons approximately one false positive was found in ∼27 kb.
Diploid genomic results
We assessed discovery of heterozygous SNPs in diploid genomes by applying this method to a computationally simulated mixture of reads from overlapping pairs of human BACs. In total, the three BAC pairs tested contained 222 heterozygous positions in the ∼215 kb of overlap. Again we found no false positives in the test region at any coverage level. As shown in Figure 2, at 20× diploid coverage (i.e., an average of 10× per allele, stochastically varying) we find ∼93% sensitivity for heterozygous SNPs. We found sensitivity of 84%, 92%, and 98% for the three overlap regions, confirming the expectation that sensitivity depends significantly on the sequence context of SNP placement in the target genome. Higher coverage gives somewhat better results, e.g., at 30× we found 88% sensitivity for the most difficult region. About half of the ∼7% of SNPs not found at 20× are in difficult-to-align regions (repetitive or highly polymorphic), while most of the rest are lost to coverage variation. The systematic loss of coverage quality described above made little contribution for this set of SNPs.
Note that the 215 kb of overlap regions serves as the simulated genome for this test. Sensitivity depends, among other factors, on the repeat content of the genome. As an example, we also analyzed these regions in the context of the entire genome (removing reads ambiguous in that context). For two of the three regions, sensitivity at 20× coverage was unchanged, but for the most difficult one, sensitivity dropped from 84% to 74%.
SNP discovery in high coverage PCR amplicons
The cost of a project can be reduced by sequencing targeted regions of the genome in PCR amplicons rather than the entire genomic DNA. To evaluate the effectiveness of our method on 454 reads from PCR amplicons, we designed PCR amplicons to cover known heterozygous SNPs in human DNA. We sequenced 208 SNPs in 27 kb of non-overlapping amplicons from two human individuals (Methods), pooling all amplicons for sequencing. Inevitable variation in concentration of the pooled amplicons caused very significant loss of efficiency: Even after careful normalization, we found that the best-covered 80% of amplicons spanned more than a threefold range of coverage.
In this case, therefore, for realism, we analyzed all amplicons together, defining the coverage as the ratio of SNP-calling-accepted bases in accepted reads to the total number of bases in all amplicons. As before, we randomly selected appropriate numbers of reads to simulate different levels of coverage. Because we observed a high error rate in the 454 reads, we also adjusted SNP calling parameters to reject noisy reads (Methods).
With these adjustments, we still found a higher false-positive rate than we had expected from genomic data, an average of one false positive in 27 kb at 20× coverage. We replicated PCR and sequencing, and found a similar false-positive rate. However, no false positives were found when we required that SNP calls be made in both replications, indicating that these errors likely arise as PCR errors.
At 20× coverage, as shown in Figure 2, our method gave sensitivity of ∼83% in each of the independent replications. The missing 17% of SNPs arose largely from uneven pooling. The sensitivity was ∼77% when we required that SNP calls be made in both replications.
Discussion
In this article, we have described methods for quality scoring and SNP detection for SBS technology, and a series of controlled experiments that provide an accurate assessment of both methods in the context of 454 GS20 sequence data.
Quality scores
The new quality scores for 454 sequence data presented here provide several key advantages over existing ones:
They more accurately reflect the true error rate.
They accurately predict undercalls, which is critical, given that these comprise ∼30% of the errors.
They identify many more high-quality bases: ∼60% of the bases are accurately classified as Q30 (or one error in 1000) in an average run, and up to 25% can be identified as Q35.
They identify a large proportion of the bases as being in high-quality neighborhoods: More than 75% of the bases fall into a 20/15 neighborhood, compared with only ∼41% using the old scores, and 41% are in 30/30 neighborhoods, versus 0% with the old scores. We showed that these high-quality neighborhoods are very reliable for polymorphism calling.
The construction of quality scores described in this work should apply to any SBS system, with the particular SBS system affecting only the choice of predictors. While there is no formula for choosing these predictors, they can generally be arrived at through careful examination of the data and systematic testing of candidate predictors. To be effective, predictors must capture diverse aspects of the system operation. For the 454 GS20 system, we found that the most effective predictors relied on the flowgram data, as they capture the varying levels of system noise. Generally, we expect that predictors based on “raw” data will yield the best results.
SBS technologies are frequently updated, so quality scoring will need to be periodically recalibrated in order to keep accuracy high. This will involve both generation of new data from genomes of known sequence and revisiting of the choice of predictors. Fortunately, as sequencing costs drop, so does the cost of this operation.
SNP discovery
We have demonstrated an effective method for highly specific SNP discovery, appropriate for any sequencing system that has suitable quality scores. With high coverage by 454 GS20 reads, whose average quality is lower than Sanger chemistry reads, it achieves exceptional specificity: no false positives in ∼215 kb of diploid sequence at 20× coverage. With low coverage the performance depends on the read quality in NQS windows, but it is still far better than the individual base qualities; e.g., in 454 GS20 reads, we find ∼65 false positives/Mb called in NQS 20/15 windows.
Sensitivity is of course dependent upon coverage. With 20× coverage by accepted bases, we find sensitivity above 90% in diploid shotgun data. The sensitivity obtained in a particular application will depend on the character of the data set: how the chosen sequencing technology interacts with the target genomic sequence (e.g., the nature of the repeats present), and the evenness of coverage available (e.g., the accuracy of pooling of PCR products).
In conclusion, we used controlled experiments to demonstrate a general framework for defining quality scores that incorporate diverse types of information about SBS data. The resulting highly accurate quality scores provide a rigorous quantitative framework for assessing data quality. This should enable applications such as polymorphism detection to be done with high accuracy, helping the new sequencing technologies fulfill their promise.
Methods
Quality scores
Training and testing data
The training data (Supplemental Table 1) consisted of four regions (each being half of a 60 × 60 mm 454 plate) collected from three different machines in our laboratory over a period of 14 mo, for a total of 58 Mb. They included data from different species with different genome compositions: a mixture of three human BACs, Burkholderia thailandensis strain E264, Francisella tularensis strain 257, and Listeria monocytogenes strain 10403s.
Test sets included 13 regions and also came from all three machines and from the same four species, with the addition of Pseudomonas aeruginosa and other strains of L. monocytogenes, for a total of 200 Mb. All reads used for training and testing are available for download from the NCBI trace server (see Supplemental Tables 1, 2).
Error predictors
Many different error predictors were initially evaluated. We describe in detail here those that produced the best combination of results and were adopted into the final version of the quality score generation. The “current base” refers to the base for which a quality score is being generated. The predictors are ordered by (decreasing) importance.
1 and 3. Local noise: A measure of the noise seen in a flow is abs[flow − round(flow)]. Importantly, this measure is informative about possible undercalls (e.g., a flow value of 0.45, which rounds to 0). We used two predictors based on this measure. Predictor 3 is this measure for the current base’s flow. Twenty bins were used for this predictor. Predictor 1 is the maximum of the measure in a radius of 10 flows around the current base’s flow. Twelve bins were used for this predictor.
Read noise: A measure of the overall reliability of basecalls in the read was obtained as follows. All calls of 0 were grouped together, and all calls of 1 were grouped together. The mean and standard deviation of the height of the flow for each group (m0 and s0, m1 and s1) were calculated. Reads for which these distributions had greater overlap were likely to have more errors. The predictor used is −(m1 − m0)/(s0 + s1). This predictor increases with the amount of overlap and therefore with the probability of error in the read. Sixteen bins were used for this measure.
Homopolymer count: The number of consecutive bases that are identical to the current base, including itself. Six bins were used for this predictor.
Incomplete extension: the number of bases identical to the current base in the previous flow cycle. For example, if the sequence were GAAAAAATA, where the final A is current, the value of this predictor would be six. The current base is likely to have additional loss-of-synchrony noise because some of the DNA strands will not have completed the incorporation of all six As. Seven bins were used for this predictor.
Position on read: abs(40 − current base position). Read quality is slightly worse at the beginning of the read, improves up to base 30–40, and then declines all the way to the end of the read. Thus observed quality is, approximately, a monotonic function of the absolute value of (40 − base position). Fifteen bins were used for this predictor.
Alignment and read selection
The accuracy of the quality scores depends on correct training data; in particular, it is critical to avoid misplaced or misaligned reads. We used the QueryLookupTable aligner developed for ARACHNE (Jaffe et al. 2003). The wrapper script evalfastaNum.pl records the parameter settings we developed for 454 reads (Supplemental materials). This aligner performs a multipass heuristic alignment based on k-mer seeds. We optimized the alignments found using a banded Smith-Waterman alignment with the cost of an indel (9) less than that of a mismatch (17). (Matches were treated as 0 cost.)
We rejected alignments with <80% identity and required that reads used for training and testing the quality scores have only one accepted alignment to the reference. In addition, for genomes for which finished reference was not available, we only used reads whose best alignments were completely contained within a high-quality (NQS 40/40) part of a draft assembly that did not rely upon 454 data. Finally, aligners are less consistent at the ends of reads because there are many different ways to assign an error if there is little context to one side of it. Therefore, we required that there be at least three bases with no errors at each end of the read. If that condition was not met, we trimmed the read from each side, until the condition was met.
This selection process results in elimination of the worst reads from both the training and testing sets. Thus, our results overestimate the average quality of the 454 data. However, the proportion of reads rejected for poor alignments was only 3%, so this overestimate is not serious. So the key points about accuracy of the quality scores are not affected.
Binning and quality scoring
To generate the training data, each base used was marked correct or incorrect based on the read alignment; undercall errors were charged to one of the neighboring bases at random. Each of the six predictor values were binned into six to 20 bins; each base, together with its correct/incorrect status, was assigned to one bin in the resulting six-dimensional space containing ∼2.4 million bins. To infer quality scores from these data without excessive computation or over-fitting, we used a version of the algorithm developed for phred (Ewing and Green 1998) to reduce this space to ∼1000 non-overlapping sets of bins, relying on approximate monotonicity of the individual predictors. Each set is defined by a single bin. The quality table consists of a list of defining bins in priority order, along with their observed qualities. The observed quality is −10log10(estimated error rate), rounded to the nearest integer, where the estimated error rate for a set of bins with w wrong and c correct bases is (1 + w)/(1 + w + c). Because the phred algorithm, as described in Ewing and Green (1998), requires compute time quadratic in the number of bins, we developed an equivalent algorithm that works in linear time.
The quality table was used to assign quality scores to bases by identifying the highest-priority bin in the table for which the base qualifies and assigning the corresponding quality. A base qualifies for a bin in the table if all of its predictor values are at least as high as those of the bin. Scores beyond the limits of the training data were assigned to the closest bin. In order to accelerate the quality scoring, we changed the way quality scores were looked up in the table. Rather than looking through the whole table for each base as described in Ewing and Green (1998), which takes time proportional to (number of bases) × (number of lines in the table), we precomputed the quality values for all ∼2.4 m bins, allowing quality scoring in time proportional to (number of bases).
Evaluating quality scores
To evaluate quality scores on the testing data, we used the same read alignment methods but assigned undercall errors to the neighboring base with the lowest quality. When evaluating the old quality scores we used only the forward-aligning reads, because in that orientation our aligner places gaps at the ends of homopolymer runs, respecting the directionality of the old quality scores. To create Figure 1C, we recomputed observed quality in the testing data, penalizing one type of error at a time, and counting the other errors as correct. We adjusted quality score predictions according to the prevalence of each type of error in the aggregate data. For example, to estimate the prediction for miscall errors alone, which are 4% of all errors, we added −10log10(0.04) = 14 to the predicted qualities.
SNP detection
Alignment and read selection
The alignments were generated and filtered as described above. For SNP calling, we relaxed the uniqueness criterion, to allow for calls in imperfect repeats. We allowed reads with more than one alignment provided that the second best alignment had at least four times more errors than the best (counting 0 errors as 1 for this purpose).
To determine accepted bases for SNP calling, we used the NQS criterion (Results; Altshuler et al. 2000), with one variation. In the standard implementation, the five bases at each end of the read cannot pass NQS. For a 100-base read, this would represent a loss of 10% of the bases. Therefore, we used these end bases but continued to trim to a three-base perfect match at each end of the read.
Sensitivity and specificity were evaluated at 0× to 33× coverage; specifically, 0–1× in 0.1× increments, 1–16× in 0.5× increments, and 16–33× in 1× increments. Reads were selected at random, with probability (target coverage)/(available coverage).
Single read data
We used 36 Mb of sequence data: five regions from three 454 runs (Supplemental Table 3), each region containing one of two pools of non-overlapping human BACs. Pool A contained BACs AC005865, AC018698, AC027763; Pool B, BACs AC005912, AC090531, AC040977. The BAC pools were chosen so that each BAC in pool A overlaps with exactly one BAC in pool B, and the BAC pairs were selected for a higher-than-average SNP density in the overlaps. We created mutated references for assessing SNP-calling sensitivity using the UCSC snp126 database (Kuhn et al. 2007). We selected SNPs at random from those whose flanking sequences exactly matched the corresponding BAC sequence.
Bidirectional read haploid data
The data used were the pool-B reads from the single-read case, 23 Mb of sequence. Unambiguous reads were assigned to the BAC to which they aligned, and the available coverage was computed separately for each BAC to eliminate effects of uneven pooling. For each desired coverage level, 20 subsets of the reads were selected independently at random. The reported fraction of reference callable is the weighted (by number of bases) mean value observed in the 20 subsets for each of the three BACs.
Bidirectional read diploid genomic data
In this case, we computed accepted reads with the same criteria but aligned against an expanded set of nine targets: the non-overlapping part of each of the six BACs together with the pool-A sequence of each of the three overlap regions. The analysis was performed on the ∼120 kb of overlap region. Based on complete alignments of the finished sequences of the BAC pairs, the overlap region contains 222 positions at which the A and B sequences differ by a substitution whose 11-base neighborhood contains at most one other substitution and no indels. The data used were the same runs used in the single-read case, with the addition of the BAC pool-A region used for training the quality scores; because the pool-A reads carry no helpful information in this experimental design, no unfair benefit is obtained from reusing the training data. Subsampling to obtain reduced coverage levels was performed as for the haploid genomic data, except that the reads were selected at random from the two pools with equal probabilities to simulate diploid data.
Bidirectional read diploid PCR data
Two individual humans from the ENCODE set (NA18558 and NA18564) were chosen arbitrarily, and a random subset was selected of heterozygous positions for those individuals according to the ENCODE release r19-2005-10-24 (nonredundant) whose SNPs were in dbSNP release 123 (b34) and were bi-allelic according to that dbSNP release. Positions that lay in RepeatMasker-tagged regions (per the UCSC genome browser) were removed, as were positions at which primer design for 100 base amplicons failed. We used read filtering, SNP calling criteria and subsampling as in the diploid genomic case. To reduce the impact of PCR-introduced mutations, we required 95% identity for the best read alignment and ignored SNP calls in the five bases at each end of each read. We also ignored SNP calls in primers or in a base adjacent to a primer, and rejected a single primer pair that proved to amplify two genomic regions.
Acknowledgments
We thank Mike Zody for insightful comments on the manuscript; the Broad Institute Sequencing platform for sequence generation and processing, in particular the Technology Development and Sequencing Informatics groups; and Jim Knight of Roche/454 Life Sciences and Klaus Maisinger of Illumina/Solexa for technical discussions. This research was supported by grants from the National Human Genome Research Institute and the National Institute of Allergy and Infectious Diseases.
References
- ↵D. Altshuler,V. Pollara,C. Cowles,W. Van Etten,J. Baldwin,L. Linton,E. Lander,(2000) An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 407:513–516.
- ↵Applied Biosystems, Inc. (2004) User Bulletin. FAQ, KB Basecaller 1.2. docs.appliedbiosystems.com/pebiodocs/04362968.pdf.
- ↵B. Ewing,P. Green,(1998) Basecalling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8:186–194.
- B. Ewing,L. Hillier,M. Wendl,P. Green,(1998) Basecalling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8:175–185.
- ↵International HapMap Consortium (2003) The International HapMap Project. Nature 426:789–796.
- ↵International SNP Map Working Group (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933.
- ↵D.B. Jaffe,J. Butler,S. Gnerre,E. Mauceli,K. Lindblad-Toh,J.P. Mesirov,M.C. Zody,E.S. Lander,(2003) Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 13:91–96.
- ↵R.M. Kuhn,D. Karolchik,A.S. Zweig,H. Trumbower,D.J. Thomas,A. Thakkapallayil,C.W. Sugnet,M. Stanke,K.E. Smith,A. Siepel,(2007) The UCSC genome browser database: Update 2007. Nucleic Acids Res. 35:D668–D673.
- ↵M. Margulies,M. Egholm,W.E. Altman,S. Attiya,J.S. Bader,L.A. Bemben,J. Berka,M.S. Braverman,Y.J. Chen,Z. Chen,(2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380.
- ↵D. Walther,G. Bartha,M. Morris,(2001) Basecalling with LifeTrace. Genome Res. 11:875–888.
Notes
[1] [Supplemental material is available online at www.genome.org.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.070227.107.