
Abstract
We describe the details of a serial analysis of gene expression (SAGE) library construction and analysis platform that has enabled the generation of >298 high-quality SAGE libraries and >30 million SAGE tags primarily from sub-microgram amounts of total RNA purified from samples acquired by microdissection. Several RNA isolation methods were used to handle the diversity of samples processed, and various measures were applied to minimize ditag PCR carryover contamination. Modifications in the SAGE protocol resulted in improved cloning and DNA sequencing efficiencies. Bioinformatic measures to automatically assess DNA sequencing results were implemented to analyze the integrity of ditag structure, linker or cross-species ditag contamination, and yield of high-quality tags per sequence read. Our analysis of singleton tag errors resulted in a method for correcting such errors to statistically determine tag accuracy. From the libraries generated, we produced an essentially complete mapping of reliable 21-base-pair tags to the mouse reference genome sequence for a meta-library of ∼5 million tags. Our analyses led us to reject the commonly held notion that duplicate ditags are artifacts. Rather than the usual practice of discarding such tags, we conclude that they should be retained to avoid introducing bias into the results and thereby maintain the quantitative nature of the data, which is a major theoretical advantage of SAGE as a tool for global transcriptional profiling.
Serial analysis of gene expression (SAGE) offers a particularly attractive technology for profiling eukaryotic transcriptomes (Velculescu et al. 1995) because of the digital and quantitative nature of the data, its efficient sampling of short sequence tags from known and novel mRNA transcripts, and its theoretically unlimited dynamic range. Numerous improvements to the original technology have been described (Peters et al. 1999; Saha et al. 2002; Gowda et al. 2004; Heidenblut et al. 2004; Wei et al. 2004; Kodzius et al. 2006). These include the production of longer tags, which have improved the specificity of tag-to-gene mapping (Saha et al. 2002; Matsumura et al. 2003), and modifications designed to facilitate library construction from nanogram quantities of total RNA (Peters et al. 1999; Neilson et al. 2000). Recently, the use of SAGE-like procedures to identify regions of the genome interacting with DNA-binding proteins has been described (Impey et al. 2004; Chen and Sadowski 2005; Kim et al. 2005; Loh et al. 2006; Wei et al. 2006). Such approaches represent viable alternatives to ChIP-on-chip (Ren et al. 2000).
SAGE is among the few relatively accessible digital gene expression profiling technologies capable of generating comprehensive transcriptome profiles. Nevertheless, challenges associated with laborious library construction and generally limited access to inexpensive automated DNA sequencing have restricted its application to large-scale initiatives. Experiments at our Genome Center (Smailus et al. 2005) and elsewhere have resulted in a steady decrease in DNA sequencing costs using conventional capillary electrophoresis instruments. New instrumentation (Shendure et al. 2004; Bennett et al. 2005; Margulies et al. 2005), only now just becoming available, is expected to further reduce operating costs dramatically. However, the requirement for large-scale production of SAGE libraries remains a challenge. To create a platform that could produce 100 or more libraries per year, we sought to refine the protocols to be used so that they would be sufficiently robust for execution by entry-level technical staff and to design and implement bioinformatics approaches that would measure the accuracy of the tags generated. These efforts resulted in a quality-controlled, production-scale SAGE library construction platform in an academic setting, in which six technicians could generate >100 libraries per year from microdissected samples. We have now used this pipeline to construct 298 libraries. Further increases in scale can be accommodated as required. We describe the details of our approach here, and provide in the Supplemental materials a standard operating procedure to allow implementation of the pipeline at other genome centers.
Methods
Laboratory design
Laboratory space and workflow were designed to limit the potential of PCR cross-contamination. In particular, we segregated pre-ditag work from ditag and post-ditag work. We adopted a policy of single-use aliquots for reagents, exclusive reliance on disposable plasticware and protective apparel, daily decontamination routines, and species-specific work areas. An effective biochemical measure for limiting cross-species contamination was the design and implementation of species-specific LongSAGE adapters. PCR primers corresponding to each adapter pair were designed to be incapable of amplifying ditags from any other adapter pair. We relied on the I-SAGE Long kit (Invitrogen) as the primary source of quality-tested and modularized reagents. These were supplemented as required with materials from suppliers of standard molecular biology equipment and reagents.
Tissue collection and RNA extraction
The diversity of tissue sources used for library construction required several cell and tissue collection approaches. When possible, we used snap-freezing in liquid nitrogen to preserve tissues prior to RNA extraction. Minute samples harvested using microdissection techniques from the earliest stages of mouse development were successfully processed with TRIzol reagent (Invitrogen). Alternatively, for convenient storage and transport of samples at ambient temperature, we routinely used the RNA stabilization reagent RNAlater (Ambion). RNAs from tissues known to harbor high levels of endogenous nucleases, for example spleen and pancreas, were purified successfully by rapid homogenization in RNA extraction buffer immediately following tissue dissection. A PowerGen 125 rotor-stator homogenizer (Fisher Scientific) with a 5-mm disposable generator was routinely utilized for mechanical shearing of solid tissue samples, using working volumes as little as 50 μL. Total RNA was most often isolated with TRIzol reagent in conjunction with Eppendorf brand phase lock gel tubes (Fisher Scientific). RNAs from lipid-rich and fibrous tissues were successfully purified using spin-column-based RNeasy kits developed specifically for such tissues (Qiagen). Processing of samples consisting of only a few thousand cells, such as those typically harvested using laser capture microdissection (LCM), was achieved using spin-column-based RNA isolation methods along with on-column DNase treatment.
Removal of contaminating genomic DNA was performed with Ambion’s DNA-free reagent and protocol, a method that does not require subsequent organic extraction, alcohol precipitation, heating, or the addition of EDTA to the DNase-treated RNA sample. Protocols requiring the latter conditions sometimes yielded degraded RNA following DNase treatment, possibly due to the activation of residual ribonucleases still present after RNA extraction.
Assessment of RNA quality
All RNA samples were analyzed with an Agilent 2100 Bioanalyzer prior to entering the SAGE library construction pipeline. We routinely performed total RNA quality assessments using as little as a few hundred picograms of RNA in sample volumes of 1 μL. Also, the RNA integrity number (RIN; Schroeder et al. 2006) was used to help establish an RNA quality standard. We coupled this RIN metric with a biochemical RNase assay (RNaseALERT, Ambion) to generate comprehensive assessments of RNA quality prior to global gene expression profiling. Despite some variability with RNA Pico LabChip performance, possibly due to extreme sensitivity of the assay to experimental conditions and sample contaminants, the method allowed evaluation of RNA quality at the picogram level with minimal sacrifice of sample material. Quantitation was also performed with a Victor2 fluorometer (Perkin Elmer) coupled with RiboGreen dye (Invitrogen).
LongSAGE library construction
Our library construction pipeline is presented schematically in Supplemental Figure 1. Its design is based on several previously published protocols, including those of Velculescu et al. (1995), Saha et al. (2002), and Gowda et al. (2004), with modifications that we found improved pipeline performance.
Construction of “standard” libraries initially required 2–50 μg of DNase-treated total RNA. mRNA was captured using oligo(dT) magnetic beads followed by synthesis of double-stranded cDNA using SuperScript II reverse transcriptase (Invitrogen), RNAseH, and Escherichia coli DNA polymerase (Invitrogen). The resulting bead-bound cDNA was digested with the tagging enzyme NlaIII (Invitrogen), and the product was divided into two fractions for separate ligation of two adapters with 4-bp overhangs complementary to NlaIII digestion products. The adapter–cDNA ligation products were digested with the type IIS tagging enzyme MmeI (NEB), releasing adapter-tag products with 2-nucleotide (nt) overhangs. The two adapter–tag fractions were then ligated to form ∼131-bp adapter–ditag–adapter products that served as template for scale-up PCR. Scale-up PCR used 23–39 amplification cycles with 1/20 to 1/80 dilutions of template material and 48 50-μL reactions. Gel-purified scale-up PCR products were digested with the anchoring enzyme NlaIII, yielding tail-to-tail ligated ∼36-bp cDNA ditags with CATG overhangs. These were gel-purified and ligated to form concatemers, which were then size fractionated by PAGE and subjected to a partial NlaIII digestion step prior to cloning. Concatemers were cloned into SphI-digested pZErO-1 vector (Invitrogen), and transformations were done using One Shot TOP10 electrocompetent E. coli (Invitrogen). Following screening of transformants by colony PCR, the best concatemer size fraction, defined as that fraction with the most favorable combination of insert-containing clones and insert length, was chosen for sequencing. DNA visualization and band recovery from preparative polyacrylamide gels were performed using a non-UV Dark Reader transilluminator (Clare Chemical Research), along with SYBR Green I dye (Invitrogen).
Colony picking was performed using a Q-Pix robot (Genetix), and inoculations were made into 2× YT media with 50 μg/mL Zeocin and 7.5% glycerol. Following overnight culture, glycerol stocks were used to inoculate larger-volume cultures for plasmid preparation using a standard alkaline lysis procedure adapted for high-throughput processing with microtiter plates (Yang et al. 2005). DNA sequencing was performed with BigDye v3.1 dye terminator cycle sequencing reactions (Applied Biosystems) run on Tetrad thermal-cyclers (Bio-Rad). Sequencing reaction products were purified by ethanol precipitation and then analyzed on model 3730xl capillary DNA sequencers (Applied Biosystems). DNA sequence data were collected and stored automatically using a custom DNA sequencing database, processed by trimming of low-quality bases from the sequence and removal of sequences from non-recombinant clones, vector DNA, and linker-derived tags. Following analysis of data quality from a first 384-well sequencing plate, each library was sequenced to completion, with an average sampling depth of 100,000 raw tags for most libraries.
Incorporation of published SAGE protocol modifications
We incorporated into our Standard Operating Procedure a number of protocol modifications, including published technical improvements. These included a concatemer heating step prior to gel electrophoresis (Kenzelmann and Muhlemann 1999), strict adherence to incubation of ditags on ice to avoid GC content bias (Margulies et al. 2001), and a partial NlaIII digest of SAGE concatemers (Gowda et al. 2004). Blue-white selection (Kirschman and Cramer 1988) of SAGE clones using the pZErO-1 vector improved tag yields by up to 20%. We did not find it necessary to use biotinylated PCR primers as an additional step to remove contaminating linker molecules following the NlaIII digestion step (Powell 1998). Instead, a 15% PAGE gel run at 200 volts (14.2 volts/cm) for 6 h in 1× TAE buffer was satisfactory for separation of the cDNA ditags, and linker-derived tag contamination was generally ≤0.01%. Digestion of the scale-up PCR products with the NlaIII anchoring enzyme also worked efficiently, avoiding the need for an additional post-PAGE purification step before digestion (Angelastro et al. 2000).
Sub-microgram LongSAGE
Over the period that our pipeline has operated, we have been able to consistently reduce the minimum amount of starting total RNA from which LongSAGE libraries can be constructed. For example, we recently constructed three LongSAGE libraries using only 50 ng of total RNA, without the need for an initial RNA or cDNA amplification step. This became feasible following the incorporation of a partial NlaIII digest of the SAGE concatemers prior to cloning, as described by Gowda et al. (2004), and resulted in a dramatic improvement in cloning efficiencies, likely due to linearization of unclonable circularized concatemers, a presumed common product of ditag concatenation.
Construction of SAGE-Lite libraries
Until the development described above, samples yielding <100 ng of total RNA were subjected to a cDNA amplification step according to the SAGE-Lite method (Peters et al. 1999). SAGE-Lite biochemistry is based upon the SMART (Switching Mechanism At the 5′ end of RNA Transcripts) cDNA synthesis strategy (Clontech) for the generation of full-length cDNA libraries. In SMART cDNA synthesis, only polyadenylated RNA molecules that have been full-length reverse transcribed are extended with a polyC tail by a terminal transferase property inherent to the reverse transcriptase. A synthetic oligonucleotide with a 3′ polyG stretch is hybridized to the first-strand cDNA and serves as a primer for synthesis of the second cDNA strand. Thus, each full-length first-strand cDNA molecule incorporated a synthetic 5′ priming site and a 3′ site (a pool of oligo dT primers with degenerate 3′ ends; i.e., 5′-T(30)VN-3′), which allowed the cDNA to be amplified using a subsequent PCR step. Following PCR amplification, the cDNA was processed according to the standard LongSAGE protocol.
Detection of cross-species ditag contamination
For every species analyzed using our SAGE pipeline, a list of virtual tags was generated computationally from available transcript DNA sequence resources and from genome sequences if they were available. Alternatively, in the case of species where transcript sequence resources were limited and a genome sequence was not available, the list of tags from a meta-library of all available SAGE libraries for the species under study was assembled. For each species, all tag sequences also found in any other species were subtracted, leaving a list of tag sequences exclusive to each species. To assess SAGE libraries for cross-species contamination, the proportion of tags from a library that matched sequences in the species-specific lists from other species was measured. We counted individual sequences only, not the total number of tags. This provided a sensitive assay for low levels of contamination.
Measuring duplicate ditag frequency
A diagnostic test was performed on every library to determine if the frequency of observed duplicate ditags was similar to that predicted from the frequencies of the individual tags. For ShortSAGE libraries a simulation was performed in which each tag from a library was assigned a random number. The tag list was then sorted by the random number, ditags were formed by combining tags adjacent in the list, and then the frequency of duplicate ditags was measured by sorting and counting the simulation results. To account for the specificity of tag-to-tag ligation in LongSAGE resulting from the two-base overhangs produced by digestion with MmeI, the process was the same except that ditags were formed only when the two-base overhangs at the 3′ ends matched to form a complementary pair. The simulated ditag frequencies were then compared with the observed frequencies. To determine the statistical significance of duplicate ditag frequencies, the distribution of log ratios of observed versus simulated frequencies was determined. Outliers, defined as libraries further than four standard deviations from the mean, were removed and the mean and standard deviation again computed. A library was deemed to be significantly different if significance was P < 0.01.
Error analysis and correction
DNA sequencing yielded tag sequences and a quality estimate for each sequenced base in the form of PHRED scores (Ewing and Green 1998; Ewing et al. 1998). These PHRED scores were combined to generate a probability of sequence error, a “quality factor (QF)”, for each tag (Siddiqui et al. 2005). We used methods similar to those of others (Colinge and Feger 2001; Beissbarth et al. 2004; Akmaev and Wang 2004) to determine the rate of single-base errors in each library and then cluster error tags. A detailed description of the approach can be found in the Supplemental methods.
The error rate was equivalent to the frequency at which off-by-one tag sequences of highly abundant tags occurred in a library (where off-by-one tag sequences are defined as containing a single base pair permutation, insertion, or deletion relative to a highly abundant tag sequence). Using the QF, we were able to identify tag sequences likely to originate from a sequencing error. By removing those tag sequences and measuring the frequency of single-base errors in the remaining tag sequences, we were able to determine the frequency of single-base errors introduced prior to sequencing (the “library construction” error rate).
Tag-to-gene mapping
SAGE tag-to-gene mapping was performed using publicly available sequence resources. The reference sequence database (RefSeq), the reference sequence pre-annotation database (RefSeqX), and the reference sequence predicted gene database (RefSeqGS) were obtained from the NCBI (ftp://ftp.ncbi.nih.gov/refseq). Ensembl transcripts and Ensembl genomic transcription units were obtained from www.ensembl.org. The Mammalian Gene Collection transcripts were obtained from ftp://ftp.ncbi.nih.gov/repository/MGC, and genome sequence data were obtained from ftp://ftp.ncbi.nih.gov/genomes. To maintain current versions of these databases on our local server, we established a process in which we matched our database update schedule to the release cycle of these host sites. RefSeq and MGC data were updated daily, while RefSeqX, RefSeqGS, and genome sequence data were updated at the time of NCBI updates, varying from 2 to 12 mo. With each update, a set of virtual tags was computed from each resource by searching for all NlaIII anchoring enzyme sites. Tables of tag sequence, accession number, genomic coordinate, and sense/antisense orientation were used to link observed tags to their potential positions in the genome or transcriptome.
Results and Discussion
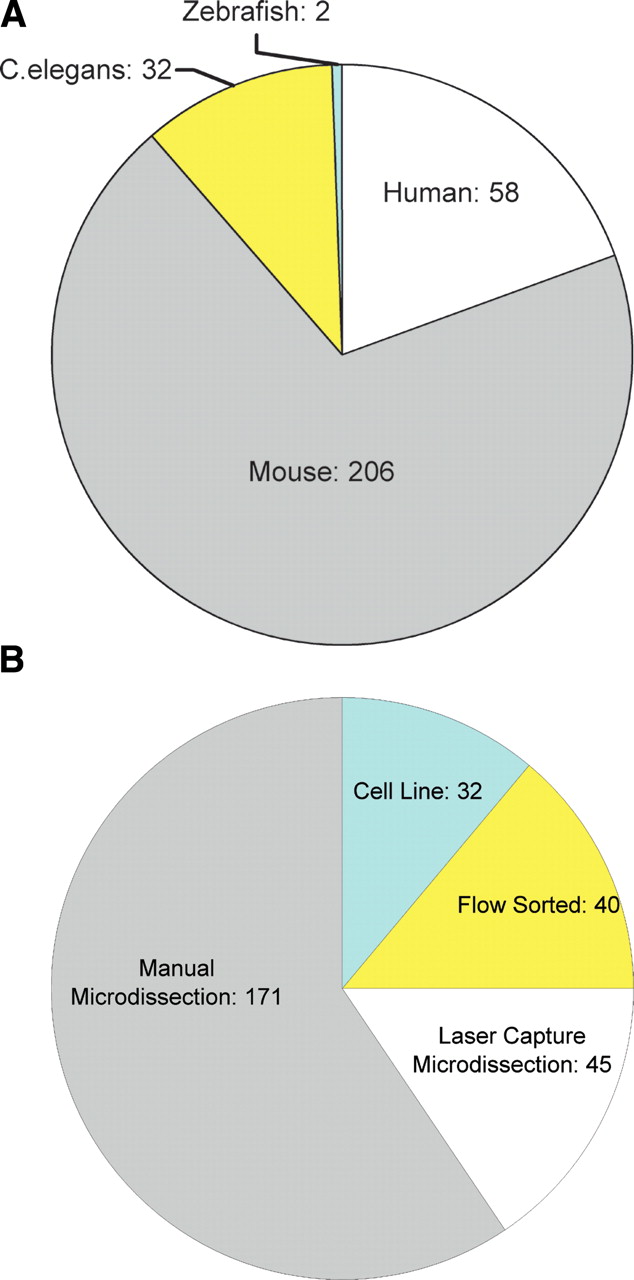
Figure 1 provides an overview of the libraries constructed using the approaches described here. As of January 2006, our platform had generated 298 libraries from four species (Supplemental Table 1), achieving a throughput of up to 12 libraries constructed per month. More than 30 million SAGE tags have been sequenced from these libraries. Fifty-eight libraries were constructed from human embryonic stem cell lines (www.transcriptomES.org) and cancer-related cell lines; 206 libraries were constructed from developing mouse tissues and cells (www.mouseatlas.org; Siddiqui et al. 2005), 32 libraries were constructed from Caenorhabditis elegans RNA samples (http://elegans.bcgsc.bc.ca; McKay et al. 2003; Halaschek-Weiner et al. 2005), and two libraries were constructed from zebrafish RNA samples.
Overview of library construction, broken down by organism (A) and by library construction method (B). Numbers of libraries are indicated.
Among the more significant adjustments to our pipeline was the incorporation of a brief NlaIII digestion prior to size fractionation and concatemer cloning (Supplemental Fig. 1). This step, suggested by Gowda et al. (2004), substantially improved cloning efficiency, which in turn yielded a cascade of improvements impacting many of the steps indicated in Supplemental Figure 1. These included a reduction in the number of ditag scale-up PCR reactions from 192 to 48, consequently reducing downstream labor and reagent costs approximately threefold. Scale-up PCR cycle numbers were likewise reduced from 30 to 25 on average, reducing the occurrence of extraneous amplicons and other PCR-derived artifacts. Library clone insert lengths improved from ∼20 ShortSAGE (14-bp) tags per clone initially to the routine generation of an average of 35–40 LongSAGE (21-bp) tags per clone, or ∼15,000 tags per 384-well plate sequenced (Supplemental Fig. 2). Higher colony titers can thus routinely yield tens of millions of tags per library. Library construction timelines have also been improved such that most libraries can now be constructed within 11 d. Most significant from the perspective of future application of LongSAGE to characterize the transcriptomes of rare cell populations (e.g., cancer stem cells, fine-needle aspirate samples, etc.) was the reduction in the requirements of total RNA to 50 ng for the construction of regular LongSAGE libraries from non-amplified starting material. For experiments involving tissue samples where accumulation of even this small amount of RNA is not possible, we have relied primarily on SAGE-Lite (Peters et al. 1999) for LongSAGE library construction. This approach, which involves an additional PCR amplification, allowed us to use our pipeline to construct 56 libraries from laser capture microdissection (LCM)-derived mouse brain regions and libraries from tissues harvested early in mouse embryogenesis (Supplemental Table 1).
RNA isolation and analysis for LongSAGE library construction was successfully performed with a variety of cell and tissue types from multiple species (Supplemental Table 1), including mouse spleen and pancreas for which purification of high-quality RNA was problematic, presumably because of the presence of elevated RNAse levels in these tissues. For degraded samples, subsequent repurification of RNA from tissues and the addition of broad-spectrum ribonuclease inhibitors such as SUPERaseIn (Ambion) were effective in producing RNA of sufficient quality. The quality of all purified RNAs was assessed using an Agilent Bioanalyzer 2100. Even so, electropherograms indicative of a high proportion of intact RNA were insufficient to guarantee that the RNA could successfully be used to generate a SAGE library. On occasion (for example, in the case of mouse spleen tissue), the RNA appeared intact, but analysis of the sample using a biochemical RNase assay (RNaseALERT) yielded a ribonuclease-positive score. Such samples usually degraded when subjected to incubation with DNase, indicating the need for an RNA re-extraction step and the use of ribonuclease inhibitors.
Continuous optimization of our SAGE library construction pipeline resulted in numerous incremental improvements affecting the rate of library construction. During the project, nearly a fourfold increase in libraries constructed per quarter was achieved during the peak library construction period (Supplemental Fig. 2), but not at the expense of library quality. For example, early efforts only rarely produced libraries yielding ≥15,000 tags per 384 clones sequenced. In contrast, such performance is typical of recently constructed libraries despite the increase in the proportion of technically challenging libraries constructed from small amounts of RNA (Supplemental Fig. 2).
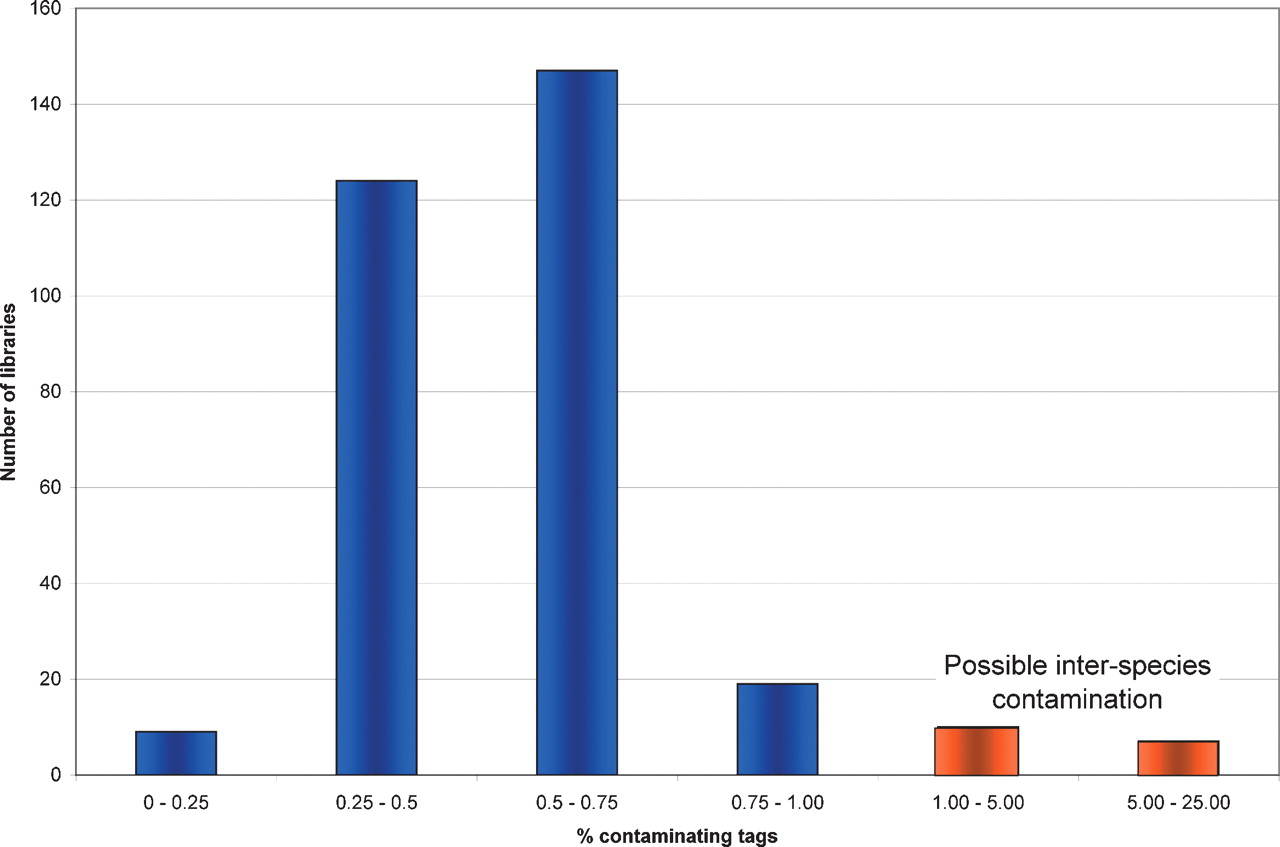
We constructed LongSAGE libraries using RNA purified from human, mouse, zebrafish, and nematode worm cells. The diversity of species analyzed using our pipeline and the PCR-intensive nature of SAGE library construction resulted in the potential for undesirable interlibrary cross-species contamination. Unless such contamination was detected, it could yield tag sequences that would fail to match sequence resources from the species under study, leading to the erroneous conclusion that previously undiscovered “novel” transcripts had been detected in the LongSAGE analysis. To address this potential problem, we sought to design a computational screen (see Methods) that could be used to analyze an initial “quality control” 384-well plate of LongSAGE sequences prior to more extensive library sequencing. In the event that the screen detected cross-species contamination, library sequencing could be aborted at an early stage. A distribution of contamination levels for all libraries analyzed is shown in Figure 2.
Distribution of potential cross-species inter-library contamination. The bars indicate the number of libraries (y-axis) contained within bins composed of libraries with contamination frequencies that fall within the ranges shown for each bin (x-axis). Percentages indicate the proportion of sequences within a library that match tag sequences we deduced to be exclusive to other species. This is a sensitive and probably maximal estimate of cross-species contamination. Five percent of the libraries assayed have contamination frequencies of ≥1%. These are the libraries we classified as potentially contaminated (red bars).
An analysis of ditags was undertaken to confirm the source and nature of cross-species tag contamination. The most prevalent contaminants were ditags from previous library preparations. In such cases, the majority of ditags in a library exhibited the property that both tags in the ditag could be mapped to the correct species, while a few ditags contained tags that could both be mapped to the “contaminating” species. We were able to distinguish such cases from those in which the contamination event occurred at an early stage prior to ditag generation and PCR amplification. In these cases, ditags should be a mixture of the three possible combinations of tags (species A/A, species B/B, and species A/B). We detected only one instance in which the majority of ditags were a mixture of two species. This was subsequently traced back to a tube mix-up in the laboratory in which two different species’ tag-adapter solutions were accidentally combined.
Many published SAGE analyses report that duplicate ditags are discarded prior to analysis under the assumption that they represent experimental (e.g., PCR) artifacts (Dinel et al. 2005). We observed that in libraries from cells and tissues known to express high levels of a few mRNA species (such as pancreatic islet cells, which express very large amounts of insulin mRNAs), the proportion of duplicate ditags was greater than in libraries with more uniform mRNA frequencies. In such libraries, we sought to assess the proportion of duplicate ditags that were due to random assortment of tags versus the proportion due to experimental artifacts. If duplicate ditags are the result of a process of random tag joining, one might expect that deeper sequence sampling from libraries would yield higher proportions of such ditags than libraries sequenced to shallower depths. A simulation based on random assortment of tags was applied to the data from all available libraries, and this was compared with the observed frequency of ditags in the libraries. In 250 out of 256 libraries analyzed in this way, the observed duplicate ditag frequency was not significantly different from the value computed in the simulation (P < 0.01; see Methods). In three libraries, the overall frequency of duplicate ditags was significantly lower than expected. These libraries each contained one or two tags that occupied 8%–17% of the total number of tag counts. In the other three out of 256 libraries, the observed ditag frequencies were significantly higher than the value produced by our simulation. These libraries were prepared with additional cycles of ditag amplification (i.e., 27, 35, and 37 cycles, instead of the 25–29 cycles normally used).
Inspection of the cloned ditag concatemer sequences revealed that in all six cases duplicate cloned concatemers, arising from process errors in the lab or in the computational pipeline, had resulted in either the resequencing or the computational re-counting of the same cloned ditag concatemer multiple times. Such multiple concatemer sequences, in which the complete sequences of the cloned ditag inserts are identical, are now removed computationally. Given our findings, we recommend that a ditag frequency diagnostic be performed as a key QC step, but we do not recommend the routine elimination or subtraction of duplicate ditags as suggested by Dinel et al. (2005).
With increasing depth of SAGE library sequencing, we observed a monotonic increase in the number of tag sequences observed only once (singletons). The majority of these singletons are likely the result of errors arising from library construction (e.g., RT and PCR errors) and DNA sequencing. However, we anticipated that the singleton class would be enriched in rare and novel transcripts as well as artifacts, and hence sought to devise a method to distinguish between these classes of singletons. We reasoned that a high-quality singleton was more likely to represent a rare transcript than to represent an artifact. We clustered our tags in a manner similar to that of Akmaev and Wang (2004) to select tags that were likely to be errors. Tags that could not be mapped to another sequence resource and which were one-off errors (a single base pair change, insertion, or deletion) of an abundant tag were counted, clustered with the more frequent parent tag, and classified as error tags. The tags in each library were analyzed in descending order of their frequency. The total error frequency in a library was estimated as the proportion of clustered one-off tags to the total number of tags.
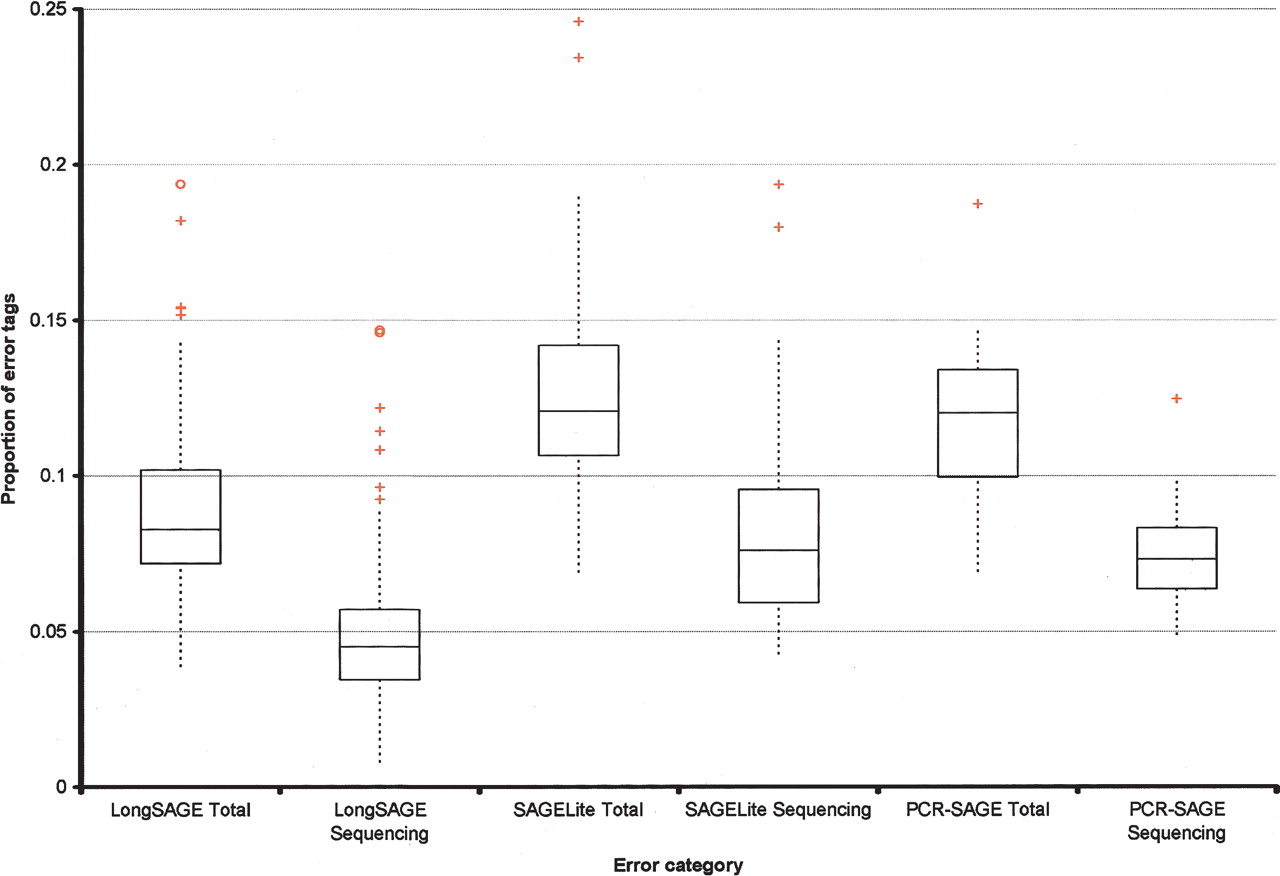
The DNA sequencing process yields phred (Ewing and Green 1998; Ewing et al. 1998) quality scores indicating the probability of error for each base. The phred scores may be combined to give a probability that a SAGE tag has a sequencing error. The total errors determined by clustering were repeated for a library at successively higher phred score cutoffs, and the asymptotic error level represented the library construction (RT, PCR) error for the library. Cumulative library construction error for all libraries is shown in Figure 3. Error rates of 3%–10% were typical, with three libraries found to contain >20% erroneous tags. SAGE-Lite LongSAGE libraries were generally found to have a higher error rate than regular LongSAGE libraries, likely due to the additional PCR-based cDNA amplification step in the former approach. Since an error probability (P-value) is computed for each tag, tag sequences may be assigned a P-value by combining the P-values of all the tags corresponding to each sequence in a library. Furthermore, tag sequences that occur in more than one library can be assigned a meta-library P-value corresponding to all the available data for a species. Note that singletons in the meta-library cannot have a very low P-value, even if the sequencing error is low, since the library construction error effectively makes the lower limit 0.03–0.06. This process reduced the number of tags with sequence errors almost completely in tag types with a frequency greater than two and substantially in tag sequences observed once or twice.
Box-and-whisker plots indicating the proportion of error tags observed in libraries constructed using LongSAGE, SAGELite, and PCR-SAGE. Error categories, on the x-axis, refer to the error attributed only to sequencing (for example, “LongSAGE Sequencing”) or to all contributing sources of error, including sequencing (for example, “LongSAGE Total”). Boxes encompass the lower and upper quartiles. (Horizontal line drawn through each box) Median error value for each category, (red crosses and circles) possible outliers. Comparing the three “Total” categories to one another and the three “Sequencing” categories to one another indicates that LongSAGE data exhibit lower proportions of error tags than the other methods. PCR-SAGE and SAGE-Lite were the methods of choice when RNA quantities were limiting. Both approaches involve additional amplification steps, compared with the LongSAGE method. It is possible that this additional amplification contributes to the observed increased proportion of error tags in these libraries.
Tag-to-genome mapping
A key aspect of SAGE-based gene expression analysis is the reliable mapping of tag sequences. If a tag maps to a transcript resource or to the genome, there is increased confidence that the tag is not an artifact. The P-value of a tag type in our mouse meta-library was observed to be improved if the tag mapped to a sequence resource. We found that >96% of error-corrected tag sequences observed more frequently than doubletons mapped to a known resource. Analysis of a sample of the remaining 4% that did not map showed that these generally corresponded to unannotated transcripts, tag sequences interrupted by splice junctions, and errors related to the evolving state of the genome sequence assembly. We noted that the probability of a tag mapping increases for highly expressed genes, perhaps reflecting a more complete state of annotation for such genes.
In a previous study (Siddiqui et al. 2005), we used a gene-specific RT-PCR approach to validate 192 high-quality mouse singleton tags selected from a meta-library of 8.55 million tags. This experiment showed that 78% of library singletons (P < 1.0 × 105) and 72% of meta-library singletons (P < 0.05) were derived from transcripts present in the RNA samples from which the tags were derived, and were not artifacts. Whether genes with such low levels of expression encode biological functions or simply represent background level transcription is unknown. The validation results above argue that consideration of P-values associated with rare transcripts may be a useful way to discriminate between experimental artifacts and rare transcripts prior to undertaking more detailed functional characterization of such transcripts.
Sequencing depth
It seems intuitive that as sequences accumulate for a SAGE library, all high-quality mRNA-derived tags will eventually be accounted for and the class of high-quality singleton tags corresponding to rare transcripts will decrease in size. At current depths of library sampling, this phenomenon was not observed.
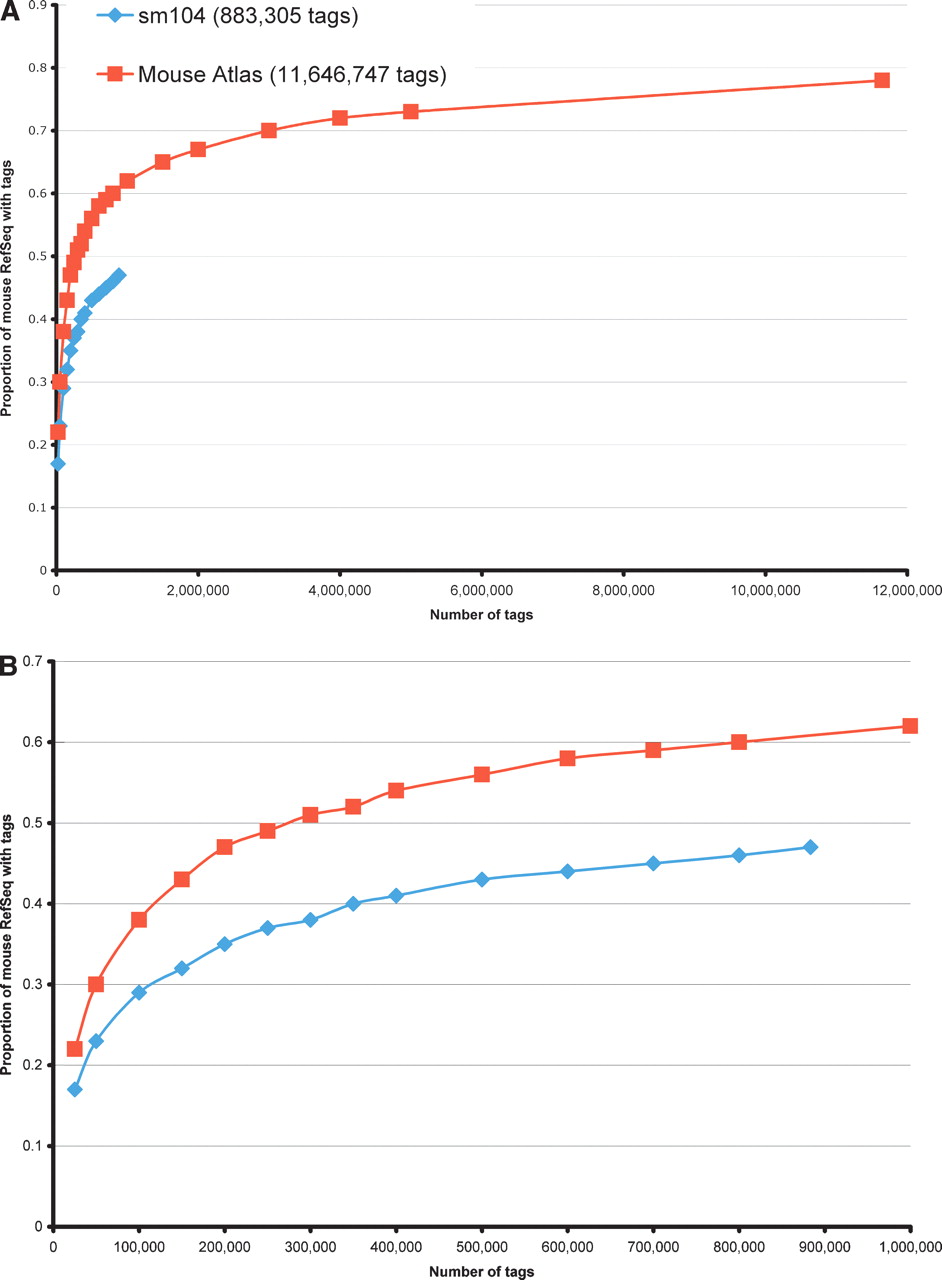
To explore the relationship between sampling depth and gene discovery, we examined the extent to which sequences in the mouse Reference Sequence database (http://www.ncbi.nlm.nih.gov/RefSeq/), a well characterized transcript database, were represented by LongSAGE tags in a 12-million-tag meta-library and in a kidney library as a function of increasing tag numbers (Fig. 4). In both libraries, we observed an initial rapid increase in RefSeq coverage with increasing tag numbers. The rate of coverage then decreased. However, for all sampling depths we analyzed (Fig. 4), the kidney LongSAGE data provided less coverage of RefSeq than the meta-library. This result was consistent with intuition and the notion that the repertoire of transcripts represented in the kidney LongSAGE data was reduced compared with the diversity of transcripts represented in the meta-library. In the case of the kidney library, Figure 4B shows that the most rapid increase in the rate of RefSeq coverage occurred in the first 100,000 tags sampled. The first 400,000 tags sampled from the kidney library represented 40% of RefSeq. The next 400,000 tags sampled provided only ∼7% of additional RefSeq coverage. Although the rate of RefSeq coverage declined, coverage of RefSeq continued to increase, indicating that even though more than 800,000 tags were generated, the kidney library was not sampled to exhaustion.
Coverage of the mouse Reference Sequence (RefSeq) data set. The proportion of entries in the mouse RefSeq database represented by a LongSAGE tag in a deeply sampled LongSAGE library (sm104 kidney, >800,000 tags; blue diamonds) and in a meta-library of Mouse Atlas tags (>11,000,000 tags; red squares) is shown. Plotted on the y-axis is the proportion of RefSeq covered. The number of tags is plotted on the x-axis. (A) With sequential sampling, performed computationally, both data sets exhibit rapid coverage of a subset of RefSeq as tag count increases. As expected, the meta-library exhibits superior coverage. (B) The same data, but with an expanded scale along the x-axis to better display the kidney library data.
Conclusions
We describe a SAGE library construction pipeline that has been devised and implemented to generate high-quality digital gene expression profiling data. The pipeline incorporates many previously published improvements and synthesizes these into a standard operating procedure (SOP) suitable for use by entry-level technical staff, following a 2-wk training period. We developed and validated our SOP by using it to generate 298 libraries yielding >30 million tags over a 4-yr period with a small group of library construction technicians (five to eight people) and variable adjustments in the scale of activity to match changing demands for libraries during that period. All data and software tools for data analysis are available at www.transcriptomES.org (embryonic stem cell data), www.mouseatlas.org (mouse data), or http://elegans.bcgsc.bc.ca (C. elegans data). These protocols should be applicable to other academic centers and facilitate the exploitation of SAGE for additional gene expression analyses and gene discovery.
Acknowledgments
This work was supported by funding from Genome Canada and the National Cancer Institute (USA). We are indebted to numerous groups at Canada’s Michael Smith Genome Sciences Centre, including the Administration, Projects, Operations, LIMS, and IT Systems teams, who have provided expert assistance in constructing and maintaining a large-scale SAGE library construction effort at our Genome Centre. We gratefully acknowledge the following individuals for providing RNA and tissue for library construction: Elizabeth M. Simpson, Cheryl Helgason, James Thomson, Martin Pera, Meri Firpo, Catherine Verfaille, Donald Riddle, Jim McGhee, Isabella Tai, Ralph Durand, Andrew Van Kessel, David Baillie, and Donald Moerman. M.M., P.H., R.H., and S.J. are scholars of the Michael Smith Foundation for Health Research. M.M. is a Terry Fox/NCIC Young Investigator. P.H. is a Canadian Institutes of Health Research New Investigator.
References
- ↵V.R. Akmaev,C.J. Wang,(2004) Correction of sequence-based artifacts in serial analysis of gene expression. Bioinformatics 20:1254–1263.
- ↵J.M. Angelastro,L.P. Klimaschewski,O.V. Vitolo,(2000) Improved NlaIII digestion of PAGE-purified 102 bp ditags by addition of a single purification step in both the SAGE and microSAGE protocols. Nucleic Acids Res. 28:E62.
- ↵T. Beissbarth,L. Hyde,G.K. Smyth,C. Job,W.M. Boon,S.S. Tan,H.S. Scott,T.P. Speed,(2004) Statistical modeling of sequencing errors in SAGE libraries. Bioinformatics 20:I31–I39.
- ↵S.T. Bennett,C. Barnes,A. Cox,L. Davies,C. Brown,(2005) Toward the $1000 human genome. Pharmacogenomics 6:373–382.
- ↵J. Chen,I. Sadowski,(2005) Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements. Proc. Natl. Acad. Sci. 102:4813–4818.
- ↵J. Colinge,G. Feger,(2001) Detecting the impact of sequencing errors on SAGE data. Bioinformatics 17:840–842.
- ↵S. Dinel,C. Bolduc,P. Belleau,A. Boivin,M. Yoshioka,E. Calvo,B. Piedboeuf,E.E. Snyder,F. Labrie,J. St-Amand,(2005) Reproducibility, bioinformatic analysis and power of the SAGE method to evaluate changes in transcriptome. Nucleic Acids Res. 33:e26.
- ↵B. Ewing,P. Green,(1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8:186–194.
- B. Ewing,L. Hillier,M.C. Wendl,P. Green,(1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8:175–185.
- ↵M. Gowda,C. Jantasuriyarat,R.A. Dean,G.L. Wang,(2004) Robust-LongSAGE (RL-SAGE): A substantially improved LongSAGE method for gene discovery and transcriptome analysis. Plant Physiol. 134:890–897.
- ↵J. Halaschek-Wiener,J.S. Khattra,S. McKay,A. Pouzyrev,J.M. Stott,G.S. Yang,R.A. Holt,S.J. Jones,M.A. Marra,A.R. Brooks-Wilson,(2005) Analysis of long-lived C. elegans daf-2 mutants using serial analysis of gene expression. Genome Res. 15:603–615.
- ↵A.M. Heidenblut,J. Luttges,M. Buchholz,C. Heinitz,J. Emmersen,K.L. Nielsen,P. Schreiter,M. Souquet,S. Nowacki,U. Herbrand,(2004) aRNA-longSAGE: A new approach to generate SAGE libraries from microdissected cells. Nucleic Acids Res. 32:e131.
- ↵S. Impey,S.R. McCorkle,H. Cha-Molstad,J.M. Dwyer,G.S. Yochum,J.M. Boss,S. McWeeney,J.J. Dunn,G. Mandel,R.H. Goodman,(2004) Defining the CREB regulon: A genome-wide analysis of transcription factor regulatory regions. Cell 119:1041–1054.
- ↵M. Kenzelmann,K. Muhlemann,(1999) Substantially enhanced cloning efficiency of SAGE (Serial Analysis of Gene Expression) by adding a heating step to the original protocol. Nucleic Acids Res. 27:917–918.
- ↵J. Kim,A.A. Bhinge,X.C. Morgan,V.R. Iyer,(2005) Mapping DNA–protein interactions in large genomes by sequence tag analysis of genomic enrichment. Nat. Methods 2:47–53.
- ↵J.A. Kirschman,J.H. Cramer,(1988) Two new tools: Multi-purpose cloning vectors that carry kanamycin or spectinomycin/streptomycin resistance markers. Gene 68:163–165.
- ↵R. Kodzius,M. Kojima,H. Nishiyori,M. Nakamura,S. Fukuda,M. Tagami,D. Sasaki,K. Imamura,C. Kai,M. Harbers,(2006) CAGE: Cap analysis of gene expression. Nat. Methods 3:211–222.
- ↵Y.H. Loh,Q. Wu,J.L. Chew,V.B. Vega,W. Zhang,X. Chen,G. Bourque,J. George,B. Leong,J. Liu,(2006) The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 38:431–440.
- ↵E.H. Margulies,S.L. Kardia,J.W. Innis,(2001) Identification and prevention of a GC content bias in SAGE libraries. Nucleic Acids Res. 29:e60.
- ↵M. Margulies,M. Egholm,W.E. Altman,S. Attiya,J.S. Bader,L.A. Bemben,J. Berka,M.S. Braverman,Y.J. Chen,Z. Chen,(2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380.
- ↵H. Matsumura,S. Reich,A. Ito,H. Saitoh,S. Kamoun,P. Winter,G. Kahl,M. Reuter,D.H. Kruger,R. Terauchi,(2003) Gene expression analysis of plant host–pathogen interactions by SuperSAGE. Proc. Natl. Acad. Sci. 100:15718–15723.
- ↵S.J. McKay,R. Johnsen,J. Khattra,J. Asano,D.L. Baillie,S. Chan,N. Dube,L. Fang,B. Goszczynski,E. Ha,(2003) Gene expression profiling of cells, tissues, and developmental stages of the nematode C. elegans. Cold Spring Harb. Symp. Quant. Biol. 68:159–169.
- ↵L. Neilson,A. Andalibi,D. Kang,C. Coutifaris,J.F. Strauss, IIIJ.A. Stanton,D.P. Green,(2000) Molecular phenotype of the human oocyte by PCR-SAGE. Genomics 63:13–24.
- ↵D.G. Peters,A.B. Kassam,H. Yonas,E.H. O’Hare,R.E. Ferrell,A.M. Brufsky,(1999) Comprehensive transcript analysis in small quantities of mRNA by SAGE-lite. Nucleic Acids Res. 27:e39.
- ↵J. Powell,(1998) Enhanced concatemer cloning-a modification to the SAGE (Serial Analysis of Gene Expression) technique. Nucleic Acids Res. 26:3445–3446.
- ↵B. Ren,F. Robert,J.J. Wyrick,O. Aparicio,E.G. Jennings,I. Simon,J. Zeitlinger,J. Schreiber,N. Hannett,E. Kanin,(2000) Genome-wide location and function of DNA binding proteins. Science 290:2306–2309.
- ↵S. Saha,A.B. Sparks,C. Rago,V. Akmaev,C.J. Wang,B. Vogelstein,K.W. Kinzler,V.E. Velculescu,(2002) Using the transcriptome to annotate the genome. Nat. Biotechnol. 20:508–512.
- ↵A. Schroeder,O. Mueller,S. Stocker,R. Salowsky,M. Leiber,M. Gassmann,S. Lightfoot,W. Menzel,M. Granzow,T. Ragg,(2006) The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 7:3.
- ↵J. Shendure,R.D. Mitra,C. Varma,G.M. Church,(2004) Advanced sequencing technologies: Methods and goals. Nat. Rev. Genet. 5:335–344.
- ↵A.S. Siddiqui,J. Khattra,A.D. Delaney,Y. Zhao,C. Astell,J. Asano,R. Babakaiff,S. Barber,J. Beland,S. Bohacec,(2005) A mouse atlas of gene expression: Large-scale digital gene-expression profiles from precisely defined developing C57BL/6J mouse tissues and cells. Proc. Natl. Acad. Sci. 102:18485–18490.
- ↵D.E. Smailus,A. Marziali,P. Dextras,M.A. Marra,R.A. Holt,(2005) Simple, robust methods for high-throughput nanoliter-scale DNA sequencing. Genome Res. 15:1447–1450.
- ↵V.E. Velculescu,L. Zhang,B. Vogelstein,K.W. Kinzler,(1995) Serial analysis of gene expression. Science 270:484–487.
- ↵C.L. Wei,P. Ng,K.P. Chiu,C.H. Wong,C.C. Ang,L. Lipovich,E.T. Liu,Y. Ruan,(2004) 5′ Long serial analysis of gene expression (LongSAGE) and 3′ LongSAGE for transcriptome characterization and genome annotation. Proc. Natl. Acad. Sci. 101:11701–11706.
- ↵C.L. Wei,Q. Wu,V.B. Vega,K.P. Chiu,P. Ng,T. Zhang,A. Shahab,H.C. Yong,Y. Fu,Z. Weng,(2006) A global map of p53 transcription-factor binding sites in the human genome. Cell 124:207–219.
- ↵G.S. Yang,J.M. Stott,D. Smailus,S.A. Barber,M. Balasundaram,M.A. Marra,R.A. Holt,(2005) High-throughput sequencing: A failure mode analysis. BMC Genomics 6:2.
Notes
[1] [Supplemental material is available online at www.genome.org.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.5488207