
Abstract
We describe a general mass spectrometry-based approach for gene annotation of any organism and demonstrate its effectiveness using the nematode Caenorhabditis elegans. We detected 6779 C. elegans proteins (67,047 peptides), including 384 that, although annotated in WormBase WS150, lacked cDNA or other prior experimental support. We also identified 429 new coding sequences that were unannotated in WS150. Nearly half (192/429) of the new coding sequences were confirmed with RT-PCR data. Thirty-three (∼8%) of the new coding sequences had been predicted to be pseudogenes, 151 (∼35%) reveal apparent errors in gene models, and 245 (57%) appear to be novel genes. In addition, we verified 6010 exon–exon splice junctions within existing WormBase gene models. Our work confirms that mass spectrometry is a powerful experimental tool for annotating sequenced genomes. In addition, the collection of identified peptides should facilitate future proteomics experiments targeted at specific proteins of interest.
The robustness and speed of genome sequencing technology have resulted in an explosion of large-scale sequencing efforts, which as of January 2008 had produced over 700 completed genomes, with an additional 2820 in progress (Genomes OnLine Database v 2.0, http://genomesonline.org). With completed genomes comes the challenge of understanding how the information stored in the DNA sequence leads to the form and function of the organism. A key requirement in understanding the functional elements of the genome is accurate annotation of protein-coding genes. Most gene structures in newly sequenced organisms are based on computational predictions, often unsupported by experimental evidence; when available, experimental validation is usually based on cDNA analysis (Waterston et al. 1992; Reboul et al. 2001, 2003). Because cDNAs may derive from RNA genes and aberrant transcripts as well as protein-coding genes, ultimate confirmation of the latter must occur at the protein level. Consequently, use of proteomics data to experimentally validate gene annotations has recently become an increasingly valuable complement to cDNA efforts (Brunner et al. 2007; Sevinsky et al. 2007).
Tandem mass spectrometry (MS/MS) allows a promising “shotgun proteomics” approach for detecting translated open reading frames (ORFs). In this approach, an unfractionated protein mixture is first digested by proteolysis or chemical cleavage. The resulting peptide mixture is then separated by microcapillary chromatography and emitted into a fast scanning tandem mass spectrometer. The mass spectrometer isolates and automatically selects (using an on-board computer making decisions in real time) peptide ions to undergo collision-induced dissociation with an inert gas, and then performs a second stage of mass analysis to generate an MS/MS spectrum of the resulting product ion fragments. In general, MS/MS spectra are predictable from the peptide sequence and, thus, sequences from a protein or translated nucleic acid sequence database can be used to generate predicted fragmentation spectra that are then matched against the experimental spectra (Eng et al. 1994).
The nematode Caenorhabditis elegans offers an excellent model system to demonstrate the use of proteomics data to experimentally annotate protein-coding regions of a multicellular organism’s genome. The genome sequence is complete, extending from telomere to telomere without gaps for all six chromosomes. Additionally, the genome sequences of the diverged species C. briggsae and C. remanei can be used to identify conserved sequences independent of gene predictions in C. elegans. ORFs in these conserved sequences can be used as putative ORFs to query with experimentally acquired MS/MS data. C. elegans has benefited from large-scale efforts to identify transcripts (Waterston et al. 1992; Reboul et al. 2001, 2003), which have resulted in a well curated set of protein-coding genes. Specifically, WormBase release WS150 contains 20,066 predicted genes with a total of 22,858 predicted spliced transcripts. However, only 6513 (28.5%) of the predicted coding sequences in these transcripts are fully confirmed (translation start and stop site, all coding exons, and exon/intron junctions) by experimental data, 11,417 (49.9%) are partially confirmed, and 4928 (21.6%) remain as unsupported gene predictions. It is also clear that numerous annotation errors remain. For example, directed annotation of the putative seven-pass chemoreceptor family revealed a substantial number of probable new genes and correction of a large number of gene predictions (Robertson 2000, 2001). Short single-exon genes also present problems for prediction and, lacking strong support from experimental evidence, tend to be ignored (Basrai et al. 1997). Putative pseudogenes and recently duplicated genes are also problematic. For all these reasons, existing analyses fall well short of a complete “parts list” of expressed C. elegans proteins.
Here, we report the first use of mass spectrometry for large-scale experimental identification of protein-coding regions of the C. elegans genome. Our data not only confirm and revise existing gene predictions but also reveal novel translated ORFs. The complete collection of identified peptides has been mapped back to the C. elegans genome and is available through http://wormbase.org as a resource for improving ab initio gene prediction algorithms as well as a basis for future discovery and targeted proteomics efforts in C. elegans. The annotated spectra have been assembled into a reference spectrum library for use in rapid assignment of peptide sequences to MS/MS spectra acquired in subsequent experiments, and are available along with software (Frewen et al. 2006) for improved peptide identifications from http://proteome.gs.washington.edu. Additionally, as reported with a recent catalog of Drosophila melanogaster proteins (Brunner et al. 2007), these experimentally observed tryptic peptides provide a basis for targeting identified proteins in future hypothesis-driven experiments.
Results
We performed a shotgun proteomics analysis of C. elegans proteins from mixed stage animals to validate existing gene predictions on the protein level, to identify missing or erroneous gene predictions, and to create a data set for use as a basis for future proteomics experiments. Our approach involves biochemical fractionation of proteins, enzymatic digestion of the separated proteins, and analysis of the resulting peptides by microcapillary multidimensional liquid chromatography tandem mass spectrometry. The resulting mass spectra were searched against a protein database that contained WormBase WS150 gene predictions, additional predictions from an updated version of GeneFinder, and ORFs greater than 30 codons that are conserved in either C. briggsae or C. remanei. Confirmed peptides were then mapped back to the genome sequence to identify translated genomic regions.
In total, 30 multidimensional microcapillary tandem mass spectrometry (μLC/μLC-MS/MS) analyses and 11 one-dimensional microcapillary tandem mass spectrometry (μLC-MS/MS) analyses were performed on 41 different biochemical protein fractions. These experiments yielded a total of 6,440,705 MS/MS spectra. Using the peptide validation software Percolator (Käll et al. 2007), we identified spectra that matched peptides with a q-value (false discovery rate) < 0.01 (Käll et al. 2008a, b). By these criteria, 1,426,236 of our MS/MS spectra were accurately matched to peptide sequences (22.1%). These peptide spectrum matches resulted in 66,489 unique peptide identifications, which partially confirmed 6350 proteins (roughly 27%) from existing WormBase WS150 gene models. The identified peptides map to proteins with widely varying molecular weights and isoelectric points (Supplemental Fig. 1), indicating that we have broad coverage of proteins spanning a large range of physiochemical properties.
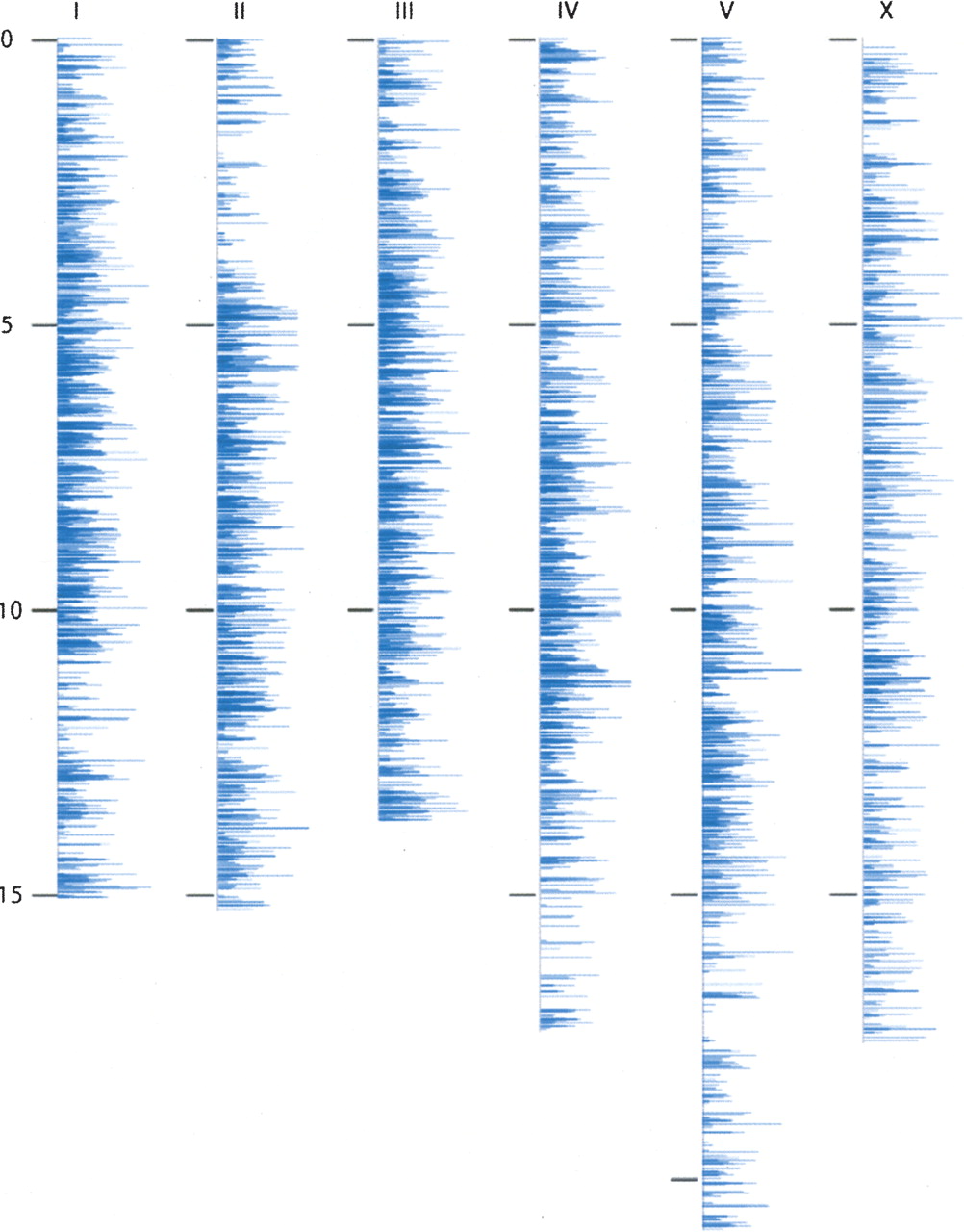
Figure 1 shows the distribution of the detected peptides across the six chromosomes. While gene density is generally uniform across autosomes (The C. elegans Sequencing Consortium 1998), we detected more spectra from peptides mapping to the central regions of autosomes. Because more abundant proteins are sampled with higher frequency by the mass spectrometer (Liu et al. 2004; Zybailov et al. 2005, 2006), the greater number of spectra for the central autosomal regions suggests that protein expression levels are higher for these genes, consistent with what has been observed using transcript data (The C. elegans Sequencing Consortium 1998). Supplemental Figure 2 illustrates the distribution similarities of peptide spectra detected for chromosome I with the number of respective EST sequences present in WormBase. Again consistent with transcript data, the density of detected peptides on the X chromosome is more uniform.
Chromsomal distribution of peptides identified by mass spectrometry in C. elegans. Shown here are the distributions of our mass spectral identifications by chromosome location. The chromosomes are binned into sections of ∼100 kb, and the length of the blue line represents the number of spectra mapping to genes in that bin. This figure shows that our peptides are sampled more frequently from genes in the center of the autosomes and more disperse on the arms of the autosomes and on the sex chromosome. Assuming that peptides are sampled more frequently for abundant proteins, these data support that proteins near the center of the autosome are, on average, expressed at a greater abundance than proteins located on the arms of the autosome.
Confirmation of genes at the protein level
Our data validate many WormBase gene predictions at the protein level, and in particular provide evidence for a number of genes having little or no prior experimental support by cDNA sequencing or alternative methods. Of the 6350 distinct genes our peptides identified from existing WormBase gene models, 384 have no prior experimental evidence, and 3538 are annotated as only partially confirmed; thus, 3922 (over half) of our protein identifications provide new experimental evidence to support gene models that had not yet been fully confirmed.
Confirmation of splice junctions
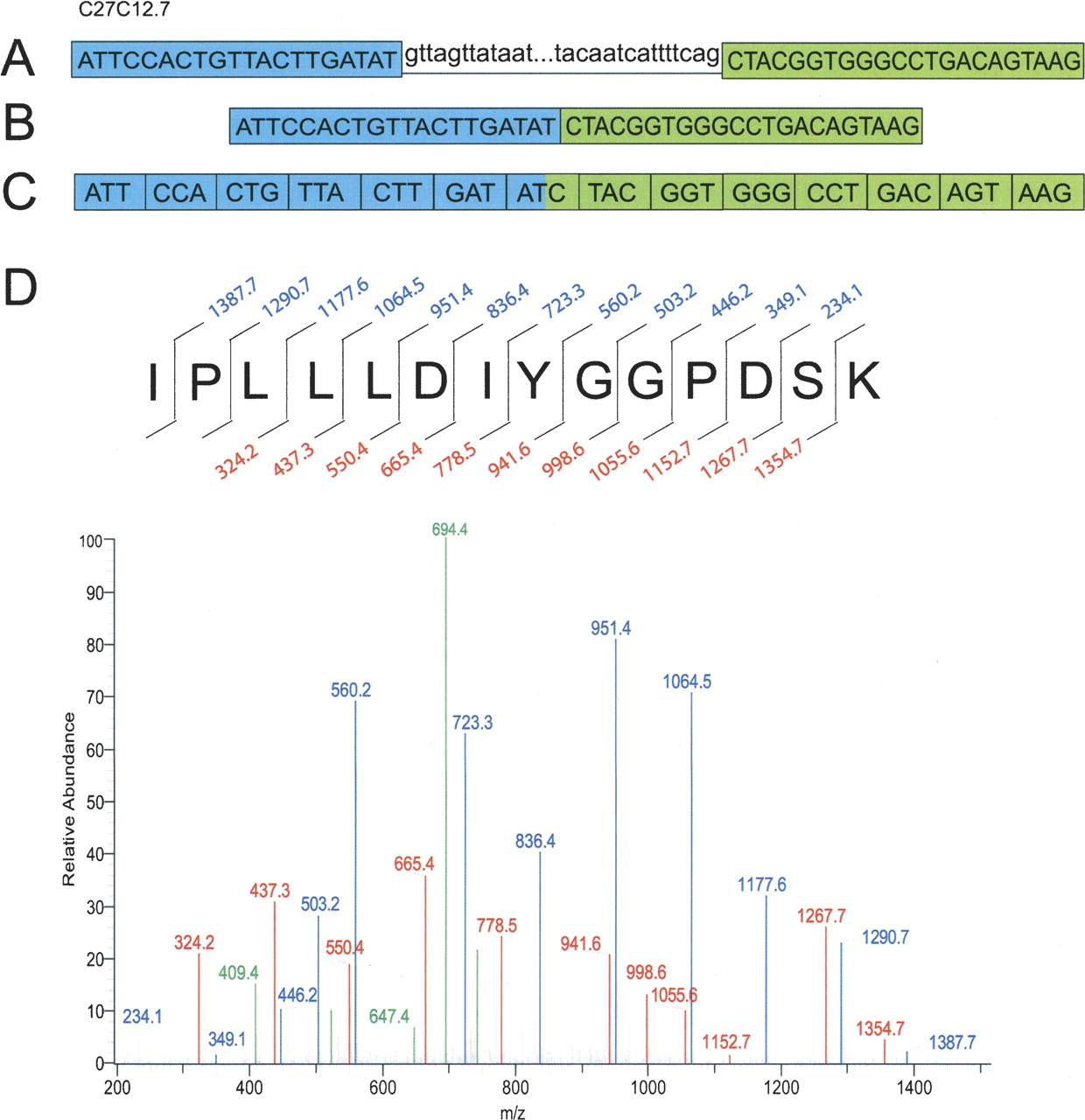
Some of the peptides identified by tandem mass spectrometry span splice junctions and thereby confirm the locations of exon boundaries. Figure 2 gives one example of the data confirming a single splice junction in the gene C27C12.7 (dpf-2), which encodes a dipeptidyl peptidase. The figure shows the DNA sequence, the predicted transcript spanning the splice junction, and the tandem mass spectrum identifying the spanning peptide sequence. The peptide sequence is unique to this locus within the query protein database. This spectrum was assigned to this peptide sequence with a q-value = 0, providing a high confidence confirmation of the splice junction. In all, ∼8% of the 67,047 nonredundant peptides we identified experimentally confirm 6010 independent splice junctions annotated in WormBase WS150, by spanning exon–exon boundaries; a list of these is provided as Supplemental Table 1.
Splice junction confirmation via mass spectrometry. The confirmation of the splice junction between exon 10 (in blue) and exon 11 (in green) for the C. elegans gene C27C12.7 encoding a dipeptidyl peptidase (DPF-2) is illustrated. (A) The unspliced DNA sequence of C27C12.7 between the end of exon 10 and the beginning of exon 11. (B) Exon 10 and exon 11 spliced together. (C) The spliced exons separated into codons. (D) The peptide sequence spanning the splice junction and the representative mass spectrum. The numbers in blue above the peptide sequence represent the C-terminal y-ions and the red numbers below the peptide sequence represent the N-terminal b-ions. (Blue) y-ions; (red) b-ions; (green) all other ions (a-ions, doubly charged ions, ions from the loss of water or ammonia, etc.).
Identification of previously unannotated genes and corrections to existing gene models
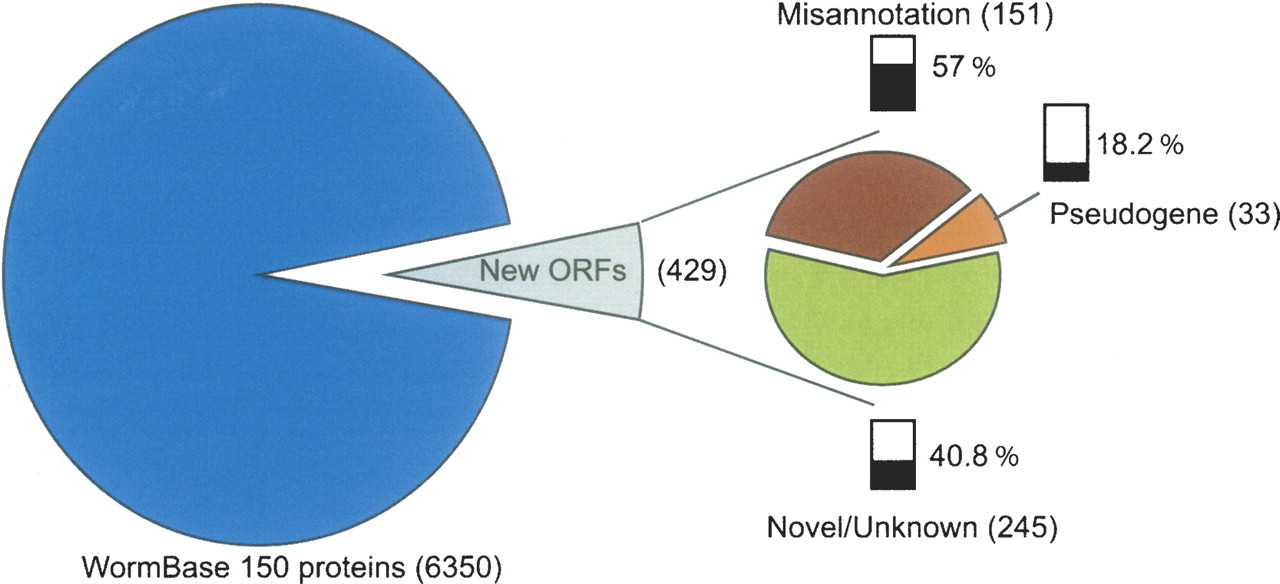
While several laboratories have used proteomics data to confirm existing gene models (Yates et al. 1995; Jaffe et al. 2004a; Desiere et al. 2006; Brunner et al. 2007), few have used these data to identify new genes or correct existing gene models (Jaffe et al. 2004b; Gupta et al. 2007), and even fewer have used proteomics data for the detection of new genes in a metazoan system (Sevinsky et al. 2007). By using new GeneFinder predictions and conserved ORFs as part of our query database, we could map peptide spectra to previously unannotated regions of the C. elegans genome. Our data provide evidence for a total of 429 new coding sequences, falling into three main categories: (1) completely novel gene models (i.e., missing from WormBase WS150), (2) incorrectly specified gene models, or (3) genes incorrectly annotated as pseudogenes (see Fig. 3).
Classification of proteins identified from existing or new coding sequences. From the total 6779 proteins identified, 6350 were identified based on the protein-coding genes from WormBase WS150, and 429 proteins were identified using either new GeneFinder predictions, the conserved intergenic data set, or both. From the 429 new proteins, 33 mapped to predicted pseudogenes in WS150. Of the 33 predicted pseudogenes, 18.2% have been confirmed by RT-PCR. We have identified 151 misannotated protein sequences, and 56.9% of these new coding sequences have RT-PCR confirmation. The last category represents 245 novel or unknown coding sequences of which 40.8% have RT-PCR confirmation.
The combination of these matches suggests a surprisingly high percentage of unanticipated gene models and/or corrections: Almost 6% of the identified protein loci (429 of 6779) were not annotated in WormBase WS150. Supplemental Table 2 shows the distribution of new or modified gene models by chromosome. Most of the unannotated genomic regions (283 of 429) correspond to updated GeneFinder predictions not present in WormBase WS150. An additional 111 translated ORFs correspond to intergenic regions conserved between C. elegans and either C. briggsae (Stein et al. 2003) or C. remanei. The remaining 35 of 429 match to both the GeneFinder predictions and the conserved intergenic set. These data suggest that shotgun proteomics as performed here could provide an important method for identifying protein-coding regions in newly sequenced genomes, complementary to cDNA sequencing.
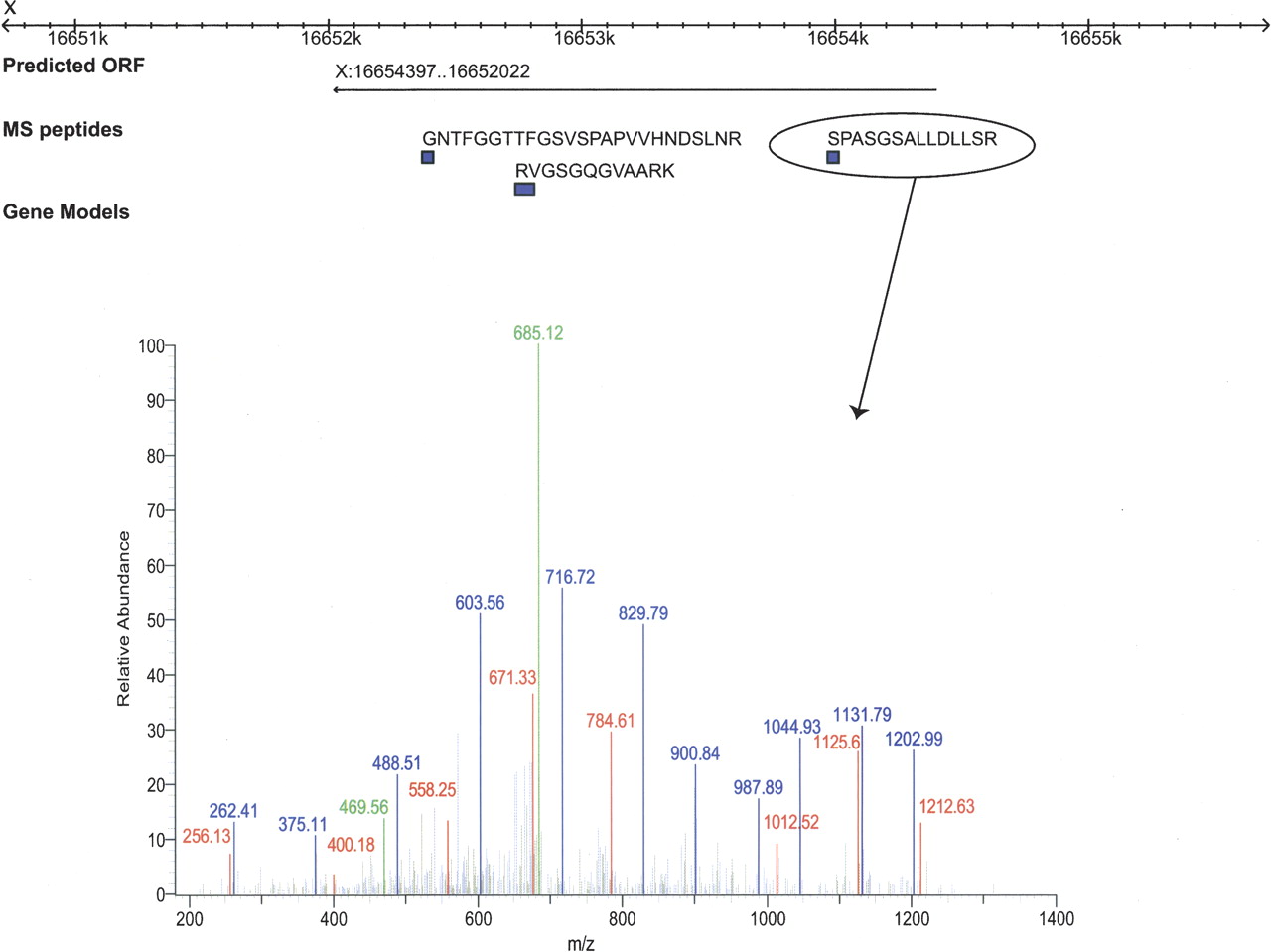
Many of the new coding sequences are located in unannotated genomic regions (245 of 429) in WS150. An example of a novel gene identification is shown in Figure 4. This chromosome X gene was predicted by the updated version of GeneFinder but absent from WormBase WS150 and was still unannotated as of manuscript submission. We identified three unique peptides with q-value ≤ 0.01 that provide evidence for this gene. The spectrum identifying the peptide SPASGSALLDLLR is shown in the figure.
Identification of a novel coding sequence by shotgun proteomics. Three unique peptides were identified in the genomic region 16,652,022–16,654,397 on the X chromosome. This genomic region represents a new ORF from the new GeneFinder prediction set. There are no gene models predicted in this region in WormBase WS150; however, several SAGE tags confirming this gene model have been added since WS150. A mass spectrum from the peptide SPASGSALLDLLSR is shown.
Incorrect or incomplete gene models
Of the 429 identified new coding sequences, 151 are likely corrections to existing known but incompletely specified gene models, rather than novel genes. These are of two types: (1) peptides mapping completely or partly within annotated introns of WS150 gene models (96 peptides); and (2) peptides mapping completely or partly within untranslated regions (UTRs) of gene models (37 peptides in 32 different regions). These peptides may reflect errors in the predicted splicing pattern or reading frame for the gene, or they may indicate an unannotated alternative splice form. In either case, these data confirm that our understanding of the existing gene model is incomplete.
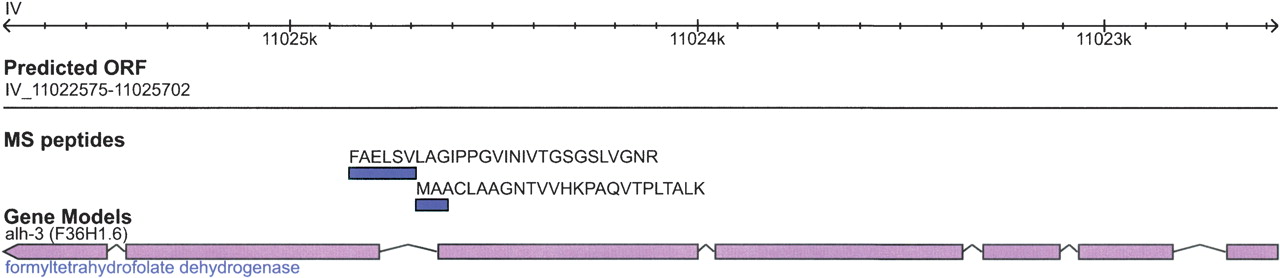
Figure 5 indicates a case of peptides falling within an annotated intron. Two peptides were identified in a single intron in the chromosome IV gene F36H1.6 (alh-3). The tryptic peptide FAELSVLAGIPPGVINIVTGSGSLVGNR spans an exon–intron boundary in WormBase WS150. In the updated GeneFinder predictions, this region of the gene is predicted as protein coding and matches the acquired tandem mass spectrometry data. The combination of the new GeneFinder prediction and our mass spectrometry data suggests the WS150 model is incorrect. Interestingly, this gene model was corrected independently in WormBase WS163 based on EST and protein homology evidence. This subsequent correction of the gene model within a later version of the WormBase gene predictions provides confirmation that our approach correctly identified an error.
Correction of a misannotated coding sequence. The gene alh-3 (F36H1.6) contains six exons (pink) and encodes a dehydrogenase in C. elegans according to WormBase 150. We have identified two unique peptides (blue) between exons 2 and 3 that span the genomic region 11,022,575–11,025,702 on chromosome IV. Both peptides lie at least partially within an intron. This gene model has since been fixed in WS180.
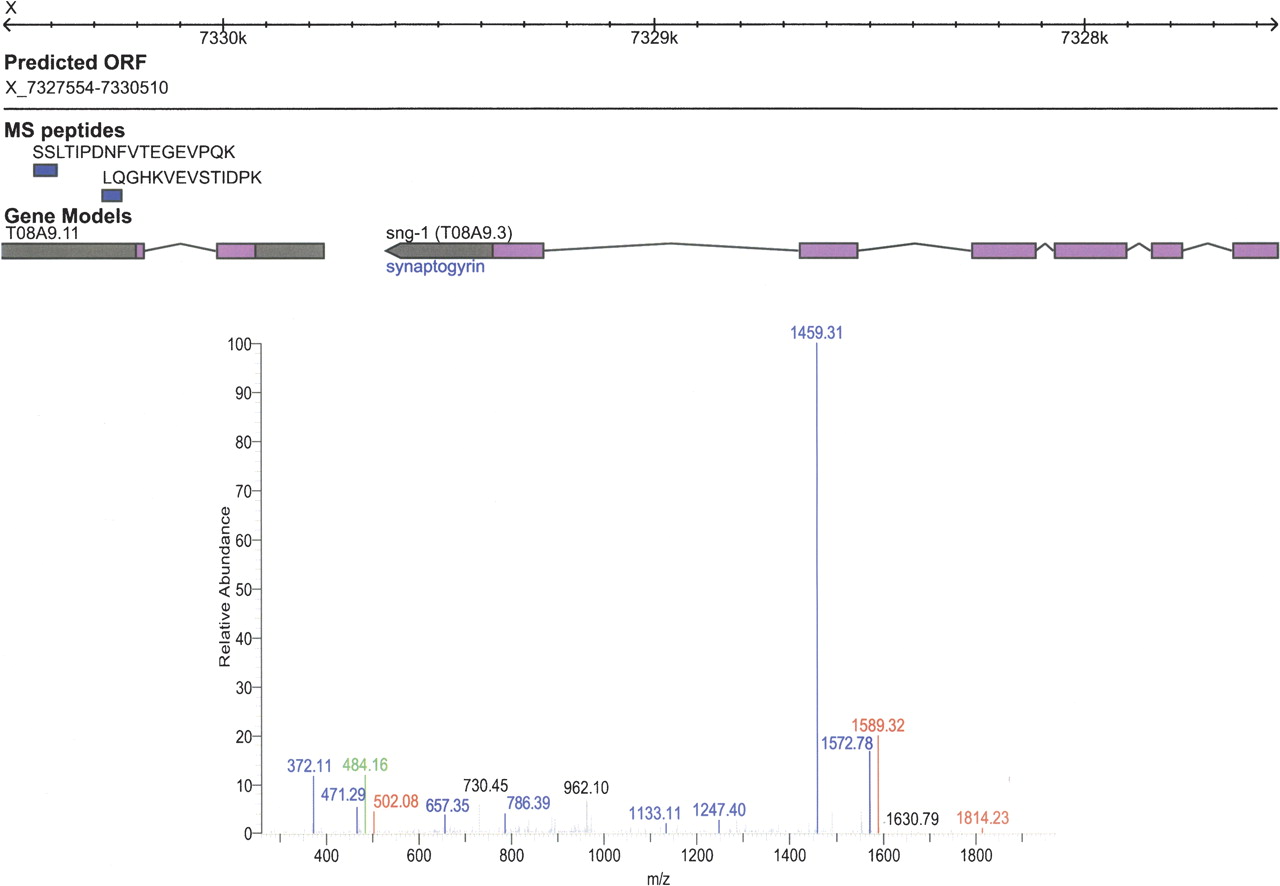
A subset of peptides found in our proteomics data map to UTRs of existing gene models. Because of the robustness and speed of DNA sequencing technology, most protein-coding gene validation has been performed on the nucleotide level. However, transcript data only indirectly indicate the translated protein and will have particular difficulty at distinguishing translated and untranslated regions of the mRNA. In total, our data set contains 37 peptides that reside within 32 different regions annotated at least partially as UTRs in WormBase. An example of the translation of a region that is annotated as an UTR is shown in Figure 6. We identified two peptides that map uniquely to the predicted 3′ UTR of the gene T08A9.11 on the X chromosome. The tandem mass spectrum for the peptide SSLTIPDNFVTEGEVPQK is shown below the WormBase gene models.
Identification of a misannotated coding sequence located in an untranslated region (UTR). WormBase gene model T08A9.11 lies within genomic region of 7,327,554–7,330,510 on chromosome X. The two unique peptides (blue) lie within 3′ UTR (gray) region of the gene in WormBase 150. A mass spectrum from the peptide SSLTIPDNFVTEGEVPQK, one of the two peptides identified within the 3′ UTR, is shown.
Peptides from predicted pseudogenes
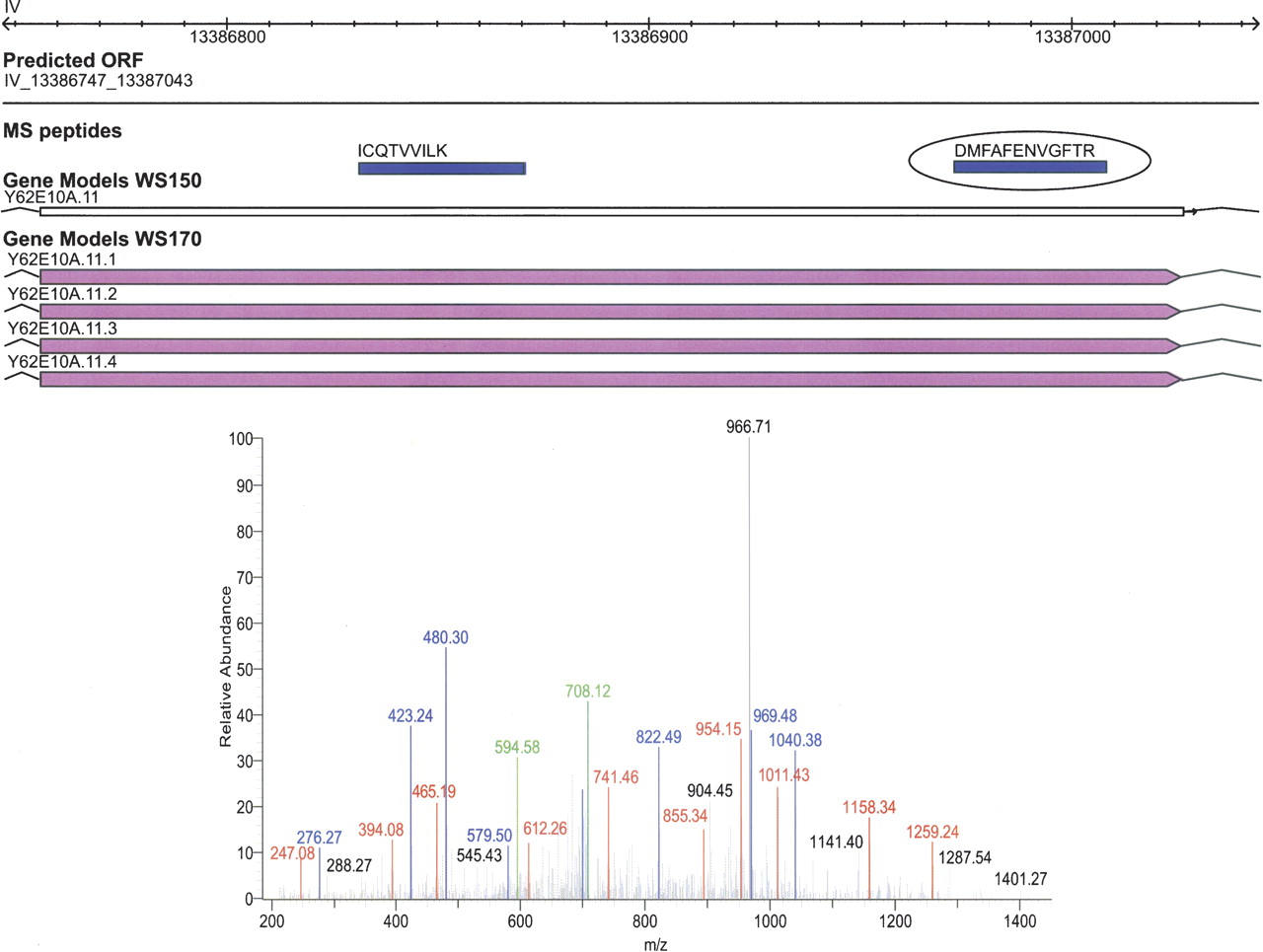
Pseudogenes complicate the prediction of protein coding genes because they share sequence elements with genes but do not encode functional proteins. In total, WS150 annotates 1237 pseudogenes in the C. elegans genome. Because most pseudogenes share ancestry with one or more functional genes, many ORFs from pseudogenes were added to our query sequence database based on sequence conservation with C. briggsae and/or C. remanei. Furthermore, the updated GeneFinder annotations contain several predicted protein-coding genes that are listed as WS150 pseudogenes. We identified 52 distinct peptides that uniquely mapped to 33 different pseudogenes. Figure 7 shows an example of peptides mapping to a WS150-annotated pseudogene on chromosome IV. During the course of our analyses, the annotation of this gene was changed to functional based on other data (WS170). Thus, proteomics data can be used to correctly identify false-negative gene predictions because of misclassification as pseudogenes.
Identification of a translated pseudogene. Two unique peptides (blue) span the conserved intergenic ORF prediction located at 13,386,747–13,387,043 on chromosome IV. In WormBase WS150 these peptides were present within a predicted pseudogene. In a later version of WormBase, this pseudogene has been corrected to a protein-coding gene. A mass spectrum of the peptide DMFAFENVGFTR, one of the two peptides confirming the translation of this pseudogene, is illustrated.
Confirmation of new gene models identified by mass spectrometry using RT-PCR
As described above, our MS/MS data identified 429 new or modified coding sequences, including 317 that mapped to new GeneFinder predictions not present in WormBase WS150. We compared our data with reverse transcriptase polymerase chain reaction (RT-PCR) data that had been collected previously (C. Davis, B. Ewing, D. Gordon, and P. Green, unpubl.), as part of a project to validate the updated GeneFinder predictions. Of the 429 ORFs, 192 (44.8%) have been confirmed to date by RT-PCR. Specifically, 146 had been selected for RT-PCR by the Green Lab (34% of our new gene models). Of the 146 gene predictions for which RT-PCR was specifically targeted, 81 were confirmed as transcribed gene elements. In addition, 115 of the 429 (27%) of the new or modified genes were not specifically attempted but were confirmed with RT-PCR data as a result of ectopic priming from the targeted confirmation of other transcripts. While these 115 genes were not specifically targeted, the mRNA transcripts were sequenced and confirmed. These data provide independent confirmation of 44.8% of the total novel ORFs identified in this study. However, because the RT-PCR data were only collected for the GeneFinder predictions, these data could only have validated the 317 gene models specific to new GeneFinder models. Using this subset list, 60.6% of the novel ORFs matching updated GeneFinder predictions were confirmed by the RT-PCR data. A table listing the new ORFs supported by RT-PCR data is available in Supplemental Tables 2–4.
Discussion
Here, we report the first use of peptides identified by shotgun proteomics for the confirmation and correction of C. elegans gene annotations. The problem of determining the genetic and protein complement of newly sequenced animal and plant genomes is staggering. Ab initio gene predictors, EST sequencing, and homology-based exon finding all help in determining the genetic complement of these new genomes, but it is well appreciated that these approaches have limitations; and because they are DNA or mRNA sequence-based they can only indirectly identify protein-coding potential. Our proteomics approach provides information at the protein level to validate directly the translated gene model. Because ∼6% of our detected proteins map to regions of the genome not present in a WormBase gene model, it is clear that transcript-level analyses should be complemented with proteomics data. Our data were acquired with a relatively modest experimental effort that included ∼30 d of mass spectrometer time (not counting time spent working out sample fractionation and analysis strategies). Thus, we estimate that a subsequent experiment in an organism of similar proteome complexity could be performed using a similar fractionation strategy in about a month with a single technician and a single mass spectrometer.
The primary approach that we have used to increase the coverage of proteins identified by shotgun proteomics is to perform extensive fractionation of either proteins or peptides prior to on-line μLC-MS/MS or μLC/μLC-MS/MS. This fractionation decreases the number of peptides entering the mass spectrometer at any one point in time and increases the number of peptides that can be sampled during the entire analysis. While the total number of peptides identified increases, this increase comes at the expense of a much greater increase in the total acquired MS/MS spectra—i.e., the fraction of identifiable spectra decreases. Because all of these spectra, regardless of quality, are searched against protein sequences using a database searching algorithm, the larger number of spectra will increase the number of false-positive peptide spectrum matches at a given threshold (Käll et al. 2008a). To control for this, our assignments of peptide sequences to spectra use q-values (Storey 2003; Storey and Tibshirani 2003) to account for the number of hypothesis tests performed. Q-values have been used extensively to report significance in gene expression studies and, more recently, in shotgun proteomics peptide identification (Käll et al. 2007, 2008a) and quantitation (Mayor et al. 2007). As mass spectrometers become faster, more robust, and cheaper we anticipate that much larger proteomics efforts will be used to annotate genome sequences, which will put an increasing burden on the statistical metrics for reporting the results to ensure data quality and continuity between laboratories.
In addition to large-scale discovery-based proteome profiling, mass spectrometry is increasingly being applied in hypothesis-driven experiments targeted at specific proteins of interest and focused on measuring tens of proteins under tens of conditions, as opposed to hundreds of proteins under a few conditions (Anderson and Hunter 2006; Mallick et al. 2007). Using a mass spectrometer in a targeted configuration to follow groups of proteins (e.g., in a pathway or complex) under numerous conditions and/or time-points opens up a host of capabilities that previously required immunological assays. However, a complicating step in targeted protein analyses is deciding which peptide(s) can be reliably monitored as a proxy of the intact protein. Peptides that provide a sensitive and reproducible measure of the respective protein are commonly referred to as “proteotypic peptides.” An important added benefit of our data is that it identifies candidate proteotypic peptides for use in targeted proteomics approaches. These can be used (for example) to design quantitative assays to measure the effect of individual alleles and/or growth conditions on peptide and protein abundance.
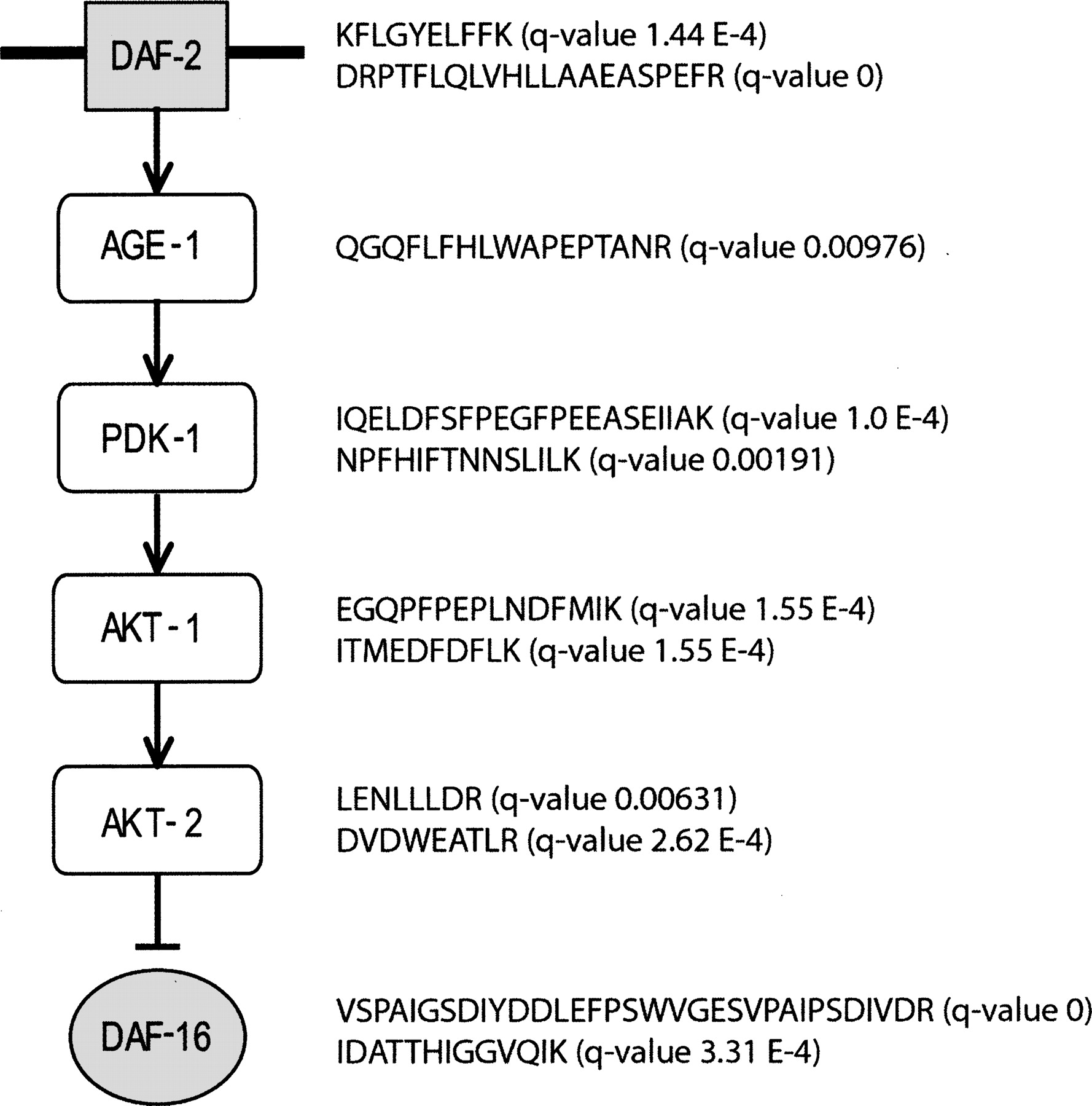
As an example, Figure 8 shows identified tryptic peptides that are potential proteotypic peptides for the targeted analysis of the insulin/insulin-like growth factor 1 signaling pathway in C. elegans. The insulin signaling pathway is highly conserved between C. elegans and humans, including orthologs of the mammalian insulin/IGF-1 receptor (daf-2) (Kimura et al. 1997), the catalytic subunit of PI-3 kinase (age-1) (Malone et al. 1996; Paradis et al. 1999), the serine/threonine kinases Akt/PKB (akt-1 and akt-2), and PDK1 (pdk-1) (Paradis et al. 1999). In both worms and vertebrates, the upstream insulin pathway negatively regulates the fork head transcription factors DAF-16 in the worm and AFX, FKHR, FKHRL1 in vertebrates (Lee et al. 2001). Supplemental Figure 3 illustrates the use of these data in the targeted analysis of DAF-16 and AKT-1 from unfractionated lysates. Because of the high protein conservation, understanding the mechanisms by which the insulin pathway is regulated in worms may provide insight into how this pathway is regulated in humans. Proteotypic peptides, in conjunction with known phenotypes of this pathway (e.g., longevity and dauer formation) can be used in quantitative assays for further investigation of pathway mechanisms.
Peptides identified in the insulin/insulin-like growth factor 1 signaling pathway can be used as proteotypic peptides in future targeted analyses. Shown here are the major proteins involved in the insulin/insulin-like growth factor 1 signaling pathway along with peptides identified from the respective proteins.
In summary, we have demonstrated the use of peptide sequences identified by tandem mass spectrometry for the annotation of the C. elegans genome. While not complete, the data collected using the described approach have contributed substantially to the correction and modification of gene models in a relatively well characterized model organism. These data indicate that a shotgun proteomics strategy is a valuable complement to more traditional efforts on the transcript level for providing experimental evidence of protein-coding regions of newly sequenced genomes. We expect that proteogenomic annotation of metazoan gene models may be even more powerful in an organism such as C. briggsae having a completed genome but lacking large-scale “ORFeome” efforts comparable to those for C. elegans.
Methods
Materials
C. elegans and bacterial strains were obtained from the CGC at the University of Minnesota. Chemicals were purchased from Sigma except HPLC-grade acetonitrile and methanol, which were purchased from VWR, and sodium carbonate and chloroform, which were purchased from Fisher Scientific.
Growth and lysis of C. elegans
C. elegans N2 (wild-type strain from Bristol) were grown on enriched peptone plates seeded with the OP50 strain of Escherichia coli at 20°C. Worms of all developmental stages were washed off plates with M9 buffer (22 mM KH2PO4, 22 mM NA2HPO4, 85 mM NaCl, 1 mM MgSO4) and sucrose-floated to remove bacterial contamination. The worms were then lysed in 50 mM ammonium bicarbonate (pH 7.8) using a small probe of a Braun Labsonic U sonicator (Braun Biotech International) for six cycles of a 30-sec continuous pulse followed by a 60-sec ice incubation. The lysate was then centrifuged at 4000 rpm for 10 min at 4°C in a microcentrifuge (Eppendorf) to remove cell debris. A second centrifugation at 14,000 rpm for 10 min at 4°C was performed to separate the soluble lysate from the insoluble lysate. Protein concentration of the soluble lysate was determined using the Bio-Rad DC Protein Assay.
Protein fractionation
The soluble lysate is biochemically fractionated either by solubility, density, charge, or hydrophobicity. The insoluble lysate was separated by molecular weight on an SDS-PAGE gel.
Biochemical fractionation of soluble proteins
Ammonium sulfate precipitation
Four milligrams of soluble N2 was precipitated using nine fractions of ammonium sulfate concentrations (15%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%) to separate based on solubility.
C8 column fractionation
Four milligrams of soluble N2 was injected onto a 4.4 × 150-mm Zorbax Eclipse XDB-C8 5-μm column (Agilent Technologies) and subjected to an increasing acetonitrile gradient (a 45-min gradient beginning with 5% acetonitrile, 95% water, 0.1% formic acid [buffer A] and ending with 95% acetonitrile, 5% water, 0.1% formic acid [buffer B]). One-milliliter fractions were collected during the entire 45-min gradient and pooled together into nine fractions of similar protein concentration. The nine fractions were decreased to a smaller volume in a Vacufuge Concentrator 5301 (Eppendorf).
Digestion of soluble fractions
Protein fractions were denatured using 0.1% RapiGest SF (Waters Corporation) in 50 mM ammonium bicarbonate (pH 7.8). The samples were vortexed and boiled at 100°C for 5 min to increase denaturation. After samples were cooled, they were reduced with 5 mM DTT (Sigma) and alkylated with 15 mM IAA (Sigma). Fractions were next digested to peptides using trypsin (sequence grade, modified, Roche Applied Science) at a substrate to enzyme ratio of 100:1 overnight at 37°C with shaking. The next day the sample was treated with 100 mM HCl to remove RapiGest from the sample.
Biochemical fractionation of insoluble proteins
Sucrose gradient
A sucrose gradient was performed to fractionate the insoluble worm lysate based on density. Ten gradients from 0% to 60% were collected and methanol-chloroform precipitated to remove lipids and other undesirables from the sample.
In-gel fractionation
Three milligrams of insoluble N2 was washed with 200 mM sodium carbonate pH 11, methanol-chloroform precipitated, and ran on a NuPAGE 10% Bis-Tris SDS-PAGE gel (Invitrogen) to separate proteins of differing molecular weight. The gel was stained with Coomassie blue G250 (Bio-Rad) and 11 bands were cut and in-gel digested as described below.
Digestion of insoluble fractions
Protein fractions from gels were washed in 100 mM ammonium bicarbonate, reduced with DTT, and alkylated with IAA. Gel bands were then shrunk with acetonitrile and speed-vacuumed to dry the gel bands. The gel bands were then digested with trypsin at a substrate to enzyme ratio of 10:1 overnight at 37°C with shaking. The next day proteins were extracted from gel bands with 60% acetonitrile, 0.1% trifluoroacetic acid, and then reconstituted in 0.1% formic acid. Each of these samples was analyzed by μLC-MS/MS using a 4-h reverse-phase chromatography gradient.
Multidimensional protein identification technology (MudPIT)
Each soluble fraction was analyzed via MudPIT. MudPIT columns of 100-μm inner diameter fused silica (Polymicro Technologies) were made in-house by pressure-loading a triphasic column as described previously (MacCoss et al. 2002a, b). The three phases consist of: 7 cm of 5-μm Luna C18 material (Phenomenex), 4 cm of 5-μm Partisphere strong cation exchanger (Whatman), and an additional 2 cm of 5-μm Luna C18 material. After column equilibration, each protein fraction was pressure-loaded offline and placed inline with an Agilent 1100 quaternary HPLC. The buffer solutions used were 5% acetonitrile–0.1% formic acid (buffer A), 95% acetonitrile–0.1% formic acid (buffer B), and ammonium acetate in different concentrations at each of 12 steps (all vol/vol). The profile for step 1 consisted of 20-min 100% A, a 5-min gradient from 0% B to 15% B, a 70-min gradient from 15% B to 40% B, a 5-min gradient from 40% B to 85% B, and a final 20-min wash in 100% A. Steps 2–11 consisted of injections of ammonium acetate in the following volumes and concentrations: 50 μL of 100 mM, 200 mM, 400 mM, 500 mM, 600 mM, 700 mM, 800 mM, 900 mM, 1 M (300 mM is intentionally omitted); and 75 μL of 1 M. The gradient profile for steps 2–11 consisted of 2-min 100% A, a 12-min gradient from 0% B to 15% B, a 90-min gradient from 15% B to 40% B, and a final 15-min wash in 100% A. Step 12 consisted of injection of 100 μL of 5 M ammonium acetate followed by 2-min 100% A, a 13-min gradient from 0% B to 15% B, a 95-min gradient from 15% B to 40% B, and a 10-min gradient from 40% B to 100% B. As peptides eluted from the microcapillary column, they electrosprayed directly into an LTQ mass spectrometer (ThermoFinnigan) with the application of a distal 2.4-kV spray voltage (Gatlin et al. 1998). A cycle of one full-scan mass spectrum (400–1400 m/z) followed by five data-dependent MS/MS spectra at a 35% normalized collision energy was repeated continuously throughout each step of the multidimensional separation. Application of mass spectrometer scan functions and HPLC solvent gradients were controlled by the Xcalibur data system (ThermoFinnigan).
Preparation of candidate ceORF30ic database
A six-frame translation of all intergenic DNA segments based on the WormBase data set in WS130 was performed, which was the most recent stable data release at the time. The entire C. elegans intergenic ORF set was used as query in a TBLASTN search of the C. briggsae and C. remanei genomes. C. briggsae and C. remanei diverged from C. elegans about 90 million years ago, and most intronic and intergenic sequences are highly divergent. The E-value for the TBLASTN search was set high (0.1) to gather complete search results, and post-processing was used to test the effects of more stringent E-values. As expected, the large majority of the intergenic ORF sequences had no detectable conservation in C. briggsae or C. remanei. Choosing an E-value cutoff for inclusion in a SEQUEST query set involved tradeoffs: A more stringent E-value better avoids false positives and improves search times, but will discard weakly conserved ORFs. For initial work we settled, somewhat arbitrarily, on an E-value cutoff of 10−3. ORFs already contained in the updated GeneFinder prediction set were removed.
Database searching
The MS/MS data are searched using SEQUEST against a fasta database containing the following components: all protein sequences from WormBase 150 that were present in the Wormpep database, unique protein coding gene predictions from the program GeneFinder (P. Green, unpubl.) not present in WormBase 150, translated intergenic ORFs with high homology with regions of the C. briggsae genome, and a shuffled decoy database of the above components. The decoy proteome was generated by a zero order Markov model adapted to each protein sequence individually. For each MudPIT the resulting peptide spectra matches were batch processed with Percolator 1.0 (Käll et al. 2007). We identified proteins by processing peptide-spectrum matches to which Percolator assigned a q-value < 0.01 with DTASelect (Tabb et al. 2002), requiring one peptide per protein and removing subset proteins.
Splice junction analysis
MS peptide identifications that matched WormBase protein-coding genes were aligned with no gaps against the sets of proteins derived from both the current set of curated coding sequence gene models and those proteins from old curated coding sequence models which have since been amended. These proteins were then mapped back onto the genome, giving mappings of the MS peptides on the genome. The MS peptides which crossed splice site boundaries in the coding sequences were noted during this mapping procedure.
RT-PCR data
C. elegans N2 strain was grown on 2% agarose 100-mm plates. The plates were seeded with 1.5 mL of OP50. Ten to twenty animals from mixed larval stages were picked to separate plates and grown for 4–5 d at 20°C. Animals were washed off plates with M9 then sucrose-floated to remove bacteria and dead eggs and dead animals. Total RNA was isolated from the worms using TRIzol (Invitrogen). C. elegans total RNA was reverse-transcribed using Superscript II (Invitrogen) and Oligo dT at 42°C for 50 min followed by 70°C for 15 min. The cDNA was incubated at 37°C for 20 min with two units of RNase H. PCR reactions were performed in 96-well plates on an MJ Research Tetrad thermal cycler. For each transcript, 200 nM each of a gene-specific forward and gene-specific reverse primer were combined with 8 μL of a PCR cocktail mix—0.1 μL of cDNA, 200 μM of each dNTP, 1.4 mM Mg2+, and 0.4 μL of Elongase enzyme mix. Reactions were heated at 94°C for 30 sec followed by 35 PCR cycles as follows: 94°C for 30 sec, 49°C (chromosome I, before optimization) or 54°C for 30 sec (chromosome II), and 68°C for 60 sec/kb of target. PCR products were diluted 10-fold and 1 μL was used as template in another round of 35 cycles of PCR amplification under the same conditions as before (rePCR). Products were visualized by running 2 μL on a 2% EGEL agarose gel (Invitrogen) and photographed under UV light.
Products from rePCR reactions were diluted 20-fold with distilled water and 2 μL was used for each sequencing reaction. Sequencing reactions were performed using ABI BigDye Terminator, version 3.1 (Applied Biosystems) in 96-well plates. The standard protocol was modified to 1/20 reactions and the gene-specific primers were used as sequencing primers in two separate reactions/template—one forward oligo reaction and one reverse. Sequencing reactions were ethanol-precipitated to remove unincorporated dyes. Reactions were resuspended in 10 μL of HI-DI formamide (Applied Biosystems) and run on either an ABI377 sequencer or an ABI Prism 3100* Genetic Analyzer using POP6 polymer and a 50-cm capillary array.
MS identifications were aligned to RT-PCR data using phred version 020425.c to call bases from the ABI trace files and cross_ match (P. Green, unpubl.) version 1.030318 to align the reads to the transcript sequences predicted by GeneFinder. The cross_ match alignment parameters were the defaults except the minimum alignment score, which was reduced to 25.
Acknowledgments
We thank Robert Waterston and LaDeana Hiller for their encouragement and helpful discussions during this project. Financial support for this work was provided from National Institutes of Health grants R21-GM074787 (J.H.T.), R01-DK069386 (M.J.M.), and P41-RR011823 (M.J.M.) and from support provided by the Howard Hughes Medical Institute (P.G.).
References
- ↵L. Anderson,C.L. Hunter,(2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5:573–588.
- ↵M.A. Basrai,P. Hieter,J.D. Boeke,(1997) Small open reading frames: Beautiful needles in the haystack. Genome Res. 7:768–771.
- ↵E. Brunner,C.H. Ahrens,S. Mohanty,H. Baetschmann,S. Loevenich,F. Potthast,E.W. Deutsch,C. Panse,U. de Lichtenberg,O. Rinner,(2007) A high-quality catalog of the Drosophila melanogaster proteome. Nat. Biotechnol. 25:576–583.
- ↵The C. elegans Sequencing Consortium (1998) Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282:2012–2018.
- ↵F. Desiere,E.W. Deutsch,N.L. King,A.I. Nesvizhskii,P. Mallick,J. Eng,S. Chen,J. Eddes,S.N. Loevenich,R. Aebersold,(2006) The PeptideAtlas project. Nucleic Acids Res. 34:D655–D658.
- ↵J.K. Eng,A.L. McCormack,J.R. Yates,(1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976–989.
- ↵B.E. Frewen,G.E. Merrihew,C.C. Wu,W.S. Noble,M.J. MacCoss,(2006) Analysis of peptide MS/MS spectra from large-scale proteomics experiments using spectrum libraries. Anal. Chem. 78:5678–5684.
- ↵C.L. Gatlin,G.R. Kleemann,L.G. Hays,A.J. Link,J.R. Yates,(1998) Protein identification at the low femtomole level from silver-stained gels using a new fritless electrospray interface for liquid chromatography-microspray and nanospray mass spectrometry. Anal. Biochem. 263:93–101.
- ↵N. Gupta,S. Tanner,N. Jaitly,J.N. Adkins,M. Lipton,R. Edwards,M. Romine,A. Osterman,V. Bafna,R.D. Smith,(2007) Whole proteome analysis of post-translational modifications: Applications of mass-spectrometry for proteogenomic annotation. Genome Res. 17:1362–1377.
- ↵J.D. Jaffe,H.C. Berg,G.M. Church,(2004a) Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics 4:59–77.
- ↵J.D. Jaffe,N. Stange-Thomann,C. Smith,D. DeCaprio,S. Fisher,J. Butler,S. Calvo,T. Elkins,M.G. FitzGerald,N. Hafez,(2004b) The complete genome and proteome of Mycoplasma mobile. Genome Res. 14:1447–1461.
- ↵L. Käll,J.D. Canterbury,J. Weston,W.S. Noble,M.J. MacCoss,(2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4:923–925.
- ↵L. Käll,J.D. Storey,M.J. MacCoss,W.S. Noble,(2008a) Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 7:29–34.
- ↵L. Käll,J.D. Storey,M.J. MacCoss,W.S. Noble,(2008b) Posterior error probabilities and false discovery rates: Two sides of the same coin. J. Proteome Res. 7:40–44.
- ↵K.D. Kimura,H.A. Tissenbaum,Y. Liu,G. Ruvkun,(1997) daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277:942–946.
- ↵R.Y. Lee,J. Hench,G. Ruvkun,(2001) Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr. Biol. 11:1950–1957.
- ↵H. Liu,R.G. Sadygov,J.R. Yates,(2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76:4193–4201.
- ↵M.J. MacCoss,W.H. McDonald,A. Saraf,R. Sadygov,J.M. Clark,J.J. Tasto,K.L. Gould,D. Wolters,M. Washburn,A. Weiss,(2002a) Shotgun identification of protein modifications from protein complexes and lens tissue. Proc. Natl. Acad. Sci. 99:7900–7905.
- ↵M.J. MacCoss,C.C. Wu,J.R. Yates,(2002b) Probability-based validation of protein identifications using a modified SEQUEST algorithm. Anal. Chem. 74:5593–5599.
- ↵P. Mallick,M. Schirle,S.S. Chen,M.R. Flory,H. Lee,D. Martin,J. Ranish,B. Raught,R. Schmitt,T. Werner,(2007) Computational prediction of proteotypic peptides for quantitative proteomics. Nat. Biotechnol. 25:125–131.
- ↵E.A. Malone,T. Inoue,J.H. Thomas,(1996) Genetic analysis of the roles of daf-28 and age-1 in regulating Caenorhabditis elegans dauer formation. Genetics 143:1193–1205.
- ↵T. Mayor,J. Graumann,J. Bryan,M.J. MacCoss,R.J. Deshaies,(2007) Quantitative profiling of ubiquitylated proteins reveals proteasome substrates and the substrate repertoire influenced by the Rpn10 receptor pathway. Mol. Cell. Proteomics 6:1885–1895.
- ↵S. Paradis,M. Ailion,A. Toker,J.H. Thomas,G. Ruvkun,(1999) A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes & Dev. 13:1438–1452.
- ↵J. Reboul,P. Vaglio,N. Tzellas,N. Thierry-Mieg,T. Moore,C. Jackson,T. Shin-i,Y. Kohara,D. Thierry-Mieg,J. Thierry-Mieg,(2001) Open-reading-frame sequence tags (OSTs) support the existence of at least 17,300 genes in C. elegans. Nat. Genet. 27:332–336.
- ↵J. Reboul,P. Vaglio,J.F. Rual,P. Lamesch,M. Martinez,C.M. Armstrong,S. Li,L. Jacotot,N. Bertin,R. Janky,(2003) C. elegans ORFeome version 1.1: Experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat. Genet. 34:35–41.
- ↵H.M. Robertson,(2000) The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res. 10:192–203.
- ↵H.M. Robertson,(2001) Updating the str and srj (stl) families of chemoreceptors in Caenorhabditis nematodes reveals frequent gene movement within and between chromosomes. Chem. Senses 26:151–159.
- ↵J.R. Sevinsky,B.J. Cargile,M.K. Bunger,F. Meng,N.A. Yates,R.C. Hendrickson,J.L. Stephenson,(2007) Whole genome searching with shotgun proteomic data: Applications for genome annotation. J. Proteome Res. 7:80–88.
- ↵L.D. Stein,Z. Bao,D. Blasiar,T. Blumenthal,M.R. Brent,N. Chen,A. Chinwalla,L. Clarke,C. Clee,A. Coghlan,(2003) The genome sequence of Caenorhabditis briggsae: A platform for comparative genomics. PLoS Biol. 1:e45, 10.1371/journal.pbio.0000045.
- ↵J.D. Storey,(2003) The positive false discovery rate: A Bayesian interpretation and the q-value. Ann. Stat. 31:2013–2035.
- J.D. Storey,R. Tibshirani,(2003) Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. 100:9440–9445.
- ↵D.L. Tabb,W.H. McDonald,J.R. Yates,(2002) DTASelect and Contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1:21–26.
- ↵R. Waterston,C. Martin,M. Craxton,C. Huynh,A. Coulson,L. Hillier,R. Durbin,P. Green,R. Shownkeen,N. Halloran,(1992) A survey of expressed genes in Caenorhabditis elegans. Nat. Genet. 1:114–123.
- ↵J.R. Yates,J.K. Eng,A.L. McCormack,(1995) Mining genomes: Correlating tandem mass spectra of modified and unmodified peptides to nucleotide sequences. Anal. Chem. 67:3202–3210.
- ↵B. Zybailov,M.K. Coleman,L. Florens,M.P. Washburn,(2005) Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal. Chem. 77:6218–6224.
- ↵B. Zybailov,A.L. Mosley,M.E. Sardiu,M.K. Coleman,L. Florens,M.P. Washburn,(2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5:2339–2347.
Notes
[1] [Supplemental material is available online at www.genome.org. The complete collection of identified peptides has been mapped back to the C. elegans genome and is available through http://wormbase.org. The annotated spectra have been assembled into a reference spectrum library available at http://proteome.gs.washington.edu.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.077644.108.