
Abstract
Approximately 90% of Arabidopsis’ unique gene content is found in syntenic blocks that were formed during the most recent whole-genome duplication. Within these blocks, 28.6% of the genes have a retained pair; the remaining genes have been lost from one of the homeologs. We create a minimized genome by condensing local duplications to one gene, removing transposons, and including only genes within blocks defined by retained pairs. We use a moving average of retained and non-retained genes to find clusters of retention and then identify the types of genes that appear in clusters at frequencies above expectations. Significant clusters of retention exist for almost all chromosomal segments. Detailed alignments show that, for 85% of the genome, one homeolog was preferentially (1.6×) targeted for fractionation. This homeolog fractionation bias suggests an epigenetic mechanism. We find that islands of retention contain “connected genes,” those genes predicted—by the gene balance hypothesis—to be resistant to removal because the products they encode interact with other products in a dose-sensitive manner, creating a web of dependency. Gene families that are overrepresented in clusters include those encoding components of the proteasome/protein modification complexes, signal transduction machinery, ribosomes, and transcription factor complexes. Gene pair fractionation following polyploidy or segmental duplication leaves a genome enriched for “connected” genes. These clusters of duplicate genes may help explain the evolutionary origin of coregulated chromosomal regions and new functional modules.
There is strong evidence for a past of tetraploidy or near tetraploidy in eukaryotic genomes (Vision et al. 2000; McLysaght et al. 2002; Vandepoele et al. 2003), including yeast (Wolfe and Shields 1997) and all flowering plant genomes (Vision et al. 2000; Blanc et al. 2000, 2003; Bowers et al. 2003; Maere et al. 2005; Paterson et al. 2006). The general evolutionary process of gene duplication, occasional retention, and subsequent divergence to new function has much case support (Lewis 1951; Ohno 1970; Li 1997; Kellis et al. 2004), although the typical fate of any gene duplicate is loss (Haldane 1933; Lynch and Force 2000). Arabidopsis thaliana, a dicot flowering plant (125-Mb genome with ∼26,000 annotated genes) (The Arabidopsis Genome Initiative 2000), has retained duplicates in overlapping syntenous blocks that have been explained by multiple tetraploidy events: Bowers et al. (2003) suggested three events, the youngest denoted by α. Vision et al. (2000) suggested five waves of segmental duplications rather than tetraploidies. Using synonymous nucleotide substitution rate data, Maere et al. (2005) called the most recent event “3R,” and dated it considerably more recently than did Bowers and coworkers. The most recent event is likely to have been a whole-genome duplication and not segmental because the comparative gene tree approach (Chapman et al. 2004) used by Bowers and coworkers covers ∼80% of the Arabidopsis genome once only. Because we begin with the unique retained pairs list provided by Bowers et al. (2003), we retain their nomenclature in referring to the most recent tetraploidy event as α.
Genes classified by different gene ontology (GO) categories are retained at different rates following tetraploidy. In yeast, Papp et al. (2003) found that genes encoding ribosomal proteins are over-retained as pairs and predicted that genes encoding transcription factors might be over-retained following tetraploidy events in higher eukaryotes. Indeed, genes encoding “transcriptional regulators” and protein kinases are significantly over-retained in Arabidopsis (Blanc and Wolfe 2004; Seoighe and Gehring 2004; Maere et al. 2005). Genes encoding rice transcription factors are vastly over-retained, at 50% compared to an average of 16%, following the most recent tetraploidy in the grass lineage (Tian et al. 2005). Each of these research groups also found GO categories of genes that are under-retained; these tended to be involved with DNA repair and modification, biochemistry involving few subunit–subunit interactions, and genes involved in particularly ancient biochemical processes.
According to the gene balance hypothesis, a gene displays dosage effects increasingly as the subunit–subunit interactions of its product increase (protein quaternary structural complexity), or from interactions with products downstream in a regulatory cascade, particularly through the interaction of positive and negative regulatory effectors (Veitia 2002; Papp et al. 2003; Birchler et al. 2005). We refer to genes showing dosage sensitivity as “connected genes.” The term “connected gene” is necessarily fuzzy in order to include a variety of dose-sensitivity mechanisms. The extreme opposite of connected genes is genes whose products work alone. Given a new autotetraploid, nonconnected genes can be removed without creating any selective consequence, but loss of a connected gene instantly creates a haploinsufficient phenotype and lowered fitness. Thus, connected genes tend to increase relative to nonconnected genes as a result of repeated tetraploidies (Seoighe and Gehring 2004; Maere et al. 2005). If connected genes are located on a chromosomal segment(s), then segmental duplications could also duplicate genes without altering dosage.
Following a duplication event, fractionation back toward the nonduplicated state is known to occur (Lockton and Gaut 2005). If the duplication is whole genome (tetraploidy) rather than segmental, this fractionation is sometimes called “diploidization.” The purpose of our study is to better understand the mechanisms that cause fractionation and the clustering of duplicate genes retained from tetraploidy, and to evaluate their significance during evolution. The trend of increasing morphological complexity during eukaryotic evolution has been explained, theoretically, as a predictable consequence of this fractionation mechanism (Freeling and Thomas 2006).
Methods
Synteny Viewer
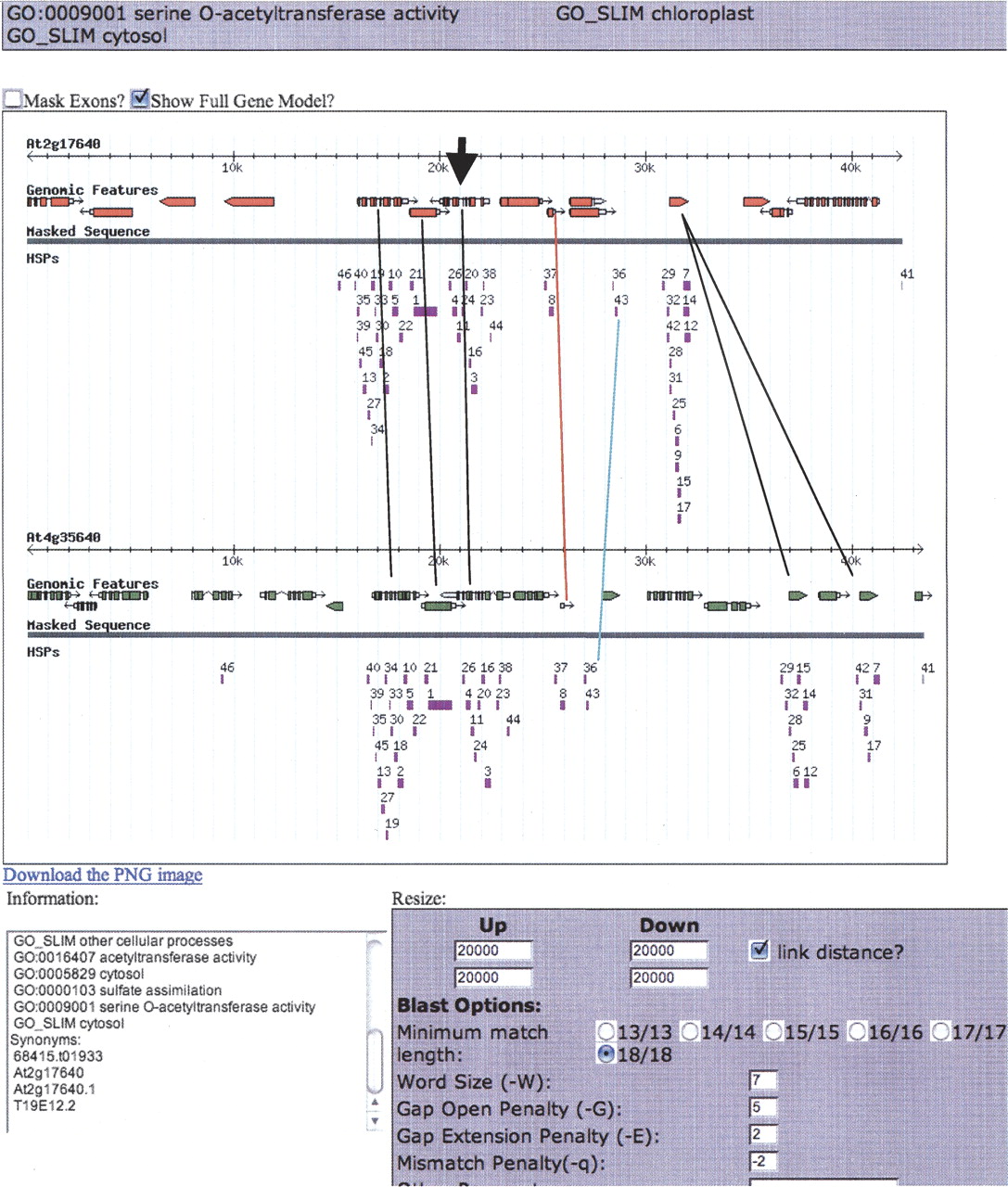
Assembly Version 5 of the annotation data for all five chromosomes in Arabidopsis was extracted from TIGR (http://www.tigr.org/tdb/e2k1/ath1). These data were parsed into a MySQL database using custom Perl language scripts. Visual inspection of syntenic regions was accomplished using a software tool we developed called Synteny Viewer. A public version of this Viewer is available at http://synteny.cnr.berkeley.edu/AtCNS. The Synteny Viewer software is a series of databases and Perl scripts that produce and display dynamic images of syntenic pairs and their BLAST high scoring pairs (HSPs). The images are displayed via a Web browser, wherein each object on the image is a link that displays information about that object, such as genomic location, sequence, orientation, GenBank annotation, EST support, GO product designations, structural information, and so on. The syntenic chromosomal pairs are compared using bl2seq (Tatusova and Madden 1999), with parameters as defined by Inada et al. (2003) in a window that is usually ±10 kb around an α-gene pair. The Viewer presents further alignment, secondary structure, and RNA expression tools to expedite analyses of DNA sequences. We used each of 3822 pairs provided by Bowers et al. (2003) to anchor the Viewer to syntenous regions and observed the patterns of retained sequences in the flanking chromosomal areas. High scoring pairs (HSPs) displayed with version 5 TIGR model annotation were initially observed in a 20-kb window (40 kb if homeologs were oriented +/−) and expanded to 100 kb or until synteny was visualized. Figure 1 is a typical screenshot of a cluster of retained genes anchored by α-pair AON075: AT2G17640–AT4G35640. Using this tool, it became visually apparent that retained genes were clustered.
Partial screenshot of a “cluster” in our Viewer aligning 42–43 kb of the α-syntenous region of chromosomes 2 and 4 anchored on a serine o-acetyltransferase gene pair (bold arrowhead). The colored rectangles are bl2seq HSPs (high scoring pairs) found using standard settings and e-value cutoffs (Inada et al. 2003) noted in the settings box. Black lines connect known α-pairs of genes. The red line connects genes into a pair whose subject genes (exons) were not called by TIGR, and is now called an “Our Additional” (_oa) gene in Supplemental material 1, Column A. The turquoise line connects two groups of syntenous HSPs that required further research to explain; these were eventually called “conserved non-coding sequences” belonging to the gene pair to the left.
Refining gene pairs
Bowers et al. (2003) published a list of gene pairs retained from the most recent tetraploidy event. This work used the version 3 GenBank annotation. The gene list is particularly useful because the authors’ investigation used a comparative phylogenetic gene tree approach to discriminate three stages of tetraploidy (we are using pairs generated by the most recent event only) and because they organized their list into 26 syntenous blocks and several smaller regions. Subsequent to the publication of their gene list, version 5 gene annotations replaced version 3. We manually proofed at least 20 kb of sequence surrounding each of the 3822 Bowers gene pairs using our Viewer. Bowers and coworkers called some, but not all, possible gene pairs within clusters of locally duplicate genes. For the purposes of this analysis, we used our Viewer to manually condense duplications, invalidating all but one among local duplicates. We considered a cluster of duplicate genes to be broken if three or more genes intervened or if the duplicate genes aligned poorly (<50% coverage of exons using bl2seq with a mismatch penalty of −1) and were also more identical to an additional α-gene. If no such “better alignment” was available, a sequence domain match was enough to condense a local duplicate into a single duplicate gene space—a situation that was very rare. We invalidated very few genes as being unalignable. Additionally, we found some new gene pairs in which both members of the pair were annotated but missed on the Bowers list, in which only one gene had annotation, or in which both genes lacked any annotation; we added these new pairs as “Our Additional” (designated OA α-pairs). In all, we condensed the original 3822 pairs to 3178 (Supplemental material 1). Of the 129 OA gene pairs, about half were actually on the Bower’s list but off by a few genes, or did not translate perfectly from version 3 to version 5. In 33 cases involving annotation error, a gene is used for defining two pairs rather than one (Supplemental material 1, Columns G and I).
Condensing the genome
In order to include all genes used in α-pairs, we needed to add “Our Additional” (OA) genes to the 29,957 total TIGR protein-coding genes for a total of 30,039 (Supplemental material 1, Column A; _oa genes are identified). This near-complete CDS genome includes local duplicates and transposons. Local duplicates were identified using Haberer et al.’s (2004) local duplication list. We used this list to condense each local cluster of genes to one duplicate gene space: The first gene in a local duplicate set was selected, unless we used a different gene in our α-pair editing (above). Transposon genes were removed using the following annotation keywords: retrotransposon, retro, Mutator, hAT-like, hobo, mutator-like, CACTA-like, transposase, reverse, copia-like, retroelement, Athila, non-LTR, IS-element, IS4, and hAT dimerization. The result of condensation and transposon removal is to reduce the 30,039 genes in Supplemental material 1, Column A to the 25,219 genes, the “Minimized Genome,” of Supplemental material 1, Column C.
Preparing an α-genome
Genes not falling within α-blocks and transposons were omitted from the genome in order to prepare alignments and run moving averages (Supplemental material 1, Column D).
The 22,209 genes that remain are referred to as the minimized genome, which is one measure of a total genome that includes all of the paired regions from the most recent tetraploidy event. These regions comprise ∼88% of the minimized genome, and are color-coded in Supplemental material 1.
Bowers et al. (2003) did not treat each homeolog in the syntenic region identically. Referring to the components of the homeologous pair as a and b, respectively, the a homeolog (Table 1) does not have alignment gaps of >20 genes. For the purposes of applying moving averages, gaps >20 genes in length were also removed from the b homeolog.
Gene content and retention data for homeologous chromosomal segments and gene clusters
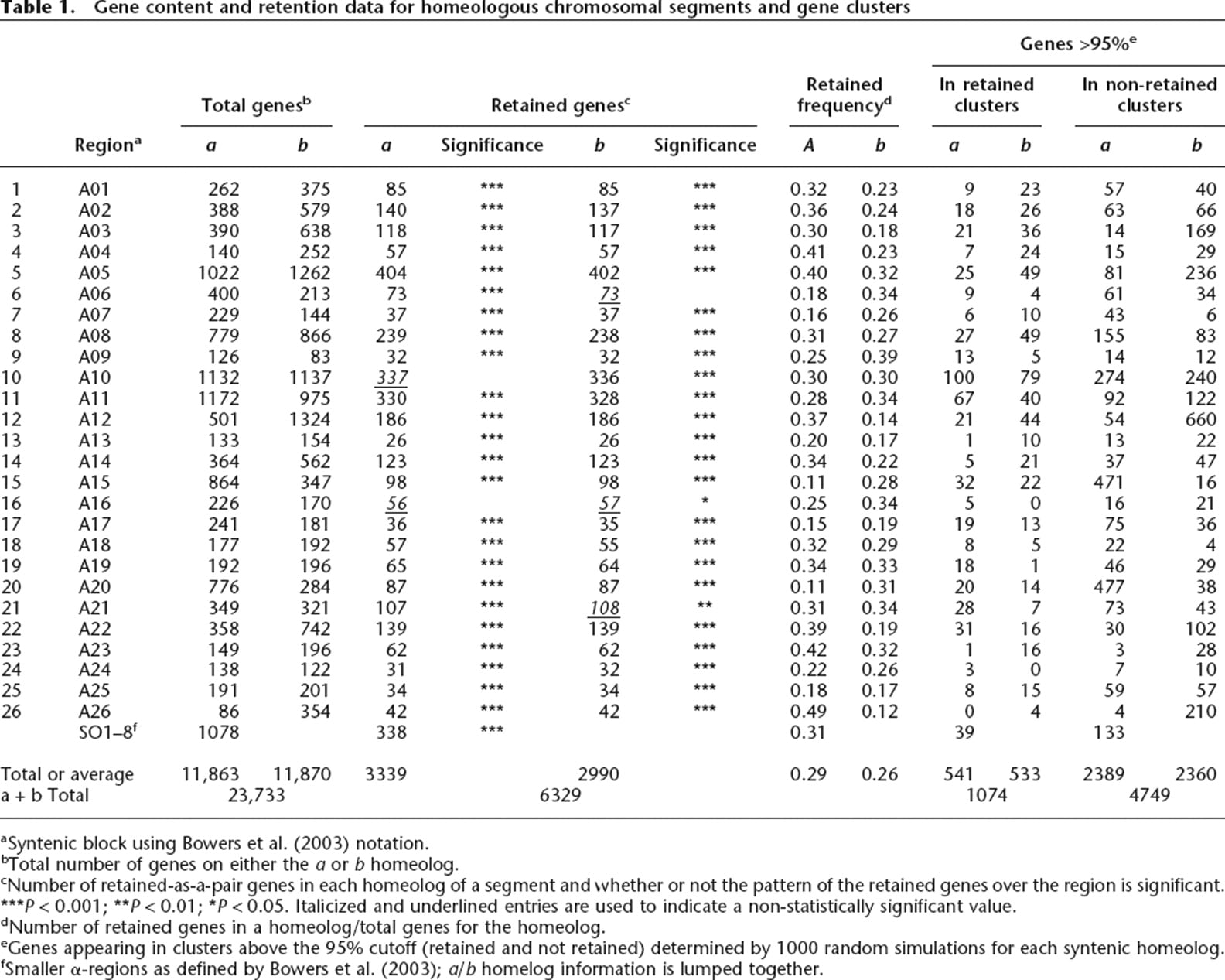
aSyntenic block using Bowers et al. (2003) notation.
bTotal number of genes on either the a or b homeolog.
cNumber of retained-as-a-pair genes in each homeolog of a segment and whether or not the pattern of the retained genes over the region is significant. ***P < 0.001; **P < 0.01; *P < 0.05. Italicized and underlined entries are used to indicate a non-statistically significant value.
dNumber of retained genes in a homeolog/total genes for the homeolog.
eGenes appearing in clusters above the 95% cutoff (retained and not retained) determined by 1000 random simulations for each syntenic homeolog.
fSmaller α-regions as defined by Bowers et al. (2003); a/b homelog information is lumped together.
Clustering statistics
Test for randomness (global)
The purpose of this test is to determine if the pattern of gene retention within the minimized genome is random. We begin with the null hypothesis that the pattern of retention is random (even though Viewer observations indicated otherwise). Each of the syntenic blocks was represented as a string of 1s and 0s, 1 for retained, 0 for non-retained. This binary sequence was then divided into non-overlapping bins of 10 genes. The mean value of each bin was calculated and stored. If the mean value in each bin is, on average, close to the mean rate of retention for the region, the sequence is random. However, if many bins have a high mean value and the others have a low mean value, this indicates clustering of retention, and the sequence is not random. The randomness of the sequence was tested for each block with the G-test (Sokal and Rohlf 1995) using the calculated mean values for all bins and the expected mean value, which is the retention rate for the entire α-region being surveyed. The G-statistic is then compared to a χ2 table to determine the probability that the sequence is random.
Test for clustering (local)
In order to find local clusters of retention, we used the same 1s and 0s notation and generated 1000 random sequences with the same length and, on average, the same rate of retention (same number of 1s) as the actual sequence for each α homeolog. A window size of 10 genes was used to calculate a moving average for each of these 1000 random sequences. In this test, the windows were overlapping. This set of averages was then sorted, and the fifth and 95th percentile values were stored. We used the average percentile levels of the 1000 simulations to delineate confidence intervals for assessing significant clustering in the data from the real α-regions. To find these clusters, we then calculated the moving average on the actual data to find locations where the real moving average was greater than (less than) the 95th (fifth) percentile values derived from the simulations. After calculating the moving average for the actual sequence, significant clusters were identified as those having a moving average outside of the confidence interval. The genes at those locations are in local clusters of retention, and may be retained or not retained.
Aligning α-regions
Each α-region has a query and a subject designation. The manner in which α-regions were identified prevented query-to-query and subject-to-subject overlap. However, in some cases, subject-to-query overlap exists. Infrequently, a gene is contained within the boundaries of two distinct α-regions. This redundancy is apparent in the difference between the total of homeolog genes from Table 1 (23,733) and the actual value of 22,209 genes from α-space.
Were it not for the 33 cases of double gene usage (Methods; Supplemental material 1, Column G), the two homeologs of a syntenic region would have identical numbers of retained genes (1s) but different numbers of non-retained genes (0s). Knowing this, we wrote a Perl script to align the 1s and distribute the 0s for each region. Because the number of non-retained genes on one α homeolog is often not the same as the other, we introduced gaps (_) on the homeolog with fewer non-retained genes to indicate this disparity and to maintain the alignment of the retained genes.
The alignment starts with the first retained gene on the a homeolog and the first retained gene on the b homeolog. For example, if between the first and second retained genes there are four non-retained genes on a (100001) and one non-retained on b (101) existed between the first and second retained genes, we introduce three gaps (___) on the b homeolog to maintain the alignment. Since it was not possible to know where the non-retained (singlet) genes were positioned relative to each homeolog, the 0s were lumped together at the left-most (lower-numbered) boundary of the gap. Therefore, the beginning of the alignment will be 100001 for the a homeolog, and 10___1 for the b homeolog. This alignment process continued to the end of the α-region, filling in the 0s and _s between retained genes. This alignment was then graphically displayed using the 0/1 designations to color-code the α-region alignment figures (see Fig. 3 below; Supplemental material 2). We used the HSP strand orientation in relation to the direction of transcription to note where changes in block orientation occurred—this located inversions. We use a color code to denote the components of our alignment on our graphic representations: blue is a retained gene, gray is a non-retained (singlet) gene, white is a gap, and inversion breakpoints are where red meets green.
We estimated the significance of fractionation bias by calculating the ratio of non-retained genes in the a and b homeologs using the side with the greater value as the numerator so that the ratio is always >1. We first reduced any run of non-retained genes longer than 20 down to 20. This was necessary to apply the same gaps rules to both homeologs; the original methods of Bowers et al. (2003) defined an α-region by requiring that the lower-numbered homeolog have no gap longer than 20 bp—this reduction actually reduces the bias. Since homeologs are treated identically, the null hypothesis is that each homeolog region carries an equal number of singleton genes, and also that gaps of any value, from one to 20 genes, should be distributed equally between homeologs. We test our data for departure from the 1:1 expectation using a two-tailed binomial distribution to assess significance.
Gene Ontology
Each gene in the minimized genome was categorized according to its most recent GO annotations, obtained from The Arabidopsis Information Service (TAIR, June 2005). We recorded the number of times each GO term was associated with a retained gene, a non-retained gene, or a gene above the 95th percentile (in a local cluster). If a gene had a particular GO term annotation, we would increment that term’s count based on whether the gene was retained, non-retained, or in a retained cluster. In this manner, we calculated total frequencies of GO terms in syntenic regions and in retained clusters. It is important to note that these are frequencies of appearance of GO terms, not frequencies of appearance of genes, since a single gene may be associated with many GO terms.
Results
Clustering: Genome-wide
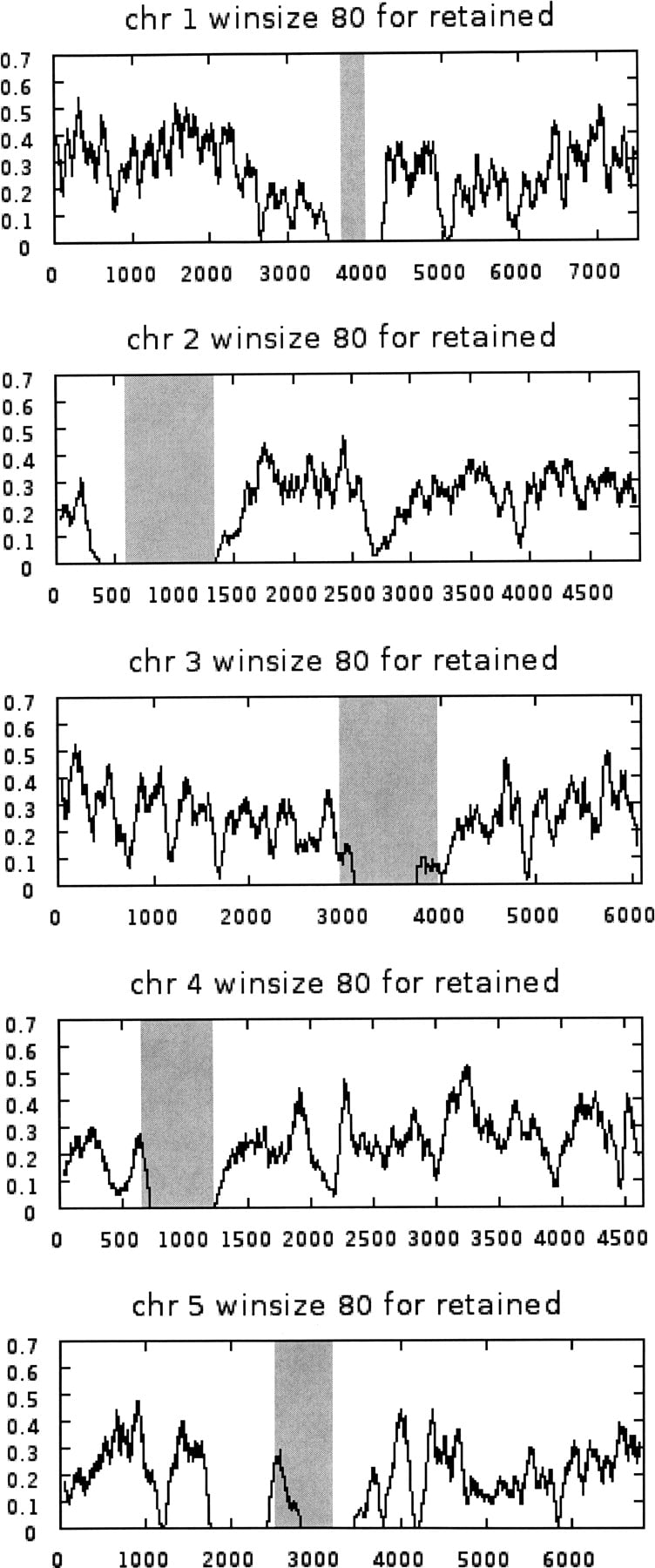
Figure 2 shows the results of running a moving average (80-gene window size) over the entire genome (Supplemental material 1, Column A), including local duplications and transposons, plotting average frequency of retained genes (Supplemental material 1, Column E) on the y-axis. Note that the centromeric regions (shaded) are either void or very low in retained genes.
Moving average gene retention frequency (y-axis) in an 80-gene window for each of the five Arabidopsis chromosomes. Chromosomes are represented by all genes encoding protein (Supplemental material 1, Column A), including genes duplicated locally and genes within transposons. The gray bands cover centromeric regions delineated by the most proximal genes with a known mutant phenotype (Meinke et al. 2003).
Alignment diagrams for homeologs and fractionation bias
Figure 3 shows the alignment diagrams for a representative three of the 26 larger and eight smaller (SO) α-regions identified by Bowers and coworkers. Two of these example alignments illustrate a surprising result: One of the homeologs has lost significantly more genes than the other during the process of fractionating the tetraploid back toward the diploid. Figure 3 A14, α-region 14, exemplifies a region where fractionation is significantly biased (Table 2A). Note that much of the white space, gaps inserted to maintain this alignment (Methods), is on one of the homeologs—a sign of biased fractionation. Fractionated genes (gray) are now singletons, and retained genes are the blue pairs. Detailed inspection of the alignment diagram of Figure 3 A14 found no indication that the preferred homeolog switched from one to the other chromosome, as one might expect if fractionation rates were set soon after tetraploidy, and then limited homeologous chromosomal recombination occurred. The red/green bar at the top of A14 shows the breakpoints for four small inversions. Following the procedure of Bowers et al. (2003), we inverted the red regions on the larger-numbered (b) homeolog in order to obtain the syntenous A14 block (Methods).
Summary of fractionation bias by α-region
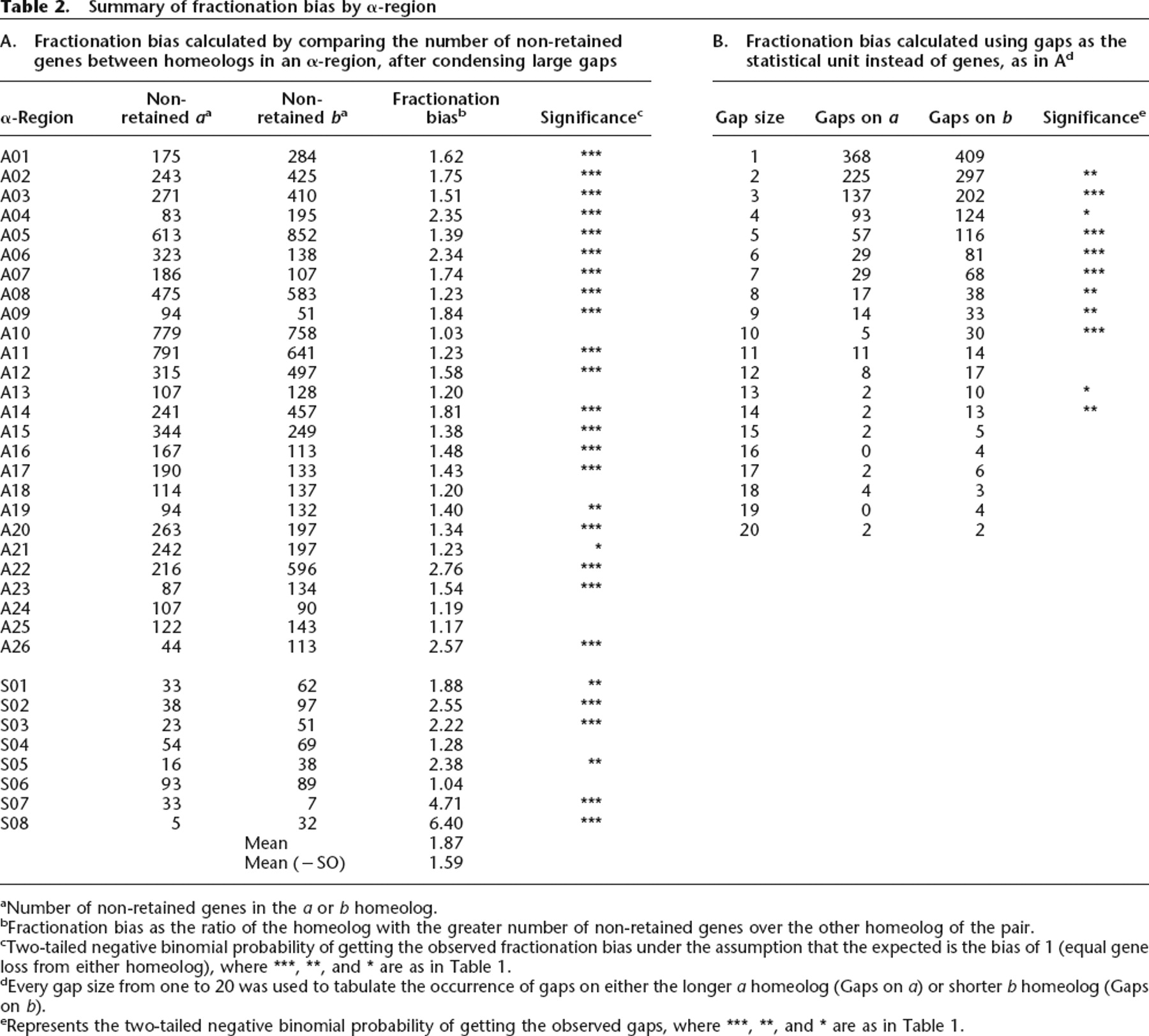
aNumber of non-retained genes in the a or b homeolog.
bFractionation bias as the ratio of the homeolog with the greater number of non-retained genes over the other homeolog of the pair.
cTwo-tailed negative binomial probability of getting the observed fractionation bias under the assumption that the expected is the bias of 1 (equal gene loss from either homeolog), where ***, **, and * are as in Table 1.
dEvery gap size from one to 20 was used to tabulate the occurrence of gaps on either the longer a homeolog (Gaps on a) or shorter b homeolog (Gaps on b).
eRepresents the two-tailed negative binomial probability of getting the observed gaps, where ***, **, and * are as in Table 1.
Three representative homeolog alignments showing different levels of fractionation bias. (A23) Typical α-region showing significant fractionation bias. (A14) Very significant fractionation bias. (A13) Insignificant fractionation bias. Each diagram is color-coded: retained genes are blue vertical lines, non-retained genes are gray vertical lines, and gaps are white space. The green-red bar above each block denotes the strand of the BLAST HSP, +/+ (green) and +/− (red) using the convention that the lower chromosome number of the pair is defined as intact, with the homeolog inverted to reconstitute synteny. The overfractionated homeolog has fewer genes than the underfractionated homeolog (Table 1), as expected and noted to the right of each alignment. There are no gaps >20 bp in these alignments. Gaps (white space) indicate the disparity between the numbers of non-retained genes on homeolog pairs. Ovals enclose particularly obvious clusters of retained genes that are much closer together that they were in the ancestors. The thin lines crossing over A13 illustrate how homeologous recombination could generate this segmentally scrambled alignment from two precursors displaying fractionation bias.
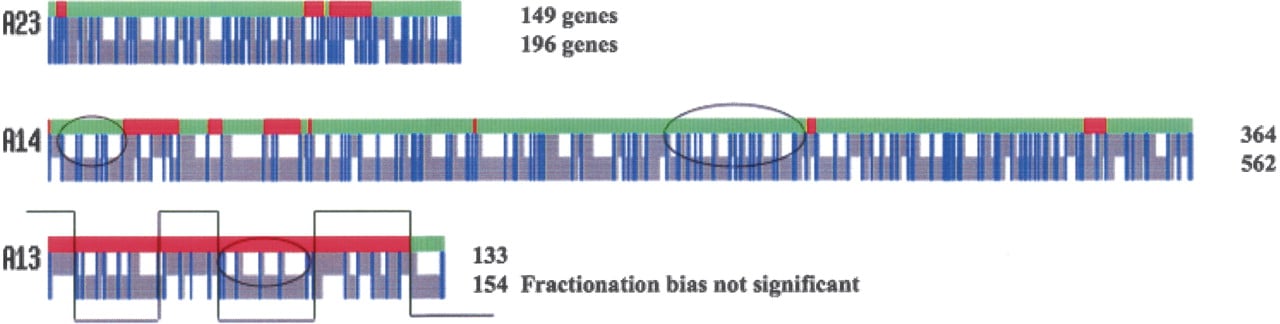
The two additional exemplary alignments in Figure 3, A23 and A13, should be read exactly as was A14. A23 is a typical α-region showing fractionation bias. A13 exemplifies the infrequent α-region not displaying a significant fractionation bias. Supplemental material 2 shows alignment diagrams like these for all 26 larger and eight smaller (SO) α-regions. In general, simply looking at an alignment finds regions where genes have been repositioned closer together, into clusters, on the overfractionated homeolog. Ovals in Figure 3 represent examples of clusters. The inversion breakpoints, where red touches green, are not associated with fractionation bias. If there had been homeologous recombination after bias was set, then biased chromosomal stretches would get switched around. The thin crossing-over line in Figure 3 A13 illustrates such imaginary switchpoints, but we do not consider such switchpoints in our analyses (Discussion).
Significance and extent of fractionation bias
Immediately following tetraploidy, each gene is paired. Fractionation reduces most of these pairs by removing one gene from one or the other (not both) of the homeologs. Thus, a measure of fractionation frequency is the number of singleton genes remaining of the total (singletons plus retained); singleton genes are colored gray in our alignment diagrams. Table 2A calculates fractionation bias for each α-region by obtaining the homeolog ratio of these singleton (not retained) genes. The mean fractionation bias is 1.87, and 1.59 if the eight smaller (SO) regions are excluded. As shown in the table, 27 of 34 regions are biased at the 95% confidence level, and all but four of these show very significant bias (P < 0.001; Table 2 legend). Because the SO regions are so small, we now refer to the mean fractionation bias as 1.6.
Table 1 gives total gene counts for each α-region. Of the 26 larger regions, 85% of total gene content is contained within α-regions that display significant (Table 2A) fractionation bias. (If Table 2A data for the shorter SO regions are included, the fractionation bias coverage is 82%.) Coverage would be near complete if the large α10 region were not “scrambled.” Close examination of the alignment diagram of this region (Supplemental material 2) suggests the possibility that fractionation was in segments created by homeologous recombination (as diagrammed for Fig. 3 A13). In any case, 85% coverage by biased fractionation implies that fractionation following the α event was biased genome-wide.
Table 2B estimates fractionation bias using gap size (measured in genes) as the unit of fractionation. Gaps devoid of genes (represented by white space in our diagrams) are necessary to permit homeolog alignment. Gaps of gene size 2–10 are all significantly different from 1:1 (Table 2B). Gaps of one gene are not significantly biased for unknown reasons, and gaps from 11 to 20 genes are sometimes biased significantly and sometimes not. We conclude that fractionation bias is a consequence of many smaller gap sizes, whereas the rare larger gaps do not influence fractionation bias significantly.
Local clustering
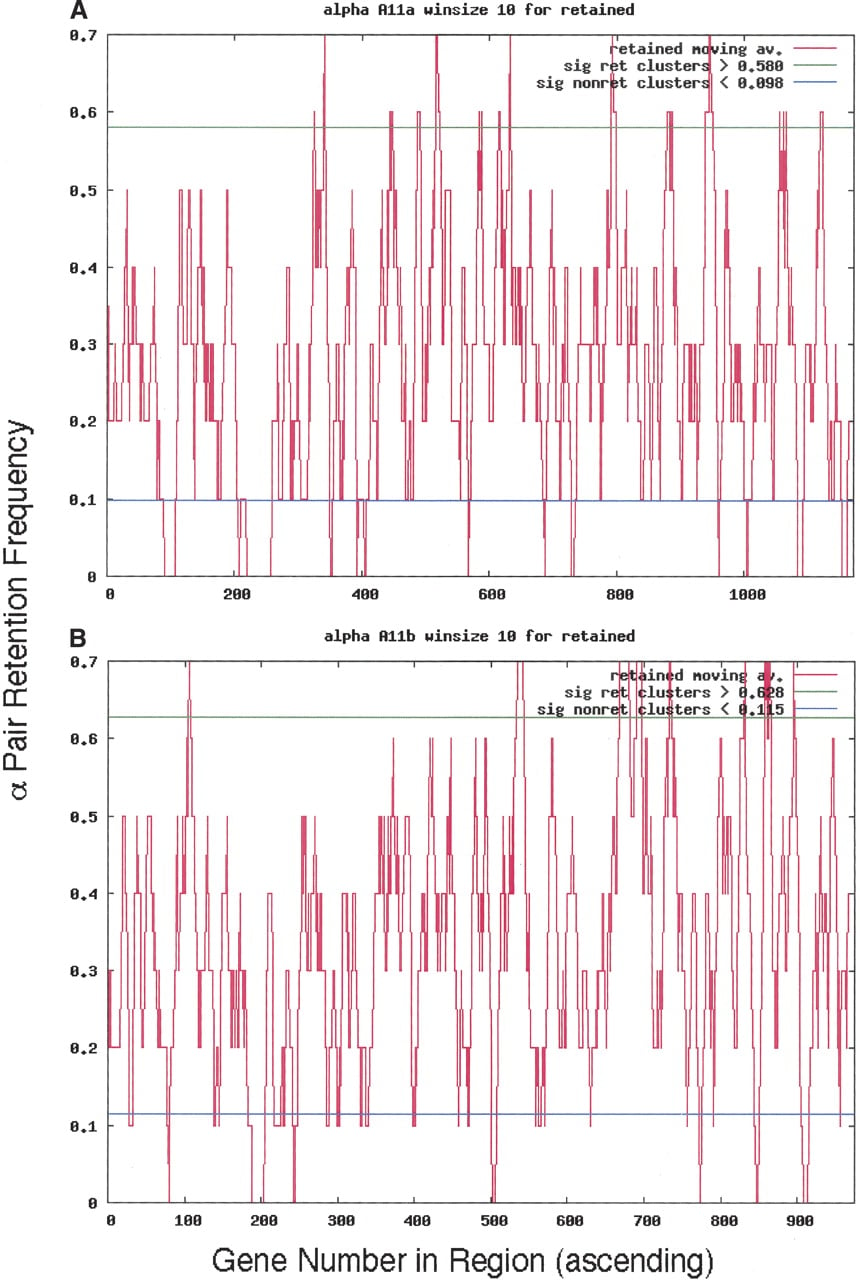
We identified clusters (usually several) of retained genes and calculated the number of genes in those clusters for each α homeolog (Table 1, “Genes >95%,” “In Retained Clusters” columns). Figure 4A and B, exemplifies moving average data for a typical pair of chromosomal segments: α11, a and b. Peaks rising above the horizontal line (95% confidence level) contain those genes in retained clusters that are significant. Thus, for the 1172 genes in α11a, 67 exist above the 95% line generated through the random simulations. Moving average data for every α-region are given in Supplemental material 3. The GO designation frequencies of genes were also tabulated. We only use data for retained gene clusters (Table 1, “In Retained Clusters” column) for the remainder of this analysis, since the retained gene content of non-retained gene clusters was almost always too small to be useful (data not shown). GO terms plotting close to the linear regression line carry genes that are retained within clusters of retention to about the same extent as they are retained in the genome.
Moving average cluster plots for both α11 homeologous chromosomal segments using a 10-gene window. (A) Homeolog 11a. (B) Homeolog 11b. The y-axis is retention frequency; 1.0 means that all 10 genes were retained in that window. The x-axis is the sequence of genes in the Minimized α-region homeolog, as explained in Methods. In α11a, for example, there are 67 and 92 genes, retained and non-retained genes, respectively (Table 1, Row 11, columns “Genes >95%” and “a”).
Genes found preferentially in clusters of retention
Some gene ontology (GO) categories appear in clusters of retention more often than expected based on their overall frequency of retention. Each GO term appearing in our minimized genome is represented in Figure 5 as a point on a plot of number of genes versus number of genes in retained clusters; the lines indicate 95% confidence limits around the linear regression line. The 40 GO terms above the 95% confidence interval are those that appear in clusters more often than expected based on their appearance in α-regions. A “y” is used in the last column of Table 3 to denote that the GO category was significantly overrepresented in clusters of retained genes.
GO categories (with >39 genes) containing genes that appeared in clusters of retained genes more than expected based on whole-genome retention frequencies
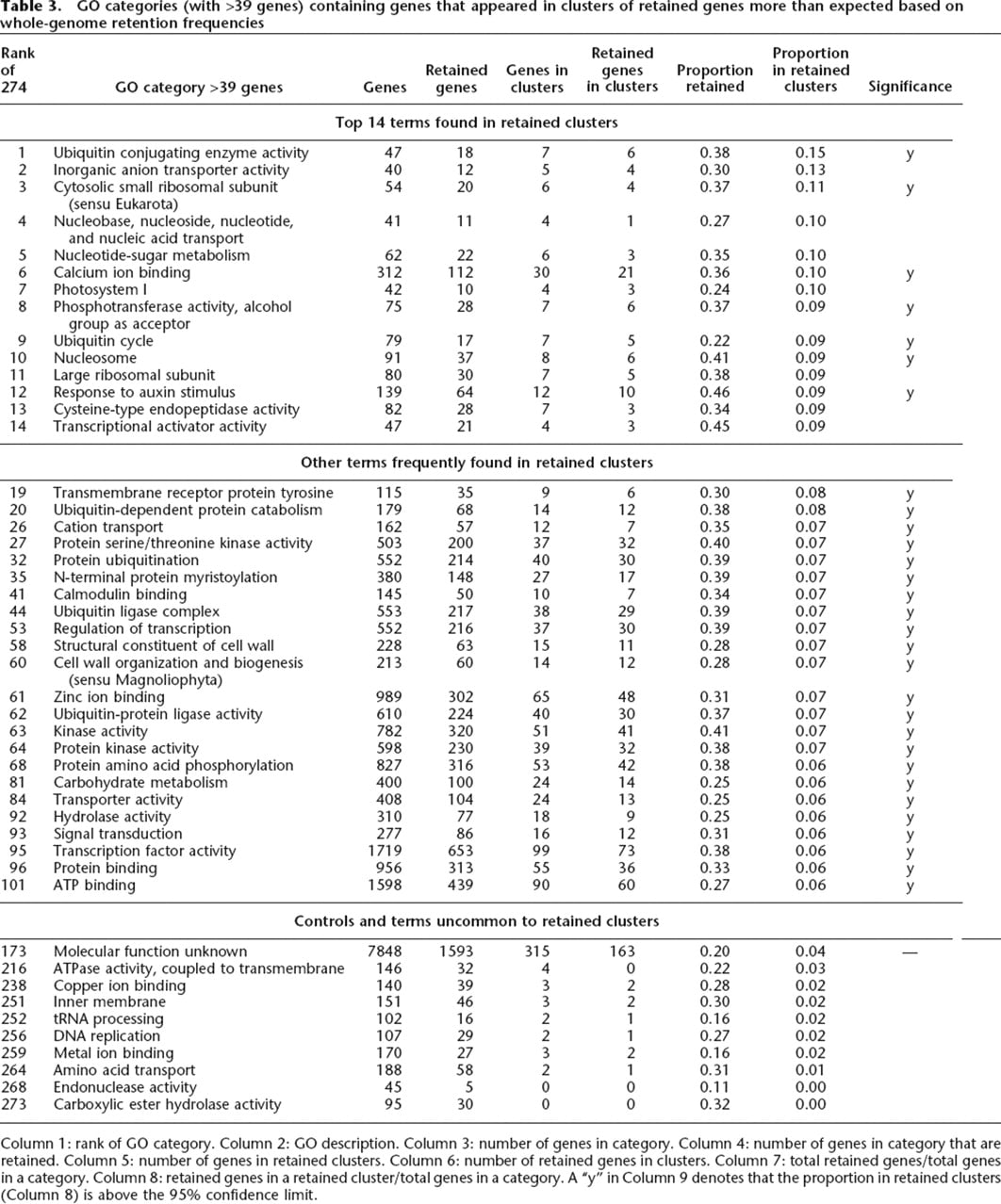
Column 1: rank of GO category. Column 2: GO description. Column 3: number of genes in category. Column 4: number of genes in category that are retained. Column 5: number of genes in retained clusters. Column 6: number of retained genes in clusters. Column 7: total retained genes/total genes in a category. Column 8: retained genes in a retained cluster/total genes in a category. A “y” in Column 9 denotes that the proportion in retained clusters (Column 8) is above the 95% confidence limit.
GO categories evaluated for overabundance in clusters of retained genes as compared to retention expectations of the GO category treated independently. GO terms with fewer than six genes in retained clusters were omitted. Linear regression analysis of the scatterplot of all GO data plotted: the x-axis is the number of genes and the y-axis is the number of these genes positioned within retained cluster space. Using column headings from Table 1: X is the “Retained Frequency” and Y is the “Genes >95%, In Retained Clusters.” Points (individual GO terms) above the upper 95% confidence interval line are those terms found significantly more often than expected in clusters of retained genes.
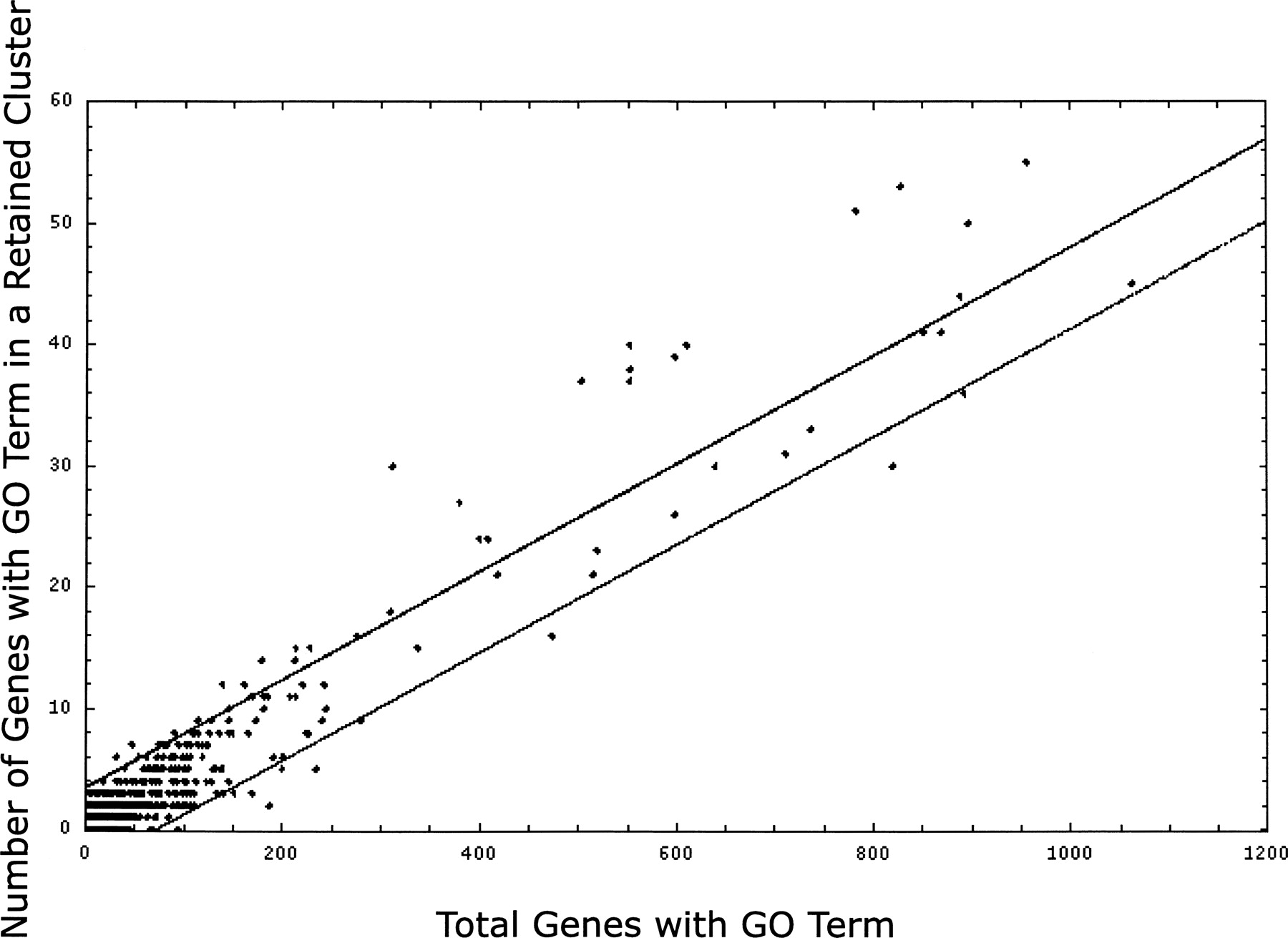
There are 274 GO categories in Arabidopsis with at least 40 genes. We refer to the ratio of frequency of retention in clusters to the frequency of genome-wide retention as the representation in clusters. A high value of that ratio indicates that a particular GO category appears in retained clusters more often than expected based on its level of retention throughout the genome. Those ratios ranged from 0.15 to 0. There were 14 GO categories with a representation in clusters >0.08 (Table 3), and seven of these GO terms were significantly clustered (from Fig. 5, denoted “y” in Table 3, last column). The middle segment of Table 3 lists those 23 GO categories with representation in clusters between 0.06 and 0.08 that were judged to be significantly overpositioned in retained clusters. Of the 40 significant GO terms, 30 are in Table 3. (We removed seven general terms, two terms with near-identical gene content to a similar term, and terms with too few genes.) The last segment of Table 3 includes 10 GO categories with lower representation in clusters.
There are particular GO categories commonly found in over-retained clusters, and likewise, there are particular GO categories absent from clusters (Table 3). The well-populated categories “transcription factor activity” (and other transcription-related) and “kinase activity” (and other signal-transduction-related) appear in clusters more frequently than expected in the whole genome. The most extreme and also significant category of Table 3 is GO: “ubiquitin conjugating enzyme activity.” Although this GO term only includes seven genes in retained clusters, there are six additional terms related to “ubiquitin” in the Top 14. The ubiquitin-proteasome pathway for specific protein degradation is known to have a particularly complicated subunit structure (Goldberg 2003).
Discussion
α-pairs are clustered
We have shown that those genes not lost following the most recent tetraploidy in Arabidopsis are frequently retained in clusters (Table 1; Figs. 3, 4; Supplemental material 2 and 3). The pattern of retention is nonrandom for 49 of 52 α homeologs as a whole (Table 1), and moving averages identify significant local clusters on almost all homeologs (Table 1; Fig. 4; Supplemental material 3). Biased fractionation is one explanation for such clusters, as readily seen in our detailed alignment graphics (Fig. 3, where obvious clusters are identified; Supplemental material 2). One homeolog, probably representing one or the other of the original parents of the tetraploid, has experienced more (1.6× on average) gene loss than its partner. The data of Table 2A indicate that 21 of 26 larger α-regions and six of eight smaller (SO) α-regions are significantly biased. This gives an α-genome (the 26 larger regions) bias coverage of ∼85%, which approximates an entire genome. This biased fractionation brings retained genes together into clusters on the overfractionated homeolog (see Fig. 3). If fractionation were not biased, then retained genes would still be brought closer together during fractionation, but not into clusters that did not already exist before fractionation. In summary, we show that retained genes are clustered, and have found a mechanism that naturally generates such clusters.
Table 2B shows that smaller gaps of two to 10 genes account for the fractionation bias. These supporting data are important because they show that larger gaps, such as might result from large deletions of >11 genes on one homeolog only, are not the explanation for fractionation bias.
Closer examination of the alignment diagrams for those few apparently “random” regions, like A13 of Figure 3 and A10 of Supplemental material 2, indicates that there are occasionally gene-orientation switchpoints that mask bias. Such switchpoints might be expected if differential homeolog mutability (bias) were established immediately after tetraploidy, and some homeologous recombination followed for a few generations. If we included the possibility of such recombination, near 100% coverage could be argued, but the validity of the argument would be difficult to test. Fractionation bias switchpoints in our alignment data weakly support the hypothesis that there was a transient period of homeologous recombination following tetraploidy. However, this period must have been short, if it existed at all, because homeologs are not significantly scrambled.
Categories of genes in over-retained clusters
The gene balance hypothesis provides general predictions as to which sorts of genes should be preferentially retained following tetraploidy. In short, some molecular machines or cascades in the organism are particularly sensitive to the balance of their constituent parts, interacting proteins, or the complicated nature of their regulatory network. According to the gene balance hypothesis, a gene displays more dosage sensitivity as its degree of interaction or connectivity increases, either during assembly or function (for reviews, see Birchler et al. 2005; Freeling and Thomas 2006).
GO categories with genes that are preferentially retained after the most recent tetraploidy in Arabidopsis have been identified by previous workers, as reviewed above. Interestingly, GO terms that appear in clusters (Table 3) include these same genes preferentially retained after the α-tetraploidy event: genes that encode transcription factors (Ranks 12, 14, 53, and 95), components of signal transduction (Ranks 19, 27, 63, 64, 68, and 93) and ribosomes (Ranks 3 and 11). These connected genes occur in clusters at frequencies significantly above that expected for the α-region as a whole. The GO categories most retained were also most represented in clusters. For example, the most generally retained GO category in Arabidopsis (n > 18) is “proteasome core complex,” with 15 of 20 genes retained, and four of four genes retained in clusters, yielding a record-high representation in clusters. Our general conclusion is that the retention potential of individual genes dictates the gene content of clusters of over-retention. This does not exclude other mechanisms—those involving resistance to loss by long deletions—that, in theory, could also generate clusters of retention (see below).
Fractionation bias requires that homeologs be differentially marked for epigenetic inheritance prior to fractionation
Fractionation from tetraploid to near diploid is biased, in at least megabase stretches (and often for an entire syntenic region), to one homeolog (Fig. 3; Table 2; Supplemental material 2). One explanation of this bias is that the parents contributing to the tetraploid were significantly different in coadapted regulatory behavior, as would be expected of allotetraploidy. In any case, tetraploidy must have resulted in one of the parents’ chromosomal sets becoming “marked” epigenetically, and thus targeted for over- or underfractionation. Here we argue that 86% biased fractionation supports the hypothesis that the two original chromosome sets (100%) were differentially marked. Perhaps overexpression of one genome in a hypothetical allotetraploid preferentially resulted in RNA-triggered silencing. Since all plants have a pollen and egg parent, an autotetraploid could also have homeologs marked differentially based on the direction of transmission. Because mis-segregations lower fitness, and are expected following any sort of tetraploidy, allotetraploidy seems generally likely because there is some argument for increased fitness via segregation and unique heterotic-type interactions in the polyploid (Stansfield 1977). Homeologs were differentially and heritably marked prior to fractionation; thereafter they mutated (fractionated) at different rates.
Fractionation bias was unexpected. That this bias is so uniformly evident after all these years is remarkable. Given the observed fractionation bias, differential homeolog mutability does generally fit into a history of nonadditive phenomena observed in hybrid and polyploid organisms. Gene expression in hybrid plants is not the sum of the gene expression of the parents, there is methylation of the underexpressed homeolog, and underexpression may often be reversed with chemicals known to demethylate DNA (Heslop-Harrison 1990). Arabidopsis thaliana and Cardaminopsis arenosa are the parents of a natural allotetraploid species that exhibits specific parental gene silencing (Lee and Chen 2001). Wang et al. (2006) also found nonadditive gene regulation in synthetic allotetraploids in the Arabidopsis genus. Synthetic allotetraploid studies in cotton found silencing to be organ-specific (Adams et al. 2004). A particularly important analysis of synthetic allotetraploids in Brassica, using reciprocal crosses found that changes in gene content occur rapidly without changing karyotype or gross chromosomal structure (Song et al. 1995). These workers found no consistent bias in silencing due to parent of origin, but did find that 5%–10% of the genome changes in sequence content over the five inbreeding generations following synthetic allotetraploidy. Cytosine methylation and sequence loss follow wheat allotetraploidies (Shaked et al. 2001). Additionally, resynthesized wheat polyploids may also initiate rapid gene loss (Kashkush et al. 2002), and rapid gene loss may follow autotetraploidy as well. As concluded in a review of gene expression in polyploids (Osborn et al. 2003), silencing of genes from one or the other parent is characterized by high levels of DNA methylation and low levels of histone acetylation on the silenced homeolog. We believe that fractionation bias, a measure of epigenetically heritable chromosomal mutation rate, can now be added to this list of special behaviors initiated when genomes merge.
Lippman and co-workers (2004) studied a region of constitutive heterochromatin in Arabidopsis as compared with the homeologous region, measuring transposon and repeat content, gene content, and gene expression. They found that transposons-repeats apparently initiated an siRNA-maintained (epigenetic) silenced chromatin state, and that genes very close to transposons could themselves be silenced. Perhaps over- or underfractionation is initiated and/or maintained in a manner similar to heterochromatin.
The Arabidopsis tetraploidy is in a phylogenetic void of sequenced genomes. On the other hand, the sequenced Baker’s yeast (Saccharomyces cerevisiae) genome carries the syntenous gene pairs of an ancient tetraploidy for which there are multiple sequenced, diploid yeast out-groups, and has two post-tetraploid sister species, Candida glabrata and Saccharomyces castellii. Scannell and co-workers (2006) studied the occurrence of the 14 patterns of fractionation possible among these three sister species given zero, one, two and three (all) losses of a previously paired gene. The distribution of these patterns implies that fractionation from tetraploid to stable near-diploids was in progress during the times when these sister genera originated. These workers found 4%–7% of the 2723 deduced ancestral yeast genes present in one sister’s genome did not have a true ortholog in another sister’s genome because of what the authors call “reciprocal gene loss,” a property of sister insipient species. When this occurs, two newly evolved sister genomes carry similar singleton genes on largely nonhomologous chromosomes; this situation is expected to create instant reproductive isolation. According to neutral population genetic theory, such isolation increases the probability of speciation (theoretical citations in Scannell et al. 2006). Scannell and co-workers found instances of “convergent losses” where one homeologous stretch of genes were nonrandomly lost; this is fractionation bias. The authors attempted to explain convergent loss using selection. We think a whole-genome marking mechanism such as that proposed here provides another possible explanation. Further, the clustering of retained pairs that naturally occurs on the overfractionated homeolog could have selective consequences.
Mechanisms of fractionation
Fractionation from tetraploid toward diploid is the expected (Haldane 1933; Lynch and Force 2000) loss of DNA sequence information (from one or the other but not both homeologs) by some combination of deletion, conversion, and/or randomization by point mutations (via the pseudogene pathway). Measuring gene conversion requires useful outgroup genome sequence (Gao and Innan 2004), which does not exist yet for Arabidopsis. If fractionation included a long-deletion mechanism, it would be expected that a connected, retained gene might protect adjacent genes from fractionation simply by linkage, leading to linkage disequilibrium. Be it deletion, point mutation or both, the mechanism(s) of fractionation must recognize the epigenetic difference between homeologous chromosomes.
The mechanisms of fractionation are not necessarily those that operate during duplicate gene divergence. There is adequate evidence from a variety of organisms that a pair of genes, once retained in some way, will diverge in function either by subfunctionalization or neofunctionalization (Gu et al. 2002, 2004; Wagner 2002; Makova and Li 2003; Raes and Van de Peer 2003; Haberer et al. 2004; He and Zhang 2005). Acceleration of divergence following duplication may be common, but there are a growing number of exceptions (see review by Koonin 2005); for Arabidopsis, retained α-pairs diverged more slowly than expected, at least over the last few million years (Chapman et al. 2006), and these pairs are often expressed in near-identical organ-specific patterns (Casneuf et al. 2006).
Subfunctionalization has provided a popular explanation for duplicate retention (Force et al. 1999; Lynch and Force 2000), and has replaced the classical neofunctionalization explanation (Lewis 1951; Ohno 1970; Li 1997). Since the gene balance hypothesis predicts the gene content of retained duplicates and over-retention, neither subfunctionalization nor neofunctionalization is necessary to explain α-retention. Genes tend to be retained when upsetting the gene dosage status quo has a selective cost. Thus, the preservation of gene balance is now the best single explanation for the retention of gene pairs following large-scale genomic duplications (Freeling and Thomas 2006).
Mechanisms explaining clusters of retained genes
Biased fractionation generates clusters on the overfractionated homeolog. However clusters of over-retention occur (albeit less frequently) on the underfractionated homeolog as well. We suggest that repeated fractionations that did not consistently favor the same chromosome lineage could achieve the clustering we have noted.
There are at least three explanations for duplicate retention that evoke mechanisms that are in place before or at the time of tetraploidy. Each of them involves selection for the status quo established before the new tetraploid existed by not fractionating certain loci. If the fractionation mechanism were long deletions, then those loci resistant to fractionation would also protect nearby genes. Candidates for such deletion-resistant loci are: (1) chromosome cis integrity regions; (2) heterotic allotetraploid homeologous gene pairs; and/or (3) duplicate genes that are particularly susceptible to a haploinsufficiency phenotype (predicted by the gene balance hypothesis). Matrix attachment regions (MARs), examples of the first explanation, have been reported as a large fraction of mammalian phylogenetic footprints that are not gene-associated (Glazko et al. 2003). A sorghum–rice chromosomal comparison found conservation of a MAR-like region in a syntenous position (Avramova et al. 1998). MAR-like positions could only explain clusters of retention if they nucleate a region that provides deletion protection because they, themselves, cannot be deleted. Similarly, a primary mechanism for retention of genes in clusters invokes heterotic gene pairs selected simultaneously with an allotetraploid event; this mechanism also helps explain the initial survival of the tetraploid. The third explanation predicts that some genes, even though they are duplicated, cannot be deleted one-by-one following tetraploidy without incurring an unfit phenotype: this is the prediction of the gene balance hypothesis. The gene content of our clusters, comprised preferentially of “connected genes” (Table 3), suggests that this third explanation is the best single explanation for preferential retention of genes after segmental or whole-genome duplication. However, any of these explanations might explain any single case of retention.
For any of these mechanisms to explain clustering of over-retained genes using the concept of linkage disequilibrium, they must operate in an environment where long deletions happen, and we have not proved that long deletions happen during fractionation. Biased fractionation explains clustering without any mechanical assumptions. A deletion-resistant gene could potentially boost the retention rate of a neighboring gene that would otherwise be lost. The test of this hypothesis is confounded, however, largely because long deletions cannot be proven as a fractionation mechanism and by the paucity of genes in the most specific GO categories. In summary, the molecular mechanism of fractionation remains unknown.
Repeated tetraploidies and gene content evolution
Repeated over-retention of genes encoding products that participate in the most complex machines (Seoighe and Gehring 2004; Maere et al. 2005) or cascades (see Birchler et al. 2005) increases regulatory potential. Repeated biased fractionation brings “connected” genes closer together. The occurrence of clusters of duplicated, connected genes that simply cannot be removed without creating genetic imbalance and loss of fitness presents an evolutionary potential toward gene coregulation and gene coadaptation and may also explain trends in morphological complexity in multicellular eukaryotic evolutionary lineages (Freeling and Thomas 2006).
There is a tendency in all studied eukaryotes for coregulated genes to be linked on the same chromosomal region (including in Arabidopsis) (Williams and Bowles 2004). These linkages can be an aid in the prediction of functional modules. Even without experimental data, it is reasonable to hypothesize a causal relationship between the clusters of duplicate genes we have identified here and the duplication of functional gene modules in multicellular eukaryotes. Recent work in yeast suggests that genome duplication can help create divergent gene coexpression networks (Conant and Wolfe 2006). Clusters of retained genes could be the precursors to “coadapted gene complexes,” suggesting a direct link to adaptation by natural selection.
Acknowledgments
We thank Eric Lyons, Lakshmi Rapaka, Damon Lisch, Keith Slotkin, Maggie Woodhouse, and George Theodoris for useful criticism. This work was funded by USA NSF DBI#0337083 and NSF DBI#0349737.
References
- ↵K.L. AdamsR. PercifieldJ.F. Wendel (2004) Organ-specific silencing of duplicated genes in a newly synthesized cotton allotetraploid. Genetics 168:2217–2226.
- ↵The Arabidopsis Genome Initiative. (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815.
- ↵Z. AvramovaA. TikhonovM. ChenJ.L. Bennetzen (1998) Matrix attachment regions and structural collinearity in the genomes of two grass species. Nucleic Acids Res. 26:761–767.
- ↵J.A. BirchlerN.C. RiddleD.L. AugerR.A. Veitia (2005) Dosage balance in gene regulation: Biological implications. Trends Genet. 21:219–226.
- ↵G. BlancK.H. Wolfe (2004) Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16:1667–1678.
- ↵G. BlancA. BarakatR. GuyotR. CookeM. Delseny (2000) Extensive duplication and reshuffling in the Arabidopsisgenome. Plant Cell 12:1093–1101.
- ↵G. BlancK. HokampK.H. Wolfe (2003) A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis thaliana. Genome Res. 13:137–144.
- ↵J.E. BowersB.A. ChapmanJ. RongA.H. Paterson (2003) Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422:433–438.
- ↵T. CasneufS. De BodtJ. RaesS. MaereY. de Van Peer (2006) Nonrandom divergence of gene expression following gene and genome duplications in the flowering plant Arabidopsis thaliana. Genome Biol. 7:R13.
- ↵B.A. ChapmanJ.E. BowersS.R. SchultzA.H. Paterson (2004) A comparative phylogenetic approach for dating whole genome duplication events. Bioinformatics 20:180–185.
- ↵B.A. ChapmanJ.E. BowersF.A. FeltusA.H. Paterson (2006) Buffering of crucial functions by paleologous duplicated genes may contribute cyclicality to angiosperm genome duplication. Proc. Natl. Acad. Sci. 103:2730–2735.
- ↵G.C. ConantK.H. Wolfe (2006) Functional partitioning of yeast co-expression networks after genome duplication. PLoS Biol. 4:e109.
- ↵A. ForceM. LynchF.B. PickettA. AmoresY.L. YanJ. Postlethwait (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151:1531–1545.
- ↵M. FreelingB.C. Thomas (2006) Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res, (this issue)..
- ↵L.Z. GaoH. Innan (2004) Very low gene duplication rate in the yeast genome. Science 306:1367–1370.
- ↵G.V. GlazkoE.V. KooninI.B. RogozinS.A. Shabalina (2003) A significant fraction of conserved noncoding DNA in human and mouse consists of predicted matrix attachment regions. Trends Genet. 19:119–124.
- ↵A.L. Goldberg (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426:895–899.
- ↵Z. GuD. NicolaeH.H. LuW.H. Li (2002) Rapid divergence in expression between duplicate genes inferred from microarray data. Trends Genet. 18:609–613.
- ↵Z. GuS.A. RifkinK.P. WhiteW.H. Li (2004) Duplicate genes increase gene expression diversity within and between species. Nat. Genet. 36:577–579.
- ↵G. HabererT. HindemittB.C. MeyersK.F. Mayer (2004) Transcriptional similarities, dissimilarities, and conservation of cis-elements in duplicated genes of Arabidopsis. Plant Physiol. 136:3009–3022.
- ↵J.B.S. Haldane (1933) The part played by recurrent mutation in evolution. Am. Nat. 67:5–19.
- ↵X. HeJ. Zhang (2005) Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics 169:1157–1164.
- ↵J.S. Heslop-Harrison (1990) Gene expression and parental dominance in hybrid plants. Dev. Suppl. 1990:21–28.
- ↵D.C. InadaA. BashirC. LeeB.C. ThomasC. KoS.A. GoffM. Freeling (2003) Conserved noncoding sequences in the grasses. Genome Res. 13:2030–2041.
- ↵K. KashkushM. FeldmanA.A. Levy (2002) Gene loss, silencing and activation in a newly synthesized wheat allotetraploid. Genetics 160:1651–1659.
- ↵M. KellisB.W. BirrenE.S. Lander (2004) Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428:617–624.
- ↵E.V. Koonin (2005) Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 39:309–338.
- ↵H.-S. LeeZ.J. Chen (2001) Protein-coding genes are epigenetically regulated in Arabidopsis polyploids. Proc. Natl. Acad. Sci. 98:6753–6758.
- ↵E.B. Lewis (1951) Pseudoallelism and gene evolution. Cold Spring Harb. Symp. Quant. Biol. 16:159–174.
- ↵W.-H. Li (1997) Molecular evolution. ( Sinauer, Sunderland, MA).
- ↵Z. LippmanA.-V. GendrelM. BlackM.W. BaughnN. DedhiaW.R. McCombeK. LevineV. MittelB. MayK.D. Kasschau (2004) Role of transposable elements in heterchromatin and epigenetic control. Nature 430:471–476.
- ↵S. LocktonB.S. Gaut (2005) Plant conserved non-coding sequences and paralogue evolution. Trends Genet. 21:60–65.
- ↵M. LynchA. Force (2000) The probability of duplicate gene preservation by subfunctionalization. Genetics 154:459–473.
- ↵S. MaereS. De BodtJ. RaesT. CasneufM. Van MontaguM. KuiperY. de Van Peer (2005) Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. 102:5454–5459.
- ↵K.D. MakovaW.-H. Li (2003) Divergence in the spatial pattern of gene expression between human duplicate genes. Genome Res. 13:1638–1645.
- ↵A. McLysaghtK. HokampK.H. Wolfe (2002) Extensive genomic duplication during early chordate evolution. Nat. Genet. 31:200–204.
- ↵D.W. MeinkeL.K. MeinkeT.C. ShowalterA.M. SchisselL.A. MuellerI. Tzafrir (2003) A sequence-based map of Arabidopsis genes with mutant phenotypes. Plant Physiol. 131:409–418.
- ↵S. Ohno (1970) Evolution by gene duplication. ( Springer-Verlag, Berlin).
- ↵T.C. OsbornJ.C. PiresJ.A. BirchlerD.L. AugerZ.J. ChenH.S. LeeL. ComaiA. MadlungR.W. DoergeV. Colot (2003) Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 19:141–147.
- ↵B. PappC. PalL.D. Hurst (2003) Dosage sensitivity and the evolution of gene families in yeast. Nature 424:194–197.
- ↵A.H. PatersonJ.E. BowersY. de Van PeerK. Vandepoele (2006) Ancient duplications of cereal genomes. New Phytol. 165:658–661.
- ↵J. RaesY. de Van Peer (2003) Gene duplication, the evolution of novel gene functions, and detecting functional divergence of duplicates in silico. Appl. Bioinformatics 2:91–101.
- ↵D.R. ScannellK.P. ByrneJ.L. GordonS. WongK.H. Wolfe (2006) Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature 440:341–345.
- ↵C. SeoigheC. Gehring (2004) Genome duplication led to highly selective expansion of the Arabidopsis thaliana proteome. Trends Genet. 20:461–464.
- ↵H. ShakedK. KashkushH. OzkanM. FeldmanA.A. Levy (2001) Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 13:1749–1759.
- ↵R.R. SokalF.J. Rohlf (1995) Biometry. ( W.H. Freeman and Co. New York).
- ↵K. SongP. LuK. TangT.C. Osborn (1995) Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proc. Natl. Acad. Sci. 92:7719–7723.
- ↵W.D. Stansfield (1977) The science of evolution. ( Collier Macmillan, New York).
- ↵T.A. TatusovaT.L. Madden (1999) BLAST 2 sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174:247–250.
- ↵C.G. TianY.Q. XiongT.Y. LiuS.H. SunL.B. ChenM.S. Chen (2005) Evidence for an ancient whole genome duplication event in rice and other cereals. Acta Genetica Sinica 32:519–527.
- ↵K. VandepoeleC. SimillionY. de Van Peer (2003) Evidence that rice and other cereals are ancient aneuploids. Plant Cell 9:2192–2202.
- ↵R.A. Veitia (2002) Exploring the etiology of haploinsufficiency. Bioessays 24:175–184.
- ↵T.J. VisionD.G. BrownS.D. Tanksley (2000) The origins of genomic duplications in Arabidopsis. Science 290:2114–2117.
- ↵A. Wagner (2002) Asymmetric functional divergence of duplicate genes in yeast. Mol. Biol. Evol. 19:1760–1768.
- ↵J. WangL. TianH.S. LeeN. WeiH. JiangB. WatsonA. MadlungT. OsbornR.D. DoergeL. Comai (2006) Genome-wide non-additive gene regulation in Arabidopsis allotetraploids. Genetics 172:507–517.
- ↵E.J. WilliamsD.J. Bowles (2004) Coexpression of neighboring genes in the genome of Arabidopsis thaliana. Genome Res. 14:1060–1067.
- ↵K.H. WolfeD.C. Shields (1997) Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387:708–713.
Notes
[1] [Supplemental material is available online at www.genome.org.]
[2] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4708406