
Abstract
Phenotype-driven genetics can be used to create mouse models of human disease and birth defects. However, the utility of these mutant models is limited without identification of the causal gene. To facilitate genetic mapping, we developed a fixed single nucleotide polymorphism (SNP) panel of 394 SNPs as an alternative to analyses using simple sequence length polymorphism (SSLP) marker mapping. With the SNP panel, chromosomal locations for 22 monogenic mutants were identified. The average number of affected progeny genotyped for mapped monogenic mutations is nine. Map locations for several mutants have been obtained with as few as four affected progeny. The average size of genetic intervals obtained for these mutants is 43 Mb, with a range of 17–83 Mb. Thus, our SNP panel allows for identification of moderate resolution map position with small numbers of mice in a high-throughput manner. Importantly, the panel is suitable for mapping crosses from many inbred and wild-derived inbred strain combinations. The chromosomal localizations obtained with the SNP panel allow one to quickly distinguish between potentially novel loci or remutations in known genes, and facilitates fine mapping and positional cloning. By using this approach, we identified DNA sequence changes in two ethylnitrosourea-induced mutants.
Until recently, genetic mapping in the mouse was performed most efficiently by analysis of simple sequence length polymorphism (SSLP) markers both for initial identification of chromosomal (Chr.) localization and for high-resolution mapping (Dietrich et al. 1994). A typical SSLP-based genome scan of 80–100 markers allowed identification of genetic intervals at low to moderate resolution. Chromosomal localization can be obtained with fewer markers and small numbers of mice if techniques such as haplotype analysis are employed (Neuhaus and Beier 1998). Although SSLP-based genotyping has been used successfully for the genetic mapping of hundreds of mutations, it is labor intensive and any single SSLP panel is generally not fully informative for crosses using a variety of strain combinations.
Advances in genome sequencing have led to the discovery of thousands of single nucleotide polymorphisms (SNPs) in the mouse genome (Lindblad-Toh et al. 2000; Wiltshire et al. 2003; Pletcher et al. 2004). Recently, several groups have demonstrated the utility of SNPs for examining the haplotype structure of the mouse genome (Wade et al. 2002; Fraser et al. 2004; Liao et al. 2004); for investigating relationships between inbred strains (Petkov et al. 2004; Pletcher et al. 2004); and for developing computational methods for mapping qualitative and quantitative trait loci (QTL) in the mouse (Grupe et al. 2001; Liao et al. 2004; Pletcher et al. 2004). On a smaller scale, a strain-specific, low-density, genome-wide SNP panel was used to identify genetic modifiers (Owens et al. 2005).
We sought to utilize SNP genotyping as an alternative to microsatellite marker analysis for mapping mutations in the mouse. We generated a whole-genome panel of 394 SNPs that can be used to map monogenic mutations in a high-throughput manner with small numbers of mice and without prior consideration of genetic background. By genotyping 28 inbred and wild-derived strains and by using the panel to identify map locations for 22 mutations, we demonstrate that the SNP panel is effective at identifying chromosomal localizations at moderate resolution by analyzing small numbers of affected progeny. Two ethylnitrosourea (ENU)-induced mutants that were mapped to subchromosomal regions using the SNP panel were further fine-mapped, and single base-pair mutations were identified by sequencing of candidate genes.
Results
SNP panel description
SNPs were selected from a larger pool of published SNPs (Wiltshire et al. 2003; Pletcher et al. 2004) based on chromosomal position and the extent of polymorphism between a few common inbred strains, primarily C57BL/6J, DBA/2J, 129X1/SvJ, and A/J. The panel contains 394 SNPs across 19 autosomes, with an average distance between SNPs of 5.9 Mb. To evaluate the informativeness of the whole-genome panel, genomic DNA from 23 inbred strains, five wild-derived inbred strains, and 10 F1 hybrids were genotyped, and the number of polymorphic SNPs between each pair-wise strain combination was determined (Supplemental data). The average informativeness between the laboratory strains we have tested (excluding those that are related substrains such as C57BL/6J and C57BL/10J) is 142 SNPs. C57BL/6J, one of the most commonly used inbred strains, is highly polymorphic in the panel, having between 179 and 314 informative SNPs among 22 other nonclosely related inbred strains.
The utility of the panel for mapping a mutation with a particular genetic cross depends not only on the number of informative SNPs but also on the distribution of those informative SNPs throughout the genome. With the exception of closely related strains, there is adequate genome coverage of informative SNPs for most strain pairs. Because of the bias in SNP selection among a few inbred strains, it is not unexpected that large gaps in informative SNPs will exist for even highly polymorphic strains; e.g., C57BL/6J and FVB/NJ have 211 informative SNPs with two large gaps between SNPs of 67 (Chr. 7) and 63 Mb (Chr. 14).
SNP-based mapping of monogenic mutations
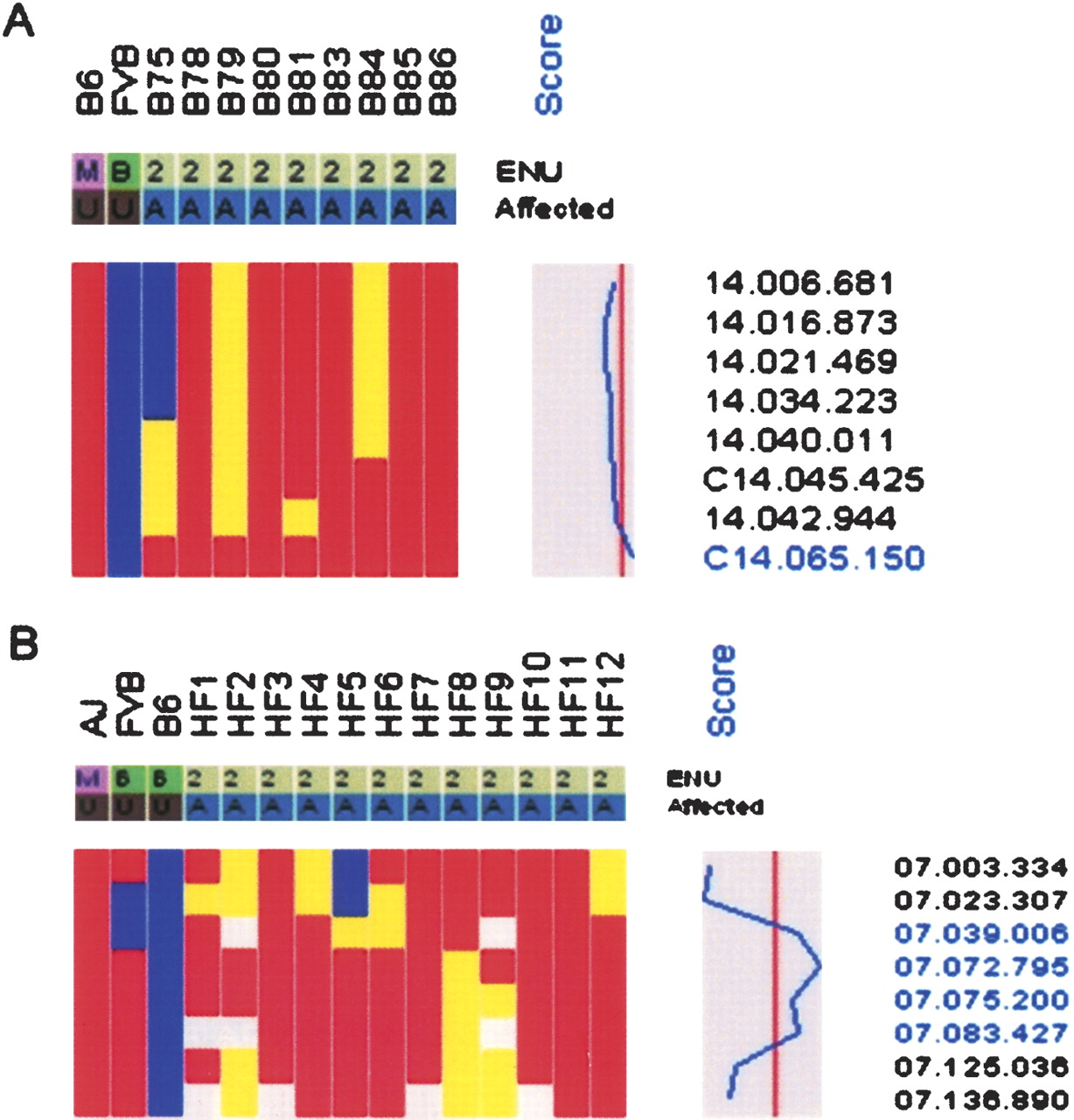
To evaluate the usefulness of the SNP panel for mapping monogenic mutations, two mutants with unrelated phenotypes were selected for whole-genome scans. baldy (bldy) is a recessive ENU-induced mutant with wrinkled skin and hair loss that progresses to complete alopecia by 5 wk of age. Nine affected (C57BL/6J × FVB/NJ) F2 mice were genotyped with the SNP panel, and linkage to distal Chr. 14 was evident from the haplotype data, which showed retention of alleles from the mutagenized strain (C57BL/6J) and statistically significant linkage (LOD = 5.22) at SNP C14.065.150 (62.3 Mb) (Fig. 1). As there are no informative SNPs for the mapping cross distal to C14.065.150, the genetic interval lies between SNP C14.045.425 (43.6 Mb) and the telomeric end of Chr. 14 (117.0 Mb).
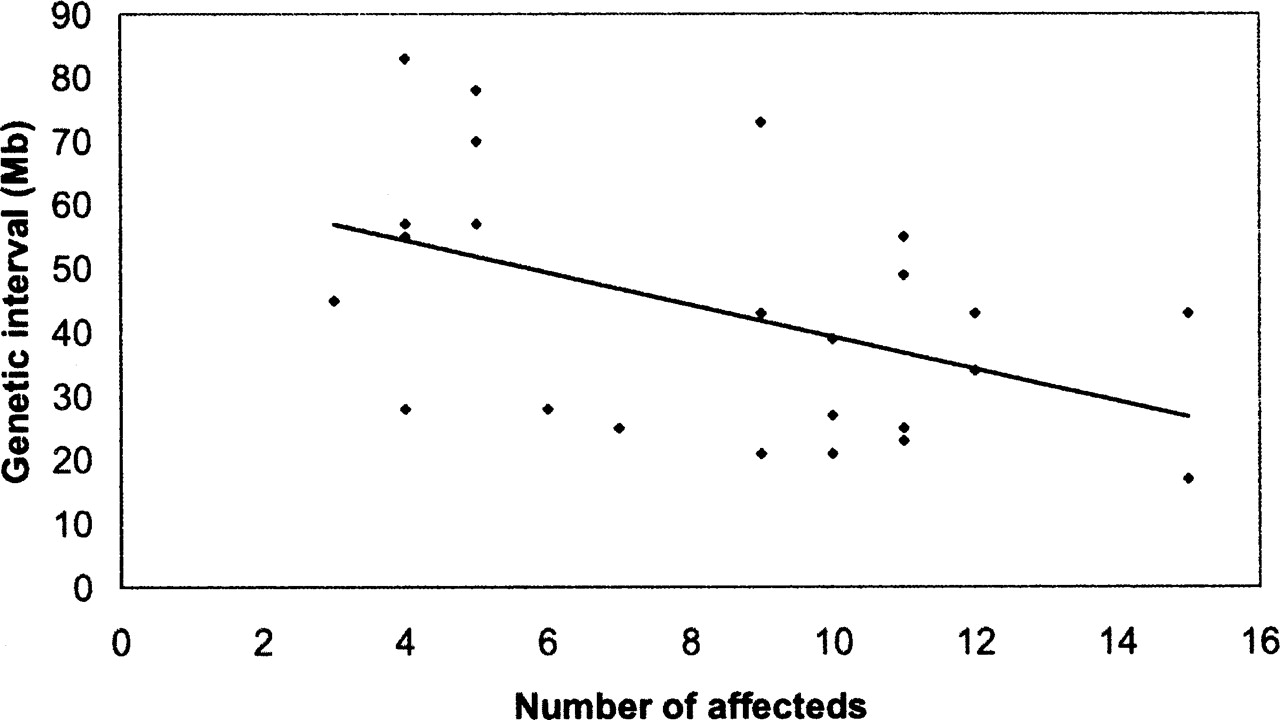
hydrops fetalis (hyft) is a recessive ENU-induced perinatal lethal mutant with dramatic embryonic growth deficiency, intravascular fluid accumulation, and frequent intrauterine demise (Herron et al. 2002). The mutation was induced on the A/J strain, maintained by backcrossing to FVB/NJ for five generations, and then outcrossed to C57BL/6J for mapping. Since the mutation is recessive, the chromosomal region containing the mutation must be heterozygous for A/J and C57BL/6J alleles in the F1 parents and homozygous for the A/J allele in the affected progeny. Although the mice contain three genetic backgrounds, allele contribution can be distinguished by comparing the individual haplotypes of the affected mice across the chromosome where SNPs are polymorphic. Localization to a 34-Mb interval on Chr. 7 (07.039.006 [33.2 Mb]–07.072.795 [67.4 Mb]) was identified by analyzing one parental F1 and 11 affected F2 progeny (Fig. 1). In addition to these analyses, we have genotyped affected mice from an additional 17 recessive and three dominant monogenic mutants; our results demonstrate that the SNP panel can be used to map mutations at moderate resolution with small numbers of affected mice (Table 1). For all mutants, all 394 SNPs were genotyped, and noninformative SNPs were not included in the analysis. The average genetic interval obtained with the SNP panel was 43 Mb, with a range of 17–83 Mb. Of particular note is that the average number of affected progeny genotyped for each mutant was nine. Map positions were identified for four mutants by genotyping as few as four affected progeny. Although the number and distribution of informative SNPs in the chromosomal region of the mutation will affect the size of the genetic interval, there is, as expected, an inverse relationship between the number of affected progeny genotyped and the size of the genetic interval (Fig. 2). For all monogenic mutations described, genotyping of SSLP markers within the genetic intervals confirmed linkage.
Monogenic mutations mapped using the SNP panel
Mutant | Mode of inheritance | Genetic backgroundsa | Mb interval | No. of affecteds |
|---|---|---|---|---|
| cincinnati | Recessive | B6, C3H/HeJ, 129X1 | 17 | 15 |
| mummy | Recessive | B6, A/J | 21 | 10 |
| Chastity | Dominant | B6, C3H/HeJ, 129S6 | 21 | 9 |
| line 11 | Recessive | B6, FVB | 23 | 11 |
| ND15 | Recessive | B6, FVB | 25 | 7 |
| ND3 | Recessive | B6, FVB | 25 | 11 |
| ND21 | Recessive | B6, FVB | 27 | 10 |
| ND94 | Recessive | B6, FVB | 28 | 4 |
| pewter-C3Hb | Recessive | B6, C3Heb/FeJ | 28 | 6 |
| hyft | Recessive | A/J, FVB, B6 | 34 | 12 |
| fe1 | Recessive | 129S6, CAST/Ei | 39 | 10 |
| tiresias | Recessive | B6, DBA | 43 | 9 |
| Bigfoot | Dominant | 129S6, B6, DBA | 43 | 12 |
| peewee | Recessive | NAW/W1, B6, C3H/HeJ, CAST/Ei | 43 | 15 |
| dill-C3Hb | Recessive | B6, C3Heb/FeJ | 45 | 3 |
| bha | Recessive | B6, CAST/Ei, 129S6 | 49 | 11 |
| ND58 | Recessive | B6, FVB | 55 | 4 |
| Bx708 | Dominant | B6, DBA | 55 | 11 |
| chaos4 | Recessive | B6, C3Heb/FeJ | 57 | 4 |
| 20884 | Recessive | B6, FVB | 57 | 5 |
| dill-FVBb | Recessive | B6, FVB | 70 | 5 |
| baldy | Recessive | B6, FVB | 73 | 9 |
| pewter-FVBb | Recessive | B6, FVB | 78 | 5 |
| ND24 | Recessive | B6, FVB | 83 | 4 |
| Average | 43.3 | 8.4 |
a B6 (C57BL/6J); 129X1 (129X1/SvJ); 129S6 (129S6/SvEvTac); FVB (FVB/NJ); DBA (DBA/2J).
b pewter and dill mutants each were mapped by using two genetic crosses.
Genetic mapping of two recessive mutations with the SNP panel. Only data from informative SNPs for each mapping cross are shown. SNP names are displayed on the right. The mutated strain (M) for each mutant is red; background strain (B), blue; heterozygotes, yellow; and No Calls, white. A plot of LOD scores is shown. The vertical line indicates a LOD score of zero, with the boundaries, –52 (left) and 7.2 (right). (A) Retention of mutated alleles (C57BL/6J; LOD 5.38) in baldy affecteds (denoted as A) on distal Chr. 14 demonstrates linkage of SNP C14.065.150 (62.3 Mb) to the baldy locus. (B) Retention of mutated alleles (A/J) demonstrates linkage of hydrops fetalis to Chr. 7. Because of limited informativeness on Chr. 7 between A/J and FVB/NJ, the strain contribution of A/J or FVB/NJ is potentially ambiguous. However, allele origin can be inferred across haplotypes by the retention of A/J alleles for two A/J and FVB/NJ polymorphic SNPs (07.023.307 and 07.039.006).
Utilizing SNP genotyping analyses to facilitate positional cloning
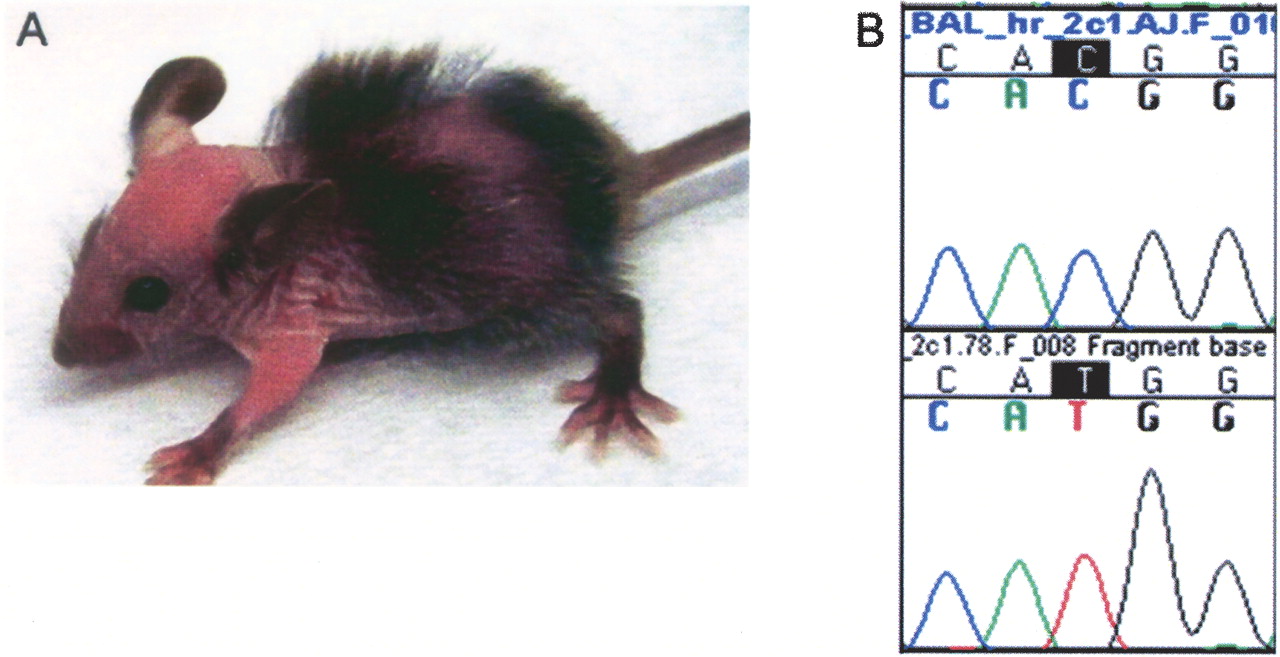
Even low-resolution chromosomal localization allows one to quickly distinguish between potentially novel loci or remutations in known genes. This is of particular utility as a means to prioritize analyses in cases where large numbers of mutants are generated, such as ENU mutagenesis screens. The SNP-based genetic mapping of both baldy and hyft suggested that both were remutations in known genes. Allelism between baldy and the hairless (hr) mutation was investigated based on the similar skin and hair phenotypes and map positions on distal 14 (Cachon-Gonzalez et al. 1994). Twenty baldy affected mice were homozygous for the mutated strain in a 70-kb interval surrounding the hr gene (62.3 Mb) (data not shown). Complementation testing between a baldy heterozygote and a rhino-hr RHJ/LeJ homozygote resulted in 12 of 22 progeny developing hair loss. The linkage of the baldy locus to hr and the failure to complement demonstrates that baldy and hr are allelic.
Sequencing of the hr gene in baldy homozygotes revealed a single base pair change from C to T in exon 2, a noncoding exon, introducing an out-of-frame ATG upstream of the normal translation initiation site (Fig. 3). hr transcript is present in baldy affecteds; however, hr protein expression in skin is dramatically reduced (data not shown). Of four alleles of hr in which the sequence change is known, this is the first known mutation in a 5′ noncoding region (Blake et al. 2003).
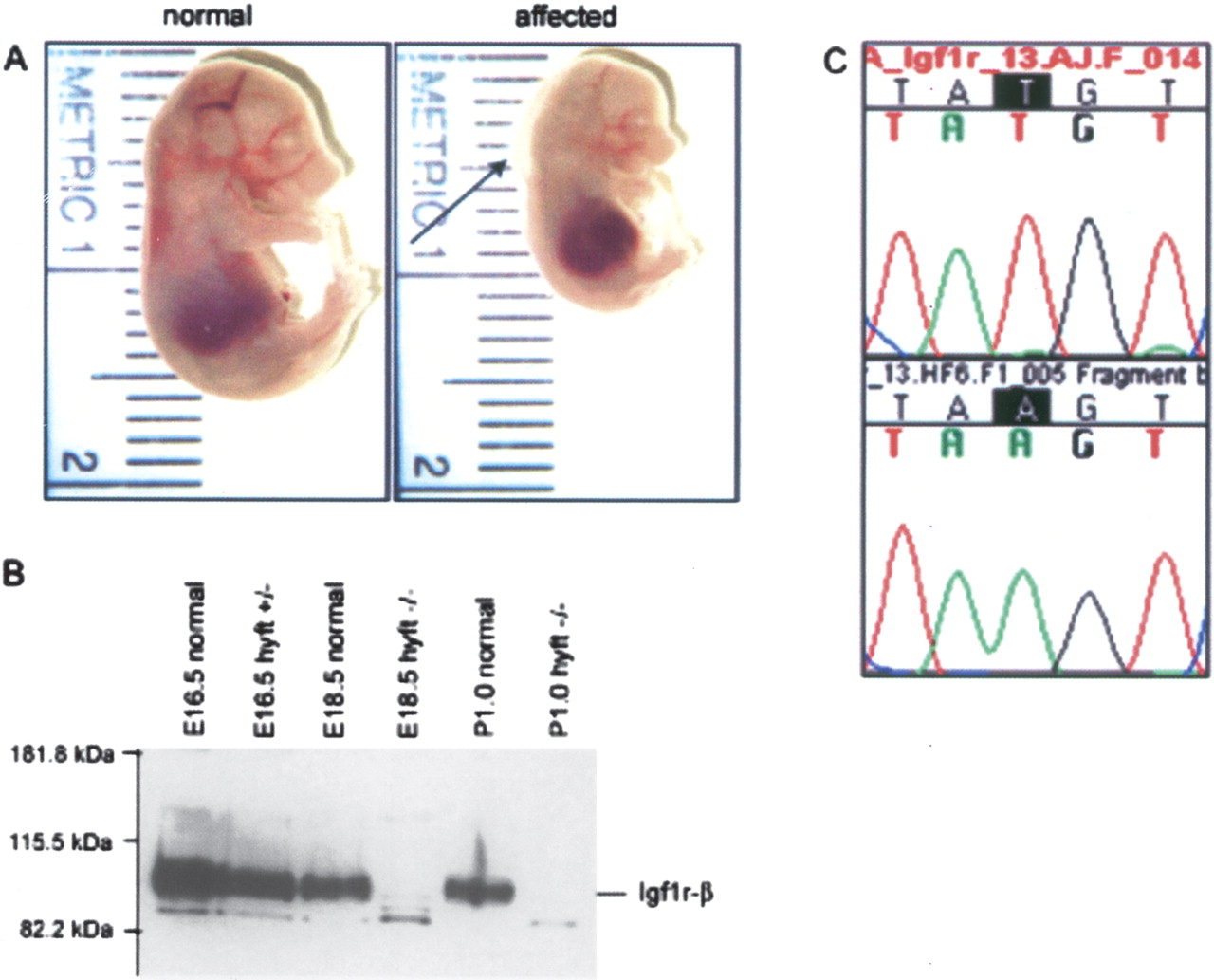
High-resolution mapping of 11 hyft affected progeny with SSLP markers narrowed the genetic interval to 10 Mb, between D7Mit89 (51.0Mb) and D7Mit182 (61.6 Mb) (data not shown). The insulin-like growth factor 1 receptor (Igf1r; 54.9 Mb) was tested as a candidate for the mutated locus because of the similarity between the embryonic growth defect in hyft and Igf1r loss-of-function mice (Liu et al. 1993). Igf1r protein expression was examined in E18.5 and P1 hyft affecteds and was found to be absent (Fig. 4). Sequencing revealed a T to A single base pair change in exon 13 (Fig. 4), which introduces a premature stop codon 5′ to the transmembrane and kinase domain of Igf1r.
We have shown that by selecting several hundred SNPs based on polymorphism in a few inbred strains, the density and informativeness of our SNP panel is sufficient to map mutations with many strain pairs, including inbred and wild-derived strain pairs. The 22 monogenic mutants were mapped utilizing crosses with a combined total of 11 different strain combinations (Table 1). Even for complex genetic studies, this SNP panel may provide a good starting point for whole-genome linkage analyses and for the design of additional panels for higher-resolution mapping. For example, preliminary results of a genetic modifier study suggests that this 394 SNP set identified a modifying locus of the DDK syndrome embryonic lethal phenotype in two wild-derived M. musculus domesticus inbred strains (LOD score = 10.1; 77 N2 mice) (F. Ideraabdullah and F. Pardo-Manuel de Villena, pers. comm.). Given the continuing advances in technology for high-throughput SNP genotyping, we anticipate that analyses using standardized genome-wide SNP panels will become a routine component of genetic studies in the mouse.
Methods
SNP assay design and genotyping
Polymerase chain reaction (PCR) primers and mini-sequencing extension primers were designed for each SNP, and SNPs were multiplexed into a maximum of five-plex pools by using Spectrodesigner software (Sequenom). Genomic DNA (2.5 ng, 1.25 ng/μL) was PCR-amplified in a 5 μL reaction containing 0.1 U HotStar Taq polymerase and 1× HotStar Taq PCR Buffer (Qiagen), 2.5 mM MgCl2, 200 μM of each dNTP (USB), and 50 nM each PCR primer. Samples were incubated for 15 min at 95°C; followed by 45 cycles of 20 sec at 95°C, 30 sec at 56°C, and 1 min at 72°C; followed by 3 min at 72°C on a GeneAmp PCR System 9700 (Applied Biosystems). Excess dNTPs were removed from the reaction by addition of 0.3 U shrimp alkaline phosphatase (SAP; USB) in Thermosequenase RCTN Buffer (Amersham Biosciences) for 20 min at 37° C followed by 5 min at 85°C. Amplified PCR products were used as a template in a second, modified, single-primer mini-sequencing reaction. Extension reactions contain 600 nM of extension primer, 50 uM dNTP, and ddNTP in Thermosequenase RCTN buffer, and 0.126 U Thermosequenase. Samples were heated at 94°C for 2 min followed by 45 cycles of 5 sec at 94°C, 5 sec at 52°C, and 5 sec at 72°C. The mini-sequencing reaction is then desalted by addition of SpectroClean resin (Sequenom) and spotted onto SpectroCHIPS (Sequenom) by using a nanoliter-plotting robot (SpectroPLOTTER, Sequenom). Chips were individually analyzed by using the Brukker Bi-flex MALDI-TOF mass spectrometer (Sequenom). The resulting spectra are converted to genotype data by using SpectroTYPER-RT software (Sequenom).
Genetic interval size is inversely correlated with the number of affecteds analyzed.
hr is mutated in the ENU-induced mutant baldy. (A) An 18-d-old baldy affected displaying patchy alopecia and wrinkled skin. (B) Chromatograms of wild-type C57BL/6J (top) and baldy affected (bottom) sequence showinga C to T mutation in baldy affecteds at nucleotide 385 of 5′ noncoding exon 2.
Genomic DNA preparation for SNP genotyping
Genomic DNA from all inbred and wild-derived strains, with the exception of 129S6SvEv (Taconic), was obtained from The Jackson Laboratory DNA Resource (Bar Harbor, ME) or from mice that were purchased from The Jackson Laboratory (JAX) and maintained on-site. 129S4SvJae DNA was obtained from the DNA stock 002497 (JAX). Genomic DNA was prepared from mouse tails either by a standard proteinase K phenol-chloroform extraction method or by using the PUREGENE Purification Kit for cells and tissue (Gentra Systems).
Polymorphism matrix
To compute the number of informative SNPs between each strain pair, only SNPs with genotype calls for each strain within a pair were considered. A list of all SNPs, megabase positions (Mb; NCBI Build 33), and genotypes of all inbred and wild-derived strains are included in the Supplemental material.
Igf1r is mutated in the ENU-induced mutant hydrops fetalis (hyft). (A) Normal littermate (left) and hyft mutant (right) at E16.5, showing reduced fetal crown-to-rump length. Arrow points to subcutaneous edema present in the affected. (B) Western blot analysis of Igf1r on protein extracts of hyft mutants and normal littermates shows an absence of Igf1r protein in affected mice. (C) Chromatograms of Igf1r sequence in wild-type FVB (top) and hyft affected (bottom) mice showing a T to A mutation, which encodes a premature stop codon in hyft mutants.
Mice
Strains C57BL/6J (B6) and FVB/NJ (FVB) used for genetic mapping of hyft and bldy were obtained from JAX. Complementation testing of baldy was performed with Hrrh-J (RHJ/LeJ, stock 001591; JAX).
Genetic mapping
baldy mice were maintained on the mutagenized strain, B6, and outcrossed to FVB for mapping. Affected F2 progeny of an F1 intercross were identified at weaning and SNP genotyped. Twenty F2 affecteds were genotyped with informative SSLP markers at the hr locus. hyft mice were generated by ENU treatment of the A/J strain (Herron et al. 2002) and maintained on FVB for five generations by serial backcrossing. N5 carriers were mated to B6, and F2 intercross affected progeny were SNP genotyped. A total of 11 hyft affecteds were genotyped with SSLP markers informative among A/J, B6, and FVB for fine-mapping.
Mouse Genome Database (Blake et al. 2003) and Mouse Genome SSR Search (http://danio.mgh.harvard.edu/mouseMarkers/musssr.html) were used as sources of SSLPs for fine-mapping or candidate gene linkage analysis.
SNP genotyping data analysis
We computed for each SNP marker the LOD score as log10 (probability of data given mutant locus/probability of data given nonmutant locus) by using only affected individuals. For an F1 intercross, assuming the mutant allele has genotype AA and the wild-type nonmutagenized strain has genotype BB, in F2 individuals a nonmutant locus expects to have genotype AA:AB:BB ratio of 1:2:1, but the mutant locus expects AA:AB:BB ratio to be 1:0:0. The appropriate allele ratios are specified for other genetic crosses accordingly. A genotyping error of 0.01 is assumed in the calculation. A hidden Markov model was used to calculate a smoothened LOD score curve by considering neighboring SNP genotypes in the inference of missing genotypes (Durbin et al. 1999; Supplemental data).
A positive LOD score provides evidence that a SNP marker is close to the mutant locus. We chose the LOD score of three as the threshold, which means the locus is 1000 times more likely to be mutant related than not. dChip software (Lin et al. 2004; www.dchip.org) is used to visualize the data, perform the computation, and convert SNP genotypes to A, B, or AB alleles.
Protein extraction and immunoblotting
Protein was extracted by homogenizing hyft+/+, hyft+/–, and hyft–/– E16.5 whole fetuses and E18.5 and P1.0 heads in non-denaturing lysis buffer (1% Triton X-100, 50 mM TrisHCl at pH 7.4, 300 nM NaCl, 5 mM EDTA, 0.02% sodium azide, 10 mM iodoacetamide, 1 mM PMSF, 2 μg/mL leupeptin). Protein extracts (100 μg) were electrophoresed on a 7% acrylamide gel, and a Western blot was performed according to standard procedures. The Western blot was hybridized with a mouse IGF-1Rβ antibody (sc-713; Santa Cruz Biotechnology, Inc.). The hybridization signal was detected by using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce).
Notes
[3] [Supplemental material is available online at www.genome.org.]
[4] Article published online ahead of print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4563306.
[5] Corresponding author. E-mail [email protected]; fax (617) 525-4705.
We thank the following individuals for their contributions: Christopher Walsh, Sally Camper, Jill Karolyi, Bill Pavan, Simon Conway, Miriam Meisler, Catherine Holland, John Schimenti, Larry Jameson, Jeffrey Weiss, Adriana Donovan, Mark Fleming, and Nancy Andrews contributed monogenic mutants for genetic mapping; Yujuan Yun, Lan Wang, Allyson Chesebro, Maura Regan, and Gwen Yarbrough contributed technical assistance; and John Hayden contributed informatics assistance. This work was supported by a grant (U01HD43430) from the National Institutes of Health.
References
- ↵Blake, J.A., Richardson, J.E., Bult, C.J., Kadin, J.A., Eppig, J.T., and Mouse Genome Database Group. 2003. MGD: The Mouse Genome Database. Nucleic Acids Res. 31: 193–195.
- ↵Cachon-Gonzalez, M.B., Fenner, S., Coffin, J.M., Moran, C., Best, S., and Stoye, J.P. 1994. Structure and expression of the hairless gene of mice. Proc. Natl. Acad. Sci. 91: 7717–7721.
- ↵Dietrich, W.F., Miller, J.C., Steen, R.G., Merchant, M., Damron, D., Nahf, R., Gross, A., Joyce, D.C., Wessell, M., Dredge, R.D., et al. 1994. A genetic map of the mouse with 4006 simple sequence length polymorphisms. Nat. Genet. 7: 220–245.
- ↵Durbin, R., Eddy, S., Krogh, A., and Mitchison, G. 1999. Biological sequence analysis: Probabilistic models of proteins and nucleic acids. Cambridge University Press, Cambridge, UK.
- ↵Fraser, K.A., Wade, C.M., Hinds, D.A., Patil, N., Cox, D.R., and Daly, M.J. 2004. Segmental phylogenetic relationships of inbred mouse strains revealed by fine-scale analysis of sequence variation across 4.6 Mb of mouse genome. Genome Res. 14: 1493–1500.
- ↵Grupe, A., Germer, S., Usuka, J., Aud, D., Belknap, J.K., Klein, R.F., Ahluwalia, M.K., Higuchi, R., and Peltz, G. 2001. In silico mapping of complex disease-related traits in mice. Science 292: 1915–1918.
- ↵Herron, B.J., Lu, W., Rao, C., Liu, S., Peters, H., Bronson, R.T., Justice, M.J., McDonald, J.D., and Beier, D.R. 2002. Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nat. Genet. 30: 185–189.
- ↵Liao, G., Wang, J., Guo, J., Allard, J., Cheng, J., Ng, A., Shafer, S., Puech, A., McPherson, J.D., Foemzler, D., et al. 2004. In silico genetics: Identification of a functional element regulating H2-Eα gene expression. Science 306: 690–695.
- ↵Lin, M., Wei, L.J., Sellers, W.R., Lieberfarb, M., Wong, W.H., and Li, C. 2004. dChipSNP: Significance curve and clustering of SNP-array–based loss-of-heterozygosity data. Bioinformatics 20: 1233–1240.
- ↵Lindblad-Toh, K., Winchester, E., Daly, M.J., Wang, D.G., Hirschhorn, J.N., Laviolette, J.P., Ardlie, K., Reich, D.E., Robinson, E., Sklar, P., et al. 2000. Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat. Genet. 24: 381–386.
- ↵Liu, J.P., Baker, J., Perkins, A.S., Robertson, E.J., and Efstratiadis, A. 1993. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75: 73–82.
- ↵Neuhaus, I.M. and Beier, D.R. 1998. Efficient localization of mutations by interval haplotype analysis. Mamm. Genome 9: 150–154.
- ↵Owens, S.E., Broman, K.W., Wiltshire, T., Belmore, J.B., Bradley, K.M., Smith, J.R., and Southard-Smith, E.M. 2005. Genome-wide linkage identifies novel modifier loci of aganglionosis in the Sox10Dom model of Hirschsprung disease. Hum. Mol. Genet. 14: 1549–1558.
- ↵Petkov, P.M., Ding, Y., Cassell, M.A., Zhang, W., Wagner, G., Sargent, E.E., Asquith, S., Crew, V., Johnson, K.A., Robinson, P., et al. 2004. An efficient SNP system for mouse genome scanning and elucidating strain relationships. Genome Res. 14: 1806–1811.
- ↵Pletcher, M.T., McClurg, P., Batalov, S., Su, A.I., Barnes, S.W., Lagler, E., Korstanje, R., Wang, X., Nusskern, D., Bogue, M.A., et al. 2004. Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol. 2: e393.
- ↵Wade, C.M., Kulbokas III, E.J., Kirby, A.W., Zody, M.C., Mullikin, J.C., Lander, E.S., Lindblad-Toh, K., and Daly, M.J. 2002. The mosaic structure of variation in the laboratory mouse genome. Nature 420: 574–578.
- ↵Wiltshire, T., Pletcher, M.T., Batalov, S., Barnes, S.W., Tarantino, L.M., Cooke, M.P., Wu, H., Smylie, K., Santrosyan, A., Copeland, N.G., et al. 2003. Genome-wide single-nucleotide polymorphism analysis defines haplotype patterns in mouse. Proc. Natl. Acad. Sci. 100: 3380–3385.