
Abstract
We identified putative long terminal repeat- (LTR) retrotransposon sequences among the 50,000 random sequence tags (RSTs) obtained by the Génolevures project from genomic libraries of 13 Hemiascomycetes species. In most cases additional sequencing enabled us to assemble the whole sequences of these retrotransposons. These approaches identified 17 distinct families, 10 of which are defined by full-length elements. We also identified five families of solo LTRs that were not associated with retrotransposons. Ty1-like retrotransposons were found in four of five species that are phylogenetically related to Saccharomyces cerevisiae (S. uvarum, S. exiguus, S. servazzii, and S. kluyveri but notZygosaccharomyces rouxii), and in two of threeKluyveromyces species (K. lactis and K. marxianus but not K. thermotolerans). Only multiply crippled elements could be identified in the K. lactis and S. servazziistrains analyzed, and only solo LTRs could be identified in S. uvarum. Ty4-like elements were only detected in S. uvarum,indicating that these elements appeared recently before speciation of the Saccharomyces sensu stricto species. Ty5-like elements were detected in S. exiguus, Pichia angusta, andDebaryomyces hansenii. A retrotransposon homologous with Tca2 from Candida albicans, an element absent from S. cerevisiae, was detected in the closely related species D. hansenii. A complete Ty3/gypsy element was present inS. exiguus, whereas only partial, often degenerate, sequences resembling this element were found in S. servazzii, Z. rouxii, S. kluyveri, C. tropicalis, and Yarrowica lipolytica. P. farinosa(syn. P. sorbitophila) is currently the only yeast species in which no LTR retrotransposons or remnants have been found. Thorough analysis of protein sequences, structural characteristics of the elements, and phylogenetic relationships deduced from these data allowed us to propose a classification for the Ty1/copiaelements of hemiascomycetous yeasts and a model of LTR-retrotransposon evolution in yeasts.
The Génolevures project used a novel approach to evolutionary genomics (FEBS Lett. 2000, special issue 487). Comparison of approximately 50,000 random sequence tags (RSTs) from 13 yeasts selected across the entire Hemiascomycetes class (see Kurtzman and Robnett 1998 for phylogenetic relationships between these species andSouciet et al. 2000) provided a wealth of sequence information on genetic redundancy, the functional classification of genes, and the conservation of synteny.
This analysis also sought repeated sequences. Indeed, an understanding of repetitious elements can be of great value in sequence assembly. Entities such as retrotransposons are known to play a role in remodeling genomes; first when they transpose into new sites and second when they are subjected to homologous recombination, leading to chromosomal rearrangements (Zolan 1995; Kim et al. 1998) such as reciprocal translocations.
One ubiquitous group of retrotransposons contains long terminal repeats (LTRs) at both extremities of the element. Different types of LTR retrotransposons exist in a wide range of eukaryotes including insects, plants, fungi, yeasts, and fishes. Recently, fossils of LTR retrotransposons were identified in mammals at a very low copy number (Volff et al. 2001). The structure of LTR retrotransposons is comparable to that of the retroviruses that replicate via mRNA intermediates (Boeke 1989). Two genes are commonly found in LTR retrotransposons. These genes are homologs of the retroviralgag and pol genes. The gag gene of retroviruses encodes structural proteins of the viral particle and the retroviral pol locus encodes a polyprotein with protease (PR), integrase (IN), reverse transcriptase (RT), and RNAseH (RH) catalytic domains. The arrangement and functions of these entities in LTR retrotransposons correspond to those in retroviruses. Some elements, such as gypsy from Drosophila melanogaster, harbor a third gene, which is homologous with the retroviral env gene encoding the protein for the envelope of infectious viral particles.
LTR retrotransposons of fungi have been divided into two distinct groups on the basis of sequence similarities of their RTs (Xiong and Eickbush 1990) and the organization of the subunits within theirpol genes. In the Ty1/copia group, these subunits are arranged in the order PR, IN, RT, and RH, whereas in the Ty3/gypsy group the order is PR, RT, RH, and IN.
LTR retrotransposons have been extensively studied in the model yeastSaccharomyces cerevisiae. Five distinct families of retrotransposons exist in this organism: four Ty1/copiaelements (Ty1, Ty2, Ty4, and Ty5) and one Ty3/gypsy element (Ty3). Only three of these families are known to be transpositionally active in S. cerevisiae; namely, Ty1, Ty2, and Ty3. Ty5 elements from S. cerevisiae are either solo LTRs or degenerate elements that have accumulated several deleterious mutations (Voytas and Boeke 1992). However, intact and active copies of Ty5 have been found in S. paradoxus, a closely related species to S. cerevisiae (Zou et al. 1996). The complete nucleotide sequence ofS. cerevisiae revealed 52 different Ty elements (Goffeau et al. 1996) and thus provided a unique opportunity to study genome organization (Kim et al. 1998), evolution (Jordan and McDonald 1998;1999b), and the coevolution of the mobile elements and their host (Jordan and McDonald 1999a).
More recent, investigations on Candida albicans identified 34 different LTR-retrotransposon families that belong to the Ty1/copia and Ty3/gypsy groups (Goodwin and Poulter 2000). Most of these families only contain solo LTRs or LTR remnants. Only three different full-length and intact retrotransposons were identified. These are (1) Tca2 (or pCa1), which is quite unusual because it carries two open reading frames (ORFs) separated by a stop codon and produces many extrachromosomal DNA copies (Matthews et al. 1997); (2) Tca5, which has a similar structure and sequence to Ty5 (Plant et al. 2000); and (3) Tca4, which is a Ty1/copiaelement close to Tca2 (Goodwin and Poulter 2001).
Other hemiascomycetous yeasts such as Yarrowia lipolytica(Schmid-Berger et al. 1994) are known to contain LTR retrotransposons, but no transposable elements have been described in theZygosaccharomyces, Kluyveromyces, or Debaryomycesyeast genera, or in the Saccharomyces sensu lato group. The Génolevures project provided evidence for the presence of LTR retrotransposons in some of the 13 yeast species studied (Blandin et al. 2000a,b; Bolotin-Fukuhara et al. 2000; Bon et al. 2000a,b;Casaregola et al. 2000; Lépingle et al. 2000; Llorente et al. 2000b;Neuvéglise et al. 2000).
Therefore, on the basis of the data of Génolevures, we attempted to characterize fully and to compare the LTR retrotransposons present in the 13 hemiascomycetous yeasts by determining the full sequences of some of these elements. Thorough analysis of the data revealed phylogenetic relationships between some of the elements and enabled us to suggest a classification system for the Ty1/copia elements of hemiascomycetous yeasts.
Identification of New Retrotransposon Sequences
Our study was based on the Génolevures sequence data, which includes 49,199 RSTs from 13 hemiascomycetous yeasts (Souciet et al. 2000). The partial sequencing of each genome provided a coverage of approximately 0.2× (2500 RSTs) of the S. exiguus, S. servazzii, S. kluyveri, K. marxianus, D. hansenii, and C. tropicalisgenomes, and a coverage of approximately 0.4× (5000 RSTs) of theS. bayanus (syn. S. uvarum), Z. rouxii, K. lactis, Pichia angusta, P. farinosa (syn. P. sorbitophila), and Y. lipolytica genomes. All of the RSTs have been compared with a database of Ty protein sequences (H. Feldmann, unpubl.). Because the copies of Ty5 elements in S. cerevisiae are degenerate, all of the RSTs were compared with the Ty5 elements of S. paradoxus. Table1 lists the RSTs that match to Ty elements. The RSTs of interest were thoroughly analyzed and manually assembled to avoid low-quality contigs. Then, LTRs associated to the identified full-length elements were defined by comparison of their 5′ and 3′ extremities. Finally, repeated sequences were screened for the presence of solo LTRs. We used the internal repeats TGTTG…CAACA that bound the LTR and the 5 bp of target site duplication, whenever they were present, in addition to the breaking points of sequence homology between two different copies, to define the border of the LTRs.
Identification of RSTs that match SaccharomycesTy proteins
| Yeast species | Ty1/Ty2 | Ty3 | Ty4 | Ty5 | Total |
| S. bayanus | 0 | 0 | 41 (Tsu4) | 0 | 41 |
| S. exiguus | 19 (Tse1) | 15 (Tse3) | 0 | 10 (Tse5.1, Tse5.2) | 45 |
| S. servazzi | 1 (Tss1) | 3 (Tss3) | 0 | 0 | 4 |
| Z. rouxii | 0 | 5 (Tzr3) | 0 | 0 | 5 |
| S. kluyveri | 11 (Tsk1) | 1 (Tsk3) | 0 | 0 | 12 |
| K. thermotolerans | 0 | 0 | 0 | 0 | 0 |
| K. lactis | 12 (Tkl1.1, Tkl1.2) | 0 | 0 | 0 | 12 |
| K. marxianus | 32 (Tkm1) | 0 | 0 | 0 | 32 |
| P. angusta | 0 | 0 | 0 | 26 (Tpa5) | 26 |
| D. hansenii | 2 (Tdh2)[ii] | 2 (Tdh3) | 0 | 28 (Tdh5) | 32 |
| P. farinosa | 0 | 0 | 0 | 0 | 0 |
| C. tropicalis | 0 | 3 (Tct3) | 0 | 0 | 3 |
| Y. lipolytica | 0 | 2 (Tyl3) | 0 | 0 | 2 |
[i] When random sequence tags (RSTs) matched several Ty elements, we only considered the best match. The name of the newly identified element is indicated in brackets.
[ii] Further identified as Tca2-like elements.
This screening revealed a large variation in match number: from zero inK. thermotolerans and P. farinosa to 45 in S. exiguus (Table 1). No matches were detected when the Ty library was compared with the RSTs from K. thermotolerans and P. farinosa (de Montigny et al. 2000). This indicates that these strains possess few or no Ty-like elements. Alternatively, the sequence of the elements from these yeast species may be so divergent from conventional Ty elements that they were not identified. In a few species (S. servazzii, S. exiguus, S. kluyveri, and D. hansenii), the RSTs matched different types of Ty elements. This implies that, like in S. cerevisiae, different elements exist in a single host. A systematic nomenclature was used to name the newly identified LTR retrotransposons: T for Transposon followed by the initials of the genus and species of the yeast and a number referring to the S. cerevisiae homologs (1 for Ty1 or Ty2, 3 for Ty3, 4 for Ty4, and 5 for Ty5) or to the C. albicans homolog (2 for Tca2). For example, Tpa5 is a P. angusta element that is homologous to Ty5, whereas Tdh2 is from D. hansenii and is homologous to Tca2. Whenever several highly divergent copies of the same element type were identified, a decimal number was added to the name, for Tkl1.1 and Tkl1.2 and Tse5.1 and Tse5.2 (see below). In all other families of full-length elements, internal variability was less than 1.2% and did not affect the creation of the consensus sequences. We identified a total of 17 families of LTR retrotransposons in the 13 yeast species (Table 1), a family being defined by the set of copies of a particular retrotransposon, itself defined by structural features and by sequence conservation, in a given yeast species.
Elements of three of the 17 families were assembled as complete consensus sequences from RST data (Tpa5, Tkm1, Tsu4), and elements of a further seven were described after additional sequencing (Tse1, Tse3, Tse5, Tsk1, Tkl1, Tdh2, and Tdh5). The remaining seven families are described from incomplete sequence information. These seven families were present at low copy number, all (except Tss1) were Ty3 homologs and were (except Tct3) highly degenerate. Table2 lists the main characteristics of these newly identified elements in comparison with known LTR retrotransposons of hemiascomycetous yeasts.
Characteristics of LTR Retrotransposons from Hemiascomycetous Yeasts
| Species | Element | ORF1 size (AA) | ORF2 size (AA) | Frameshift | LTR | |||
| name[iii] | size (bp) | copy[iv] number | size (bp) | terminal inverted repeats | ||||
| Ty1-like elements | ||||||||
| S. cerevisiae | Ty1-br | 5917 | 32 | 435 | 1321 | CUUAGGC | 332 | TG…CA |
| S. cerevisiae | Ty2-b | 5959 | 13 | 431 | 1359 | CUUAGGC | 332 | TGT…ACA |
| K. marxianus | Tkm1 | 5994 | 10–15 | 421 | 1339 | CUUAGGC | 385 | TGTTG…CAACA |
| S. exiguus | Tse1 | 5691 | 15–20 | 383 | 1235 | CUUAGGC | 424 | TGA…TCA |
| S. kluyveri | Tsk1 | 5905 | 8 | 435 | 1320 | CUUAGGC | 322–333 | TG…CA |
| K. lactis | Tkl1.1[i] | 5425 | 1 | 202[v] | 1335[vi] | CUUAGGC | 393–398 | TGTTG…AAACA or TGTTG…CAACA |
| K. lactis | Tkl1.2[i] [ii] | 1 | 391[vii] | CUUAGGC | ?…AAACA[viii] | |||
| S. servazzii | Tss1[i] [ii] | 1–2 | ||||||
| Ty4-like elements | ||||||||
| S. cerevisiae | Ty4-j | 6227 | 3 | 363 | 1440 | CUUAGGC | 371 | TGTTG…CAACA |
| S. bayanus | Tsu4 | 6095 | 15–20 | 358 | 1433 | CUUAGGC | 321 | TGTTG…CAACA |
| Ty5-like elements | ||||||||
| D. hansenii | Tdh5 | 5349 | ∼20 | – | 1490 | – | 457 | TGTTGAA…TTCAACA |
| P. angusta | Tpa5 | 4883 | 10–15 | – | 1417 | – | 322 | TGTTG…CAACA |
| S. exiguus | Tse5.1 | 5286 | 8–10 | – | 1531 | – | 370 | TGTTG…CAACA |
| C. albicans | Tca5 | 5588 | 0–5 | – | 1477 | – | 685 | TGTTG…CAACA |
| S. paradoxus | Ty5 | 5376 | 10–15 | – | 1698 | – | 251 | TGTTGA…TCAACA |
| Tca2-like elements | ||||||||
| D. hansenii | Tdh2 | 5928 | 3–5 | 336 | 1340 | no, stop codon | 379 | TGTTGG…CCAATA |
| C. albicans | Tca2 | 6426 | 5–10 | 324 | 1576 | no, stop codon | 280 | TGTTGG…CCATCA |
| gypsy elements | ||||||||
| S. cerevisiae | Ty3-g | 5351 | 2 | 285 | 1262 | GCGAGUU | 340 | TGTTGTAT…ATACAACA |
| S. exiguus | Tse3 | 6487 | 10–15 | 257 | 1181 | AUUAGUA | 945–947 | TGTAAC…GTTACA |
| Y. lipolytica | Yltl | 9451 | 35 | 1097[ix] | 1069[ix] | ? | 714 | TGT…ACA |
| S. kluyveri | Tsk3[i] [ii] | 1–2 | ||||||
| D. hansenii | Tdh3[i] [ii] | 1–2 | ||||||
| S. servazzii | Tss3[i] [ii] | 1–2 | ||||||
| Y. lipolytica | Tyl3[i] [ii] | 1–3 | ||||||
| Z. rouxii | Tzr3[i] [ii] | 3–5 | ||||||
| C. tropicalis | Tct3[ii] | 1–3 | ||||||
[i] Defective element.
[ii] Incomplete sequence.
[iii] The name of the elements specifically identified in this study are in bold.
[iv] The copy number was determined for Ty elements; it does not include solo LTRs.
[v] The first part of gag from Tkl1.1 was deleted.
[vi] This ORF has a frameshift between positions 1909 and 1931.
[vii] This ORF has a frameshift at position 1490 (ggg instead of gg).
[viii] The beginning of the LTR was not determined.
[ix] Sizes of Ylt1 ORF1 and ORF2 are only indicative because the locus that underwent the frameshift could not be precisely determined.
[x] bp, base pairs; AA, amino acids; ORF, open reading frame; LTR, long terminal repeat.
Classification Based on Structural Features and Sequence Similarity
On the basis of their structural characteristics and sequence similarity, the 17 newly identified retrotransposon families were divided into five groups, which also included previously described elements from other yeasts (Table 2).
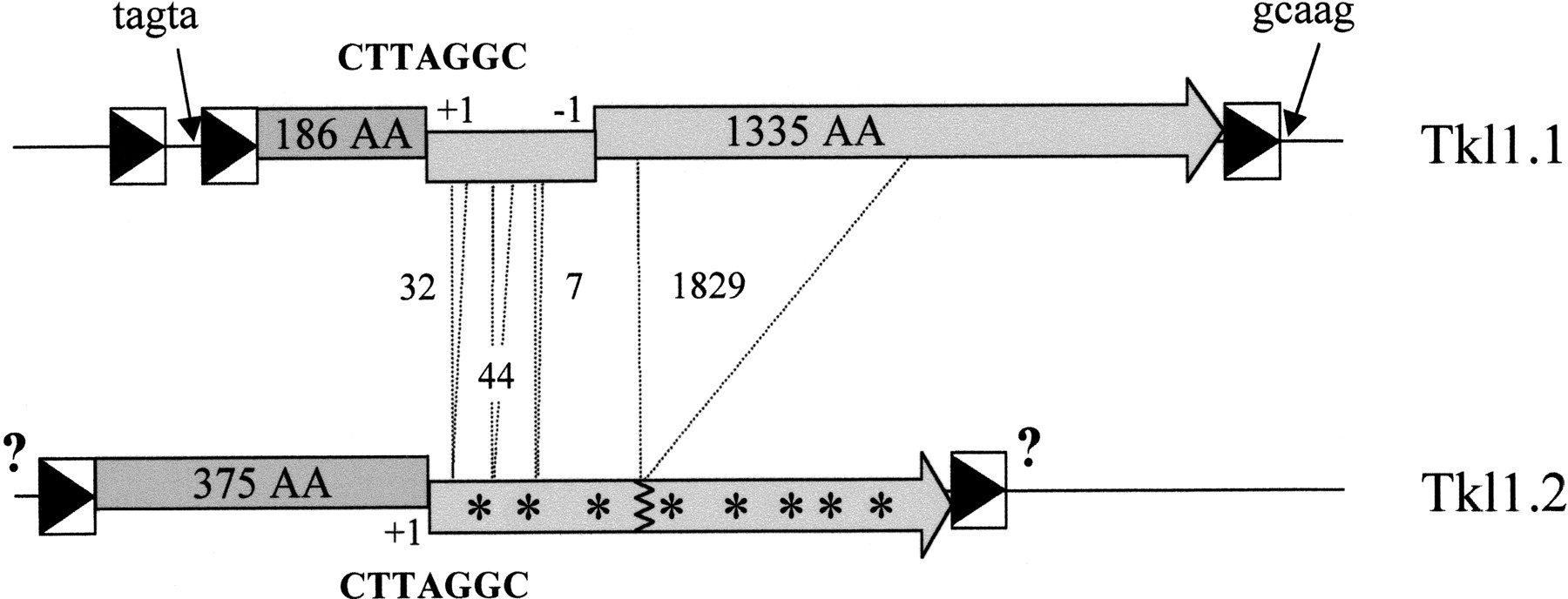
The first group (Ty1-like elements), which is a very homogeneous group, comprises Ty1 and Ty2 together with the five new homologs of Ty1. These are the partially sequenced element Tss1 and four families of completely described elements (Tse1, Tkl1, Tkm1, and Tsk1). Two different copies of Tkl1 were identified in the K. lactisstrain studied, Tkl1.1 and Tkl1.2 (Fig.1). All of the Ty1-like elements are approximately 5.9 kb long, with the exception of Tse1 from S. exiguus, which is only 5.6 kb long. Tkl1.1 is only 5425 bp in length, but the copy sequenced has lost the first 189 amino acids (aa) of gag (Fig. 1). All of the completely sequenced elements in the Ty1-like group have two overlapping ORFs separated by a +1 frameshift occurring within the highly conserved sequence CUUAGGC in the region of the overlap (Voytas and Boeke 1992). This indicates that the frameshift mechanism has also been conserved in these elements. The lengths of the LTRs are also conserved, varying from 322 bp in S. kluyveri to 424 bp in S. exiguus. All of the LTRs are terminated by the dinucleotides 5′-TG…CA-3′ or longer terminal inverted repeats. In Tkm1, the terminal inverted repeat is 5′-TGTTG…CAACA-3′.
LTR retrotransposons of K. lactis. Two copies of the same element were identified in K. lactis, namely Tkl1.1 and Tkl1.2. The 5' and 3' LTR (boxes containing a black triangle) of Tkl1.1 only share 93 % nucleotide identity and the complete element is not flanked by the expected 5 bp nucleotide duplications of the target site. Sequences adjacent to Tkl1.1 LTRs are indicated by arrows. One of the two overlapping ORFs is homologous to gag (dark gray boxes containing the size of the protein), but lacks the first 189 aa at the N-terminus, and the other one is homologous to pol (light gray boxes containing the size of the protein) but has a one nucleotide deletion between positions 1909 and 1931, which generates a –1 frameshift. Tkl1.2, contains the complete gag gene, the consensus frameshift motif CTTAGGC indicated by “+1” found in Tkl1.1 and a second gene (light gray boxes containing stars) interrupted by numerous stop codons and small (7, 32, 44 nt) or large (1829 nt) deletions (dotted lines; sizes are in nt) compared to Tkl1.1. The sequences of the flanking LTRs have not been determined (question marks). Probes for specific regions of both copies (in the 5' part ofgag for Tkl1.2 and in the part of pol gene that is deleted in Tkl1.2 for Tkl1.1) were used in Southern hybridizations (data not shown) and confirmed that only one copy of each element is present in the K. lactis strain studied.
The second group (Ty4-like elements) constitutes Ty4 and the newly described related retrotransposon Tsu4. The organization of Tsu4 and Ty4 is similar to that of the Ty1-like elements, with two ORFs subjected to the same frameshift mechanism. They have different primer binding sites (PBSs) (see below) and the sizes of ORF1 and ORF2 differ slightly: ORF1 is generally larger in Ty1-like elements and ORF2 is generally larger in Ty4 and Tsu4. The amino acid sequences of the peptides derived from ORF2 are conserved between Ty1-like and Ty4-like elements (29.6% identity and 41.1% similarity for 1240 aa), whereas their gag proteins are less well conserved and difficult to align. The nucleic acid binding motif CX2CX4HX4H of gag is found in the Ty4-like elements but not in the Ty1-like elements.
The third group (Ty5-like elements) constitutes Ty5 and Tca5 together with the newly described retrotransposons Tdh5, Tpa5, and Tse5. These elements are characterized by a single ORF corresponding to agag-pol gene fusion. The lengths of the ORFs are variable: 1417 aa in Tpa5 compared with 1698 aa in S. paradoxus Ty5. The lengths of the LTRs are also highly variable: from 251 bp in Ty5 to 685 bp in Tca5. The terminal inverted repeats are more highly conserved than in Ty1-like elements, consisting of at least 5 nt: 5′-TGTTG…CAACA-3′. Two related elements were identified inS. exiguus: Tse5.1 and Tse5.2. The sequences of theirpol genes are highly conserved (97.4% identity for 1299 amino acids), with the exception of a 34 aa deletion in Tse5.1 located within the tether region, between IN and RT. Despite these differences, we consider that Tse5.1 and Tse5.2 belong to the same family.
The fourth group (Tca2-like elements) comprises Tca2, Tca4, and the new element Tdh2. Although initially detected because of its homology with Ty1, Tdh2 is more closely related to Tca2, which is not found inS. cerevisiae. In addition to having similar nucleotide sequences, Tdh2 and Tca2 share several characteristic features. ORF1 and ORF2 are in the same phase, separated by a stop-codon (UAA in Tdh2 and UGA in Tca2). This arrangement is similar to that found in some mammalian retroviruses (Yoshinaka et al. 1985) but is unique in LTR retrotransposons. No purine-rich sequence occurs downstream from the UAA codon in Tdh2, a sequence required for the suppression of the UGA stop codon in MMLV and Tca2 (Matthews et al. 1997). Another structural peculiarity of the two elements is the occurrence of imperfect 6-bp inverted repeats at the ends of the LTRs.
The fifth group (Ty3/gypsy elements) consists of the Ty3 homologs including the seven new elements Tse3, Tss3, Tzr3, Tsk3, Tdh3, Tct3, and Tyl3. Five of these elements were found to be degenerate because of an accumulation of deletions and/or point mutations that introduce stop codons in the ORFs. For example, when the three copies of the Tzr3 element in Z. rouxii were partially sequenced, they were found to differ (3% nt divergence) and to contain a large number of stop codons. A sixth element (Tct3) present in a low copy number in C. tropicalis may be intact as no stop codons were detected in the 1360 nt sequenced. The only completely sequenced Ty3/gypsy element that appears to be structurally intact is Tse3 from S. exiguus. Its two overlapping ORFs are separated by a +1 frameshift that probably occurs within the heptanucleotide AUUAGUA, if the Ty3 model of translational frameshifting has been conserved (Voytas and Boeke 1992).
LTRs Not Associated with Complete Retrotransposons
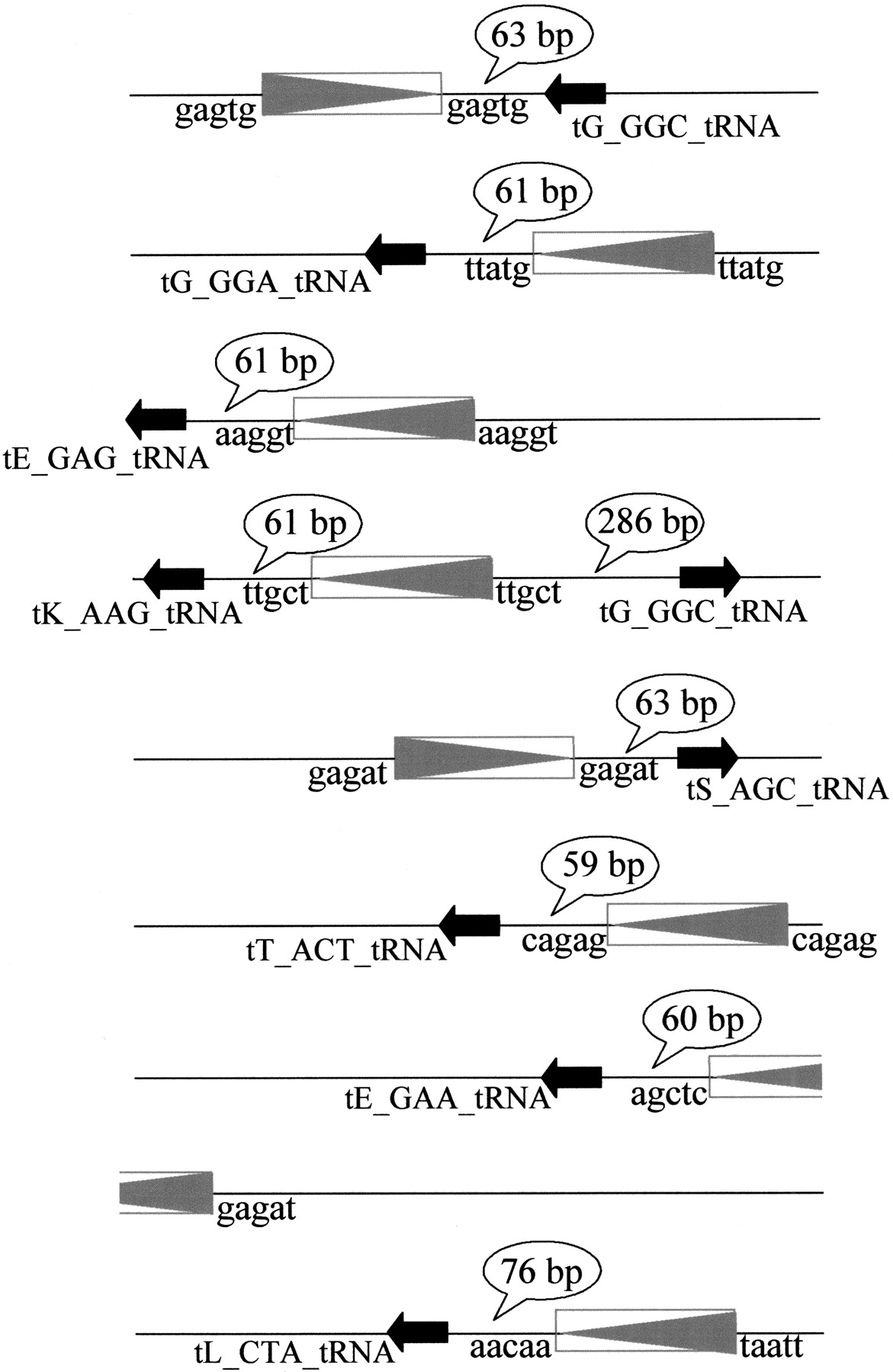
In some yeast species, LTRs not associated with complete retroelements were detected either by analysis of repeated sequences or because they were located at strategic sites such as translocation breakpoints or in the vicinity of tRNA genes or other retrotransposons. In K. thermotolerans and P. farinosa, repeated sequences were systematically screened because no homology with Ty proteins was detected. We identified two putative LTR sequences inK. thermotolerans, LTRkt1 and LTRkt2, but none in P. farinosa. The “long” version of LTRkt1 is 261 bp long and the “short” version is 243 bp long. The LTR is surrounded by 5′ TGT…ACA 3′ and was found in at least 11 RSTs corresponding to nine different loci. At each locus, the putative LTR is flanked by a 5 bp direct repeat corresponding to a duplicate of the target site (Fig.2). In addition, at each locus except one, at least one tRNA gene was located 59 to 76 bp upstream of or downstream from the LTR (Table3). This indicates that this putative LTR integrates at highly specific sites, near tRNA genes, which is consistent with the findings in S. cerevisiae (Eigel and Feldmann 1982; Kim et al. 1998).
K. thermotolerans RSTs carrying copies of LTRkt1. LTRkt1 are represented by the boxes containing a gray triangle and tRNA genes by a black arrow. The 5-bp sequences adjacent to LTRkt1, which may correspond to the target site duplication, are indicated at both extremities of each element. The distances between LTRkt1 and tRNA genes are shown in speech bubbles. Note that seven of the nine RSTs have LTRs 59–76 bp upstream of tRNA genes. The accession numbers of the corresponding RSTs are respectively (top to bottom): AL421848,AL421381, AL422269, AL420237, AL420504, AL420956, AL419815, AL420355, and AL421226.
Specific Integration of LTR Retrotransposons as Shown by Their Location on the RST
| Element | Species | LTR[i] | tRNA[ii] | Distance[iii] | Target specificity | Hot-spot for transposition |
| LTRsul | S. bayanus | 59 | 24 | 117–698 | tRNA | + |
| Tsu4 | S. bayanus | 45 | 16 | 95–540 | tRNA | + |
| Tse1 | S. exiguus | 12 | 0 | — | ? | |
| Tse3 | S. exiguus | 24 | 9 | 13–19 | tRNA | + |
| Tse5 | S. exiguus | 10 | 2 | 151–365 | ? | + |
| Tsk1 | S. kluyveri | 58 | 21 | 79–540 | tRNA | + |
| Tsk3 | S. kluyveri | 4 | 1 | — | ? | |
| LTRkt1 | K. thermotolerans | 11 | 10 | 59–79 | tRNA | |
| LTRkt2 | K. thermotolerans | 10 | 0 | — | ? | |
| Tkm1 | K. marxianus | 13 | 1 | 106 | ? | |
| Tkl1 | K. lactis | 30 | 0 | — | ? | |
| Tpa5 | P. angusta | 14 | 0 | — | ? | |
| LTRpa1 | P. angusta | 19 | 3 | — | ? | + |
| Tdh2 | D. hansenii | 4 | 0 | — | ? | |
| Tdh5 | D. hansenij | 17 | 0 | — | ? | |
| Tyl3 | Y. lipolytica | 7 | 1 | — | ? | |
| LTRyl1 | Y. lipolytica | 18 | 1 | 149 | ? |
[i] Number of RSTs carrying solo LTR or 3′ LTR.
[ii] Number of RSTs carrying both LTR and tRNA genes.
[iii] Number of nucleotides between the LTR and the tRNA gene.
LTRkt2, the second putative LTR in K. thermotolerans, was found as a repeated sequence in 10 RSTs corresponding to nine different loci. The element is 293 or 417 bp long. The longer version of LTRkt2 is often flanked by 5′ TATTG…TGACA 3′ and the shorter version is often flanked by 5′ TACGA…TGACA 3′, but some copies have accumulated point mutations.
A repeated sequence was identified as a putative LTR in Y. lipolytica (Casaregola et al. 2000). This 273-bp sequence, LTRyl1, is present in 18 RSTs and contains the characteristic TGTTG repeat at the 3′ end and the characteristic CAATA repeat at the 5′ end. Five of the 18 LTR sequences are surrounded by 5-bp repeats, which account for the duplication of the target site.
In P. angusta, a repeated sequence (LTRpa1) was found preferentially associated with other LTR sequences or Tpa5 retroelements in 19 RSTs corresponding to 17 different loci (Blandin et al. 2000a). LTRpa1 is flanked by TGTTG…CAACA and has a mean length of 265 bp.
When the synteny breakpoints of S. bayanus were sequenced, a putative solo LTR of 331 bp flanked by a 5-bp duplication site was found in S. bayanus (Fischer et al. 2001). This LTR (LTRsu1) possesses the imperfect internal repeats TGTTG…CAATA and is highly repeated in S. bayanus genome, occurring in 59 RSTs. Some of the copies seem to be intact, whereas others are degenerate or truncated. We identified tRNA genes in approximately 40% of the RSTs containing LTRsu1, indicating that tRNA genes are hotspots for LTRsu1 integration.
P. farinosa is currently the only yeast species in which no known LTR retrotransposons or remnants are known.
Insertion Sites
It is well known that all S. cerevisiae Ty elements are target specific. Ty1–4 elements insert upstream of genes transcribed by RNA polymerase III, such as tRNA genes (Chalker and Sandmeyer 1992;Kim et al. 1998), whereas Ty5 integrates preferentially into silent chromatin regions, such as at telomeres and mating loci (Zou et al. 1996). We systematically screened RSTs carrying solo LTRs or 3′ LTRs for tRNA genes and for genes that are known to be subtelomeric or telomeric in S. cerevisiae. This analysis was probably biased because we do not have the complete genome sequence but only random sequences of approximately 1 kb on average. For example, in S. cerevisiae, Kim et al. (1998) considered that Ty1 insertions were tRNA-associated if they were located within 750 bp of a tRNA gene. Thus, we probably underestimated the number of associations.
There is no proof that the Ty5-like elements considered here preferentially insert themselves into subtelomeric regions, because these regions evolve rapidly in yeasts and could not be identified during the Génolevures program (Llorente et al. 2000a).
A few retroelements follow a nonrandom mode of integration (Table 3). Five elements (LTRsu1, Tsu4, Tse3, Tsk1, and LTRkt1) are preferentially integrated into tRNA gene regions. At least one tRNA gene was identified in 35% to 40% of the RSTs containing LTRsu1, LTRs of Tsu4, Tse3, or Tsk1 and in all RSTs except one containing LTRkt1. For all other elements, we do not have enough information to draw a conclusion on their targeting specificity.
We observed two modes of integration specificity, as in S. cerevisiae. One type of targeted integration, as observed for Tse3, was found to be very precise. It always occurs 13–19 bp upstream of a tRNA gene as described for Ty3, indicating conservation of the integration mechanism of Tse3. Conversely, the four other LTR retrotransposons that carry out targeted integration have a wider target preference. Integration occurred between 58 bp and 697 bp upstream of, and sometimes downstream from, a tRNA gene. The distance is often increased if there is another LTR sequence between the LTR in question and the tRNA gene (transposition hotspot).
Copy Numbers
We tried to estimate the copy number of each retrotransposon without taking into account the solo LTRs. First, we measured the number of RSTs containing part of the full-length retrotransposons (Table 1). Considering the percentage of genes identified per genome (Génolevures 2000), and the size of the retrotransposons, we calculated the number of each element per genome. In some cases, we then used Southern hybridization with internal probes (not shown). For instance, in S. kluyveri, we identified 24% of the genes (Neuvéglise et al. 2000) and found 11 RSTs matching Ty1 or Ty2. Considering that one RST corresponds to 1/6 of Tsk1, the estimated number of elements per genome is 7.6. Interestingly, we observed eight bands on Southern blot hybridization with one Tsk1 internal probe on different digestions of S. kluyveri genomic DNA.
We found that the number of elements per genome is highly variable but lower than the number of Ty1 elements in S. cerevisiae. The retrotransposons could be classified into three groups based on their copy number: (1) highly repeated elements (Tse1, Tsu4, and Tdh5) present at 15–20 copies per genome; (2) moderately repeated elements (Tkm1, Tsk1, Tpa5, Tse5, and Tse3) with 8–15 copies per genome; and (3) weakly repeated elements (Tkl1.1 and Tkl1.2, Tdh2, Tdh3, Tsk3, Tss3, Tyl3, Tzr3, and Tct3). These highly and moderately repeated elements all seemed to be intact and to be potentially active. All identified copies of the weakly repeated elements, except Tdh2, have accumulated stop codons or deletions and are therefore defective. It is still unclear whether full-length copies of the degenerate Ty3/gypsy elements exist in the genome or not.
PBS
Most LTR retrotransposons use a specific tRNA from their host as a primer for RT. A short sequence (8–49 nt) of the retroelement located immediately downstream from the 5′ LTR, termed the PBS, is complementary to part of this tRNA molecule. In S. cerevisiae,the PBS of Ty1, Ty2, and Ty3 retroelements are complementary to the 3′ acceptor stem of the initiator methionine tRNA (tRNAiMet), whereas the PBS of Ty5 is complementary to 13 nt from an internal portion of tRNAiMet that includes the anticodon stem-loop (Voytas and Boeke 1993). S. cerevisiae Ty4 is an exception to this rule because its PBS is complementary to the 3′ end including the acceptor stem of S. cerevisiae tRNAAsn with one mismatch (Stucka et al. 1992). New types of PBS were detected inC. albicans (Goodwin and Poulter 2000). For example, the PBS of Tca1 and Tca2 are complementary to an internal fragment of the tRNAarg(UCU).
We searched the newly identified elements for potential PBS and the tRNA genes for complementary sequences. As expected, the PBS of these elements were complementary to sequences within tRNAs that were homologous to their counterparts in S. cerevisiae or C. albicans. Elements homologous to Ty1 or Ty3 contained PBS that were complementary to the 3′ acceptor stem of a tRNAiMet. We confirmed this in Tse1, Tkl1–2, and Tse3 by comparing the sequences of the tRNAiMet identified in S. exiguus and K. lactis with the sequences of the retroelements. We noticed that the PBS were longer in these elements than in Ty1, Ty2, or Ty3: 12 nt in Tse1 and Tse3 and 13 nt with one mismatch in Tkl1.2 (Fig.3). For Tsk1 and Tkm1, we had to compare the sequence with that of S. cerevisiae Ty1 PBS because no tRNAiMet genes were found in the corresponding RSTs. PBS were also found to be highly conserved among Ty5-like elements, being complementary to the anticodon stem-loop of tRNAiMet. Comparisons with the nucleotide sequence of tRNAiMet genes ofS. exiguus and P. angusta showed that the PBS of Ty5-like elements are longer in Tse5 and Tpa5 than in Ty5 or Tca5.
Primer-binding sites of LTR-retrotransposons and tRNA molecules of hemiascomycetous yeasts. The TGG trinucleotide starting most of the PBSs is complementary to the CCA trinucleotide present at the 3' end of all tRNAs and is characteristic of PBSs that utilize the 3' end of a tRNA as a primer. In contrast, the PBSs that are complementary to internal positions of tRNAs include the anticodon stem-loop (underlined). The tRNA molecule, residues that are complementary to PBS are indicated in black when located at the 3' end of the molecule and in gray when located in the internal part. Similarly, PBS sequences are written in black or gray depending on their positions. PBS sequences in bold indicate that the PBS was deduced from the comparison with the tRNA gene identified in the species. The mismatches in the PBS sequences of Tkl1.2, Tca5, Tsu4 and Ty4 are not in bold.
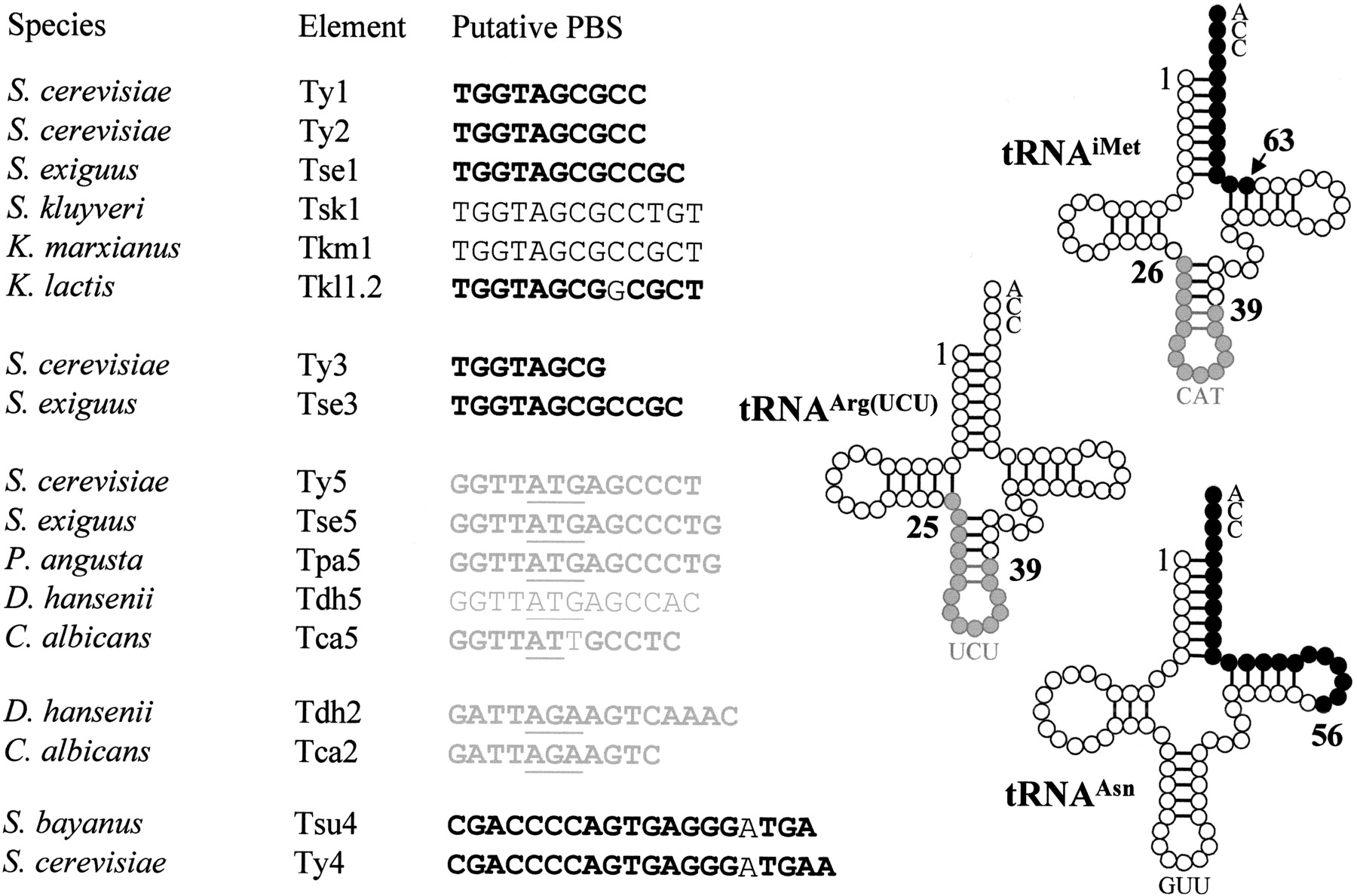
Unusual PBS were found in Tsu4 and Tdh2. The PBS of Tsu4 is complementary to 22 nt of the 3′ end of a tRNAAsn whereas Ty4 is complementary to 23 nt (Stucka et al. 1992), although both have the same mismatch at position 16 (Fig. 3). The sequence of the tRNAAsn gene from S. bayanus is identical to that ofS. cerevisiae. The PBS in Tsu4 and Ty4 are among the longest known PBS, although the longest one is the Tcn10 PBS from the basidiomycetous yeast Cryptococcus neoformans (Goodwin and Poulter 2001), which is 49 bp. In Tdh2, the PBS is complementary to an internal fragment of tRNAArg(UCU) including the anticodon stem-loop as described for its C. albicans homolog, Tca2 (Matthews et al. 1997). The identification of tRNAArg(UCU) inD. hansenii confirms that the Tdh2 PBS (15 nt) is longer than the Tca2 PBS (11 nt).
Conservation and Diversity of the ORFs of Yeast Retroelements
Sequence homologies between conserved coding regions within the ORFs of the newly identified LTR retrotransposons allowed us to align the amino acid sequences of all members of the five groups described above.
TYA is known to be extremely variable in different yeast species such that their sequences cannot be aligned. Only some of the nucleic acid-binding regions located at the carboxy terminus had homology with Ty3, Ty4, or Ty5. We found that the CX2CX4HX4H motif was conserved in all Ty5-like elements except in Tse5.1 and Tse5.2 (Fig.4A). This motif is also conserved in Ty4 and Tsu4. This motif is found in Ty3 and Ylt1, which is a Y. lipolytica Ty3/gypsy element (Schmid-Berger et al. 1994) but is quite degenerate in Tse3.
Amino acid sequence alignment of the conserved motifs in ORF1 and ORF2. (A) Sequence alignment of the RNA-binding domain located at the carboxy terminus of gag homologs in hemiascomycetous LTR-retrotransposons. The highly variable CX2CX4HX4H motif is only conserved in few hemiascomycetous retroelements including Ty3-like, Ty4-like and some Ty5-like elements. Ty1 and Ty2 lack the CX2CX4HX4H consensus motif but have a nucleic acid-binding motif homologous to a consensus prokaryotic DNA-binding sequence. Conserved residues are in white on black background. (B) Multiple sequence alignments of the protease active site. The universally conserved aspartic acid is shown in white on black background. The conservative substitutions found in more than 75% of the elements are in white on gray background. (C) Multiple sequence alignments of the zinc finger of integrase. Highly conserved residues of the HHCC zinc finger are shown in white on black background. The numbers within brackets indicate the number of residues constituting the loop between HH and CC. As the environment of the zinc finger is also conserved, the conserved K residue 4 amino acids downstream from the second C residue is shown in white on grey background. The star indicates that Tse5.1 and Tse5.2 possess exactly the same zinc finger, the sequence of the protease of Tse5.2 is not available.
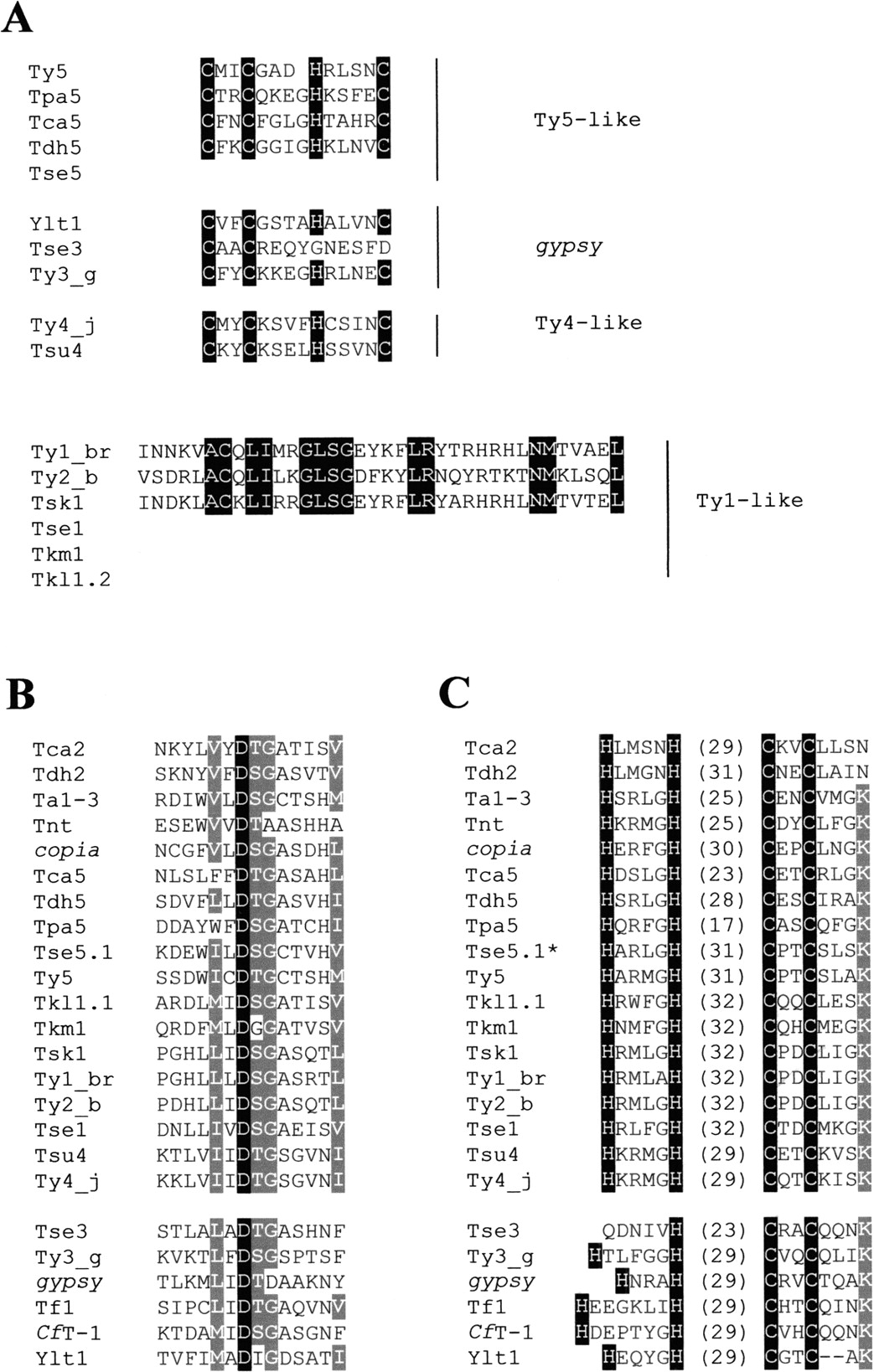
It is known that Ty1 and Ty2 elements lack the CX2CX4HX4H consensus motif (Jordan and McDonald 1999b) but possess a nucleic acid-binding motif that is homologous to a prokaryotic consensus DNA-binding sequence (Clare and Farabaugh 1985). Tsk1 is the only Ty1-like element that has a similar sequence to the putative nucleic acid-binding motives of Ty1 and Ty2. As in Tca2 (Matthews et al. 1997), no conserved motives were found in the first ORF of Tdh2.
The amino acid sequence of TYB is generally more stringently conserved than that of TYA. We aligned the sequences of the newly identified retroelements for each of the four internalTYB domains. The RT sequences were highly conserved, which allowed us to align them with other Ty1/copia and Ty3/gypsy elements. This alignment is available athttp://www.inra.fr/clib/english/genolevu.htm.
The PR, IN, and RH domains of the Ty1/copia and Ty3/gypsy elements were too divergent to allow extensive alignments. Thus, we could only align the highly conserved boxes (Fig.4B,C). The active site of the endogenous protease (D residues), which follows three to five hydrophobic acids, was found to be conserved in all hemiascomycetous retroelements and related LTR retrotransposons from other eukaryotes (Fig. 4B). The zinc finger, or HHCC domain, of the IN is highly conserved among all Ty1/copia elements, although the length of the “loop” between HH and CC varies. In contrast, Ty3/gypsy zinc fingers are less well conserved and the Tse3 zinc fingers appear to be degenerate (Fig. 4C).
Phylogenetic Relationships
The PR, IN, and RH domains were highly divergent and could not be used to deduce phylogenetic relationships between the various elements. Thus, we choose to base our phylogenetic analysis on the seven universally conserved domains of RT as described previously (Xiong and Eickbush 1990). The resulting phylogenetic tree (Fig.5) comprises the hemiascomycetous retroelements and retrotransposons belonging to other eukaryotic genomes (plants: Ta1–3 and Tnt; Drosophila: copia, gypsy,1731; fungi: CfT-1 and MAGGY; and yeasts other than hemiascomycetes: Tf1 from S. pombe, RF3, RF5, Tcn1, Tcn2, Tcn3, Tcn4, Tcn5, Tcn6, and Tcn9 from the basidiomycetous yeast C. neoformans). The tree was rooted with L1Hs, a non-LTR retrotransposon from Homo sapiens.
Phylogenetic tree showing the relationships between hemiascomycetous retrotransposons and other LTR-retrotransposons. This tree is based on RT protein sequence alignments. For bootstrap analyses, we constructed this tree by the neighbor-joining method using ClustalX. Bootstrap values based on 1000 replications are shown next to the nodes. Only bootstrap values for branches receiving >50% support are shown. Newly identified retroelements from hemiascomycetous yeasts are shown in bold. Host species are indicated within brackets. The LINE element L1Hs from Homo sapiens was used as an outgroup.
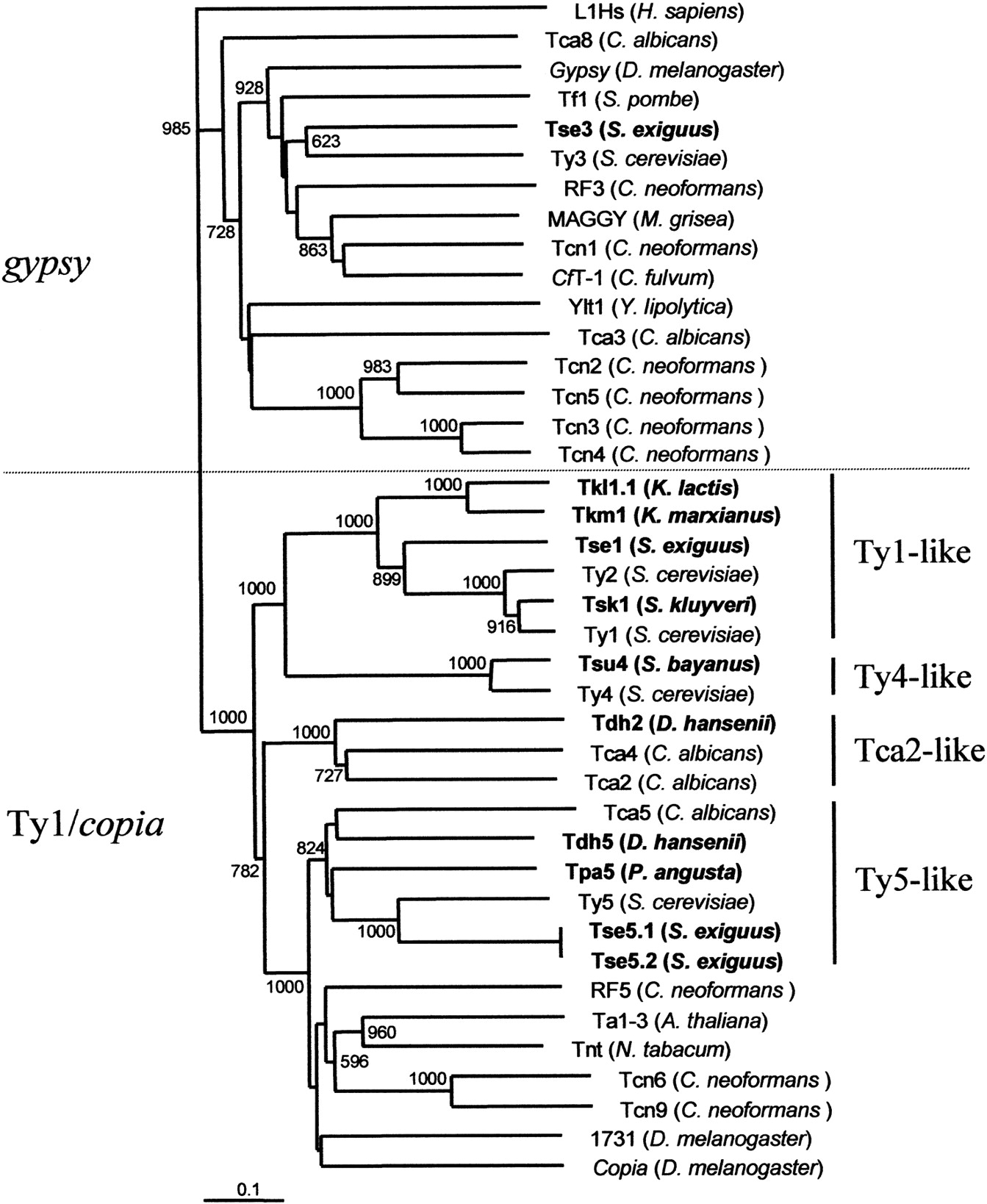
The Ty1/copia part of the tree is clearly divided into five clades corresponding to the Ty1-like, Ty4-like, Ty5-like, Tca2-like, and other Ty1/copia elements from nonhemiascomycetes. The four clades of hemiascomycetous elements support the clustering, which was established on the basis of LTRs, ORFs, frameshifting capacities, and PBS.
All known gypsy elements and Tse3 are located on a branch that is clearly distinct from the other branch, which contains all of the Ty1/copia elements (Fig. 5). Despite its original features, Tse3 is grouped with Ty3 and is phylogenetically distant from the retrotransposons of other yeasts such as Tca3 or Tca8 from C. albicans or Tcn2–5 from C. neoformans.
DISCUSSION
We used the Génolevures (2000) sequence data to identify 17 different families of LTR retrotransposons and five families of solo LTRs in 13 hemiascomycetous yeasts. We did not find any new types of LTR retrotransposon, although we identified a Tca2-like element (Tdh2) by using S. cerevisiae Ty proteins as a bait. Unlike S. cerevisiae (Goffeau et al. 1996), C. albicans (Stanford DNA sequencing and Technology Center,http://www-sequence.stanford.edu/group/candida), and C. neoformans (Stanford DNA sequencing and Technology Center,http://www-sequence.stanford.edu/group/C.neoformans/index.html), the complete genome sequences of these 13 strains are not yet available. Therefore, our genome survey remains nonexhaustive and other LTR retrotransposons or particular remnants will probably be identified when complete genomic sequences become available. Nonetheless, we managed to obtain a reliable picture of transposable elements within the hemiascomycetes class.
Host Response toward LTR Retrotransposons
A first interesting conclusion that emerges from this work is that the studied yeast species all contain less LTR retrotransposon families than S. cerevisiae. Some species, such as P. farinosa, appear to be devoid of LTR retrotransposons or to possess only remnants of solo LTRs like K. thermotolerans. Several of the other yeast species harbor a limited number of families of nonfunctional or degenerate elements. This is particularly true for species that only contain Ty3/gypsy elements: Z. rouxiiand S. servazzii. S. exiguus and, to a lesser extent,D. hansenii contain a similar pattern of LTR retrotransposons to S. cerevisiae: S. exiguus harbors at least three different families of retroelements (Ty1-like, Ty3/gypsy, and Ty5-like), all of which seem to be intact and potentially active. Surprisingly, the differences were greatest between C. albicans and its most closely related species, C. tropicalis. Only one family was detected in C. tropicalis, whereas C. albicanscarries 34 distinct families of LTR retrotransposons, as well as non-LTR retrotransposons and class II transposable elements (Goodwin and Poulter 2000; Goodwin et al. 2001).
This indicates that the host response to transposable elements is species-dependent rather than being linked to their phylogenetic position or to their ability to reproduce sexually. Clearly, genomes interact in various manners with LTR retrotransposons. Some species such as S. cerevisiae, S. exiguus, S. kluyveri, K. marxianus,and D. hansenii tend to conserve intact and probably active transposons, but this propensity is clearly not congruent with their phylogenetic relationship. Other species, such as K. lactis, S. bayanus, or S. servazzii tend to reduce the copy number and, moreover, to inactivate particular (or all) copies until only remnants of solo LTRs remain. Considering the paucity of elements inS. bayanus compared with S. cerevisiae, it seems that several transposons may have been lost simultaneously after recent speciation events of the host.
There seem to be two mechanisms for retrotransposons removal in hemiascomycetous genomes. The first mechanism involves the gradual erosion of LTR retrotransposons as a result of accumulation of point mutations or minor deletions, as observed in Z. rouxii andK. lactis. In these cases, the copy number per genome is low and the retrotransposons are getting lost. There is no evidence that this mechanism corresponds to repeat-induced point mutations (RIP) or methylation-induced premeiotically (MIP). These mechanisms are known to occur in fungi, in Ascobolus (Goyon and Faugeron 1989) andNeurospora (Cambareri et al. 1989), in which they silence or inactivate retrotransposons by generating an accumulation of point mutations as a result of G-C to A-T transitions.
The second mechanism, the most documented in S. cerevisiae(Jordan and McDonald 1999a), delete elements through LTR–LTR recombination, leaving only LTR remnants. In these cases, the copy number is sometimes high probably because of a dynamic equilibrium between the frequency of the transposition events, which tend to increase the number of elements, and the host's regulatory mechanisms, which tend to decrease the number of high copy number elements.Nakayashiki et al. (2001) recently introduced MAGGY into a naive genome of Magnaporthe grisea and showed that after equilibrium was reached at 20–30 copies, MAGGY was repressed by a mechanism that is not directly dependent on methylation and that appeared rapidly after the genome invasion. The retrotransposons copy number in the hemiascomycetous yeasts studied does not seem to exceed 20, whereas there are 32 in the sequenced S. cerevisiae strain. Several other methylation-independent mechanisms that repress the transposable elements have been described in Drosophila, but it is not known whether such mechanisms of transposon repression and inactivation exist in yeasts.
A New Subdivision of Ty1/copia Elements in Hemiascomycetes
We used the sequence data for full-length elements accumulated during this study to propose a phylogeny of the hemiascomycetous LTR retrotransposons. This phylogeny is also supported by sequence similarities and conservation of structural features (LTR conservation, organization of the ORF2 domains, frameshifts, and PBS) among the Ty1/copia hemiascomycetous elements. Therefore, we propose a subdivision of the hemiascomycetous Ty1/copia elements into four clades. Each of these clades is defined by a typical element, namely the first one historically described, such as Ty1 (Ty2 belongs to the Ty1 clade), Ty4 and Ty5 from S. cerevisiae, and Tca2 from C. albicans. The clade names refer to their typical elements, that is, Ty1-like, Ty4-like, Ty5-like, and Tca2-like (Fig.5). These four clades are clearly distinct, each of them being monophyletic and only including retrotransposons from hemiascomycetous yeasts (see Fig. 5). In addition, the phylogenetic distances between the elements are consistent with the phylogeny of the host species on the basis of rDNA sequences, with the exception of Tsk1 from S. kluyveri (discussed below). In contrast, all other Ty1/copia LTR retrotransposons, including elements from plants, basidiomycetous yeasts, and insects, are grouped together in a separate clade.
The phylogeny of Ty3/gypsy elements in hemiascomycetous yeasts is much more complex than that of Ty1/copia elements. Ty3/gypsy elements in hemiascomycetes are widely spread over the phylogenetic tree of the gypsy group of retrotransposons (Fig. 5). Most of these Ty3/gypsy elements are degenerate, truncated, or remnants of solo LTRs. Contrary to the Ty1/copiaelements, there are no monophyletic clades containing hemiascomycetous elements alone. Conversely, we showed that host-species with parts of Ty3/gypsy transposons are present in all branches of the hemiascomycetous phylogenetic tree on the basis of rDNA sequence data. In some yeast species, several divergent Ty3/gypsy elements were found, such as Ylt1 and Tyl3 in Y. lipolytica or Tca3 and Tca8 in C. albicans. In addition, the amino acid sequences of the RT of elements from closely related yeast species, such as Ty3 and Tse3 (45.4% identity and 53.6% similarity on 209 aa), differed enormously. These findings indicate that the acquisition of Ty3/gypsy elements by hemiascomycetes took place long enough ago to allow such a divergence and evolutionary events. This means that the modern hemiascomycetous Ty3/gypsy members can probably be further subdivided into different clades, like the hemiascomycetous Ty1/copia elements, although we need more sequence data on these elements.
Retrotransposon Evolution: Sequence Evolution or Horizontal Transfer?
Previous evolutionary studies on retrotransposons (Malik and Eickbush 2001) show that LTR retrotransposons were present in early eukaryotes, and a fortiori in the yeast ancestor. Ty1/copiaand Ty3/gypsy elements are derived from an ancient and diverse group that radiated into modern yeast species, mostly via vertical inheritance. However, the ancestral LTR retrotransposons have undergone evolutionary events. Some of these events (exceptional events) provided some retrotransposons a selective advantage over less active elements, leading to better proliferation and rapid invasion of the host. For example, the acquisition of a PBS complementary to a tRNAAsnmight give Ty4 in S. cerevisiae an advantage because it is not in competition with Ty1 and Ty2 for tRNAiMet molecules. Similarly, the acquisition of a frameshift mechanism is probably an advantage during expression.
Given the conservation of structural features, the congruent evolution between hosts and elements within a clade, and the phylogenetic distribution of retrotransposon types, we propose that the actual members of the four Ty1/copia clades of hemiacomycetes diverged from a common ancestor after three major exceptional events during the evolution of LTR retrotransposons (Fig.6).
Evolution of LTR-retrotransposons and phylogeny of hemiascomycetous yeasts based on 26S rDNA sequences. This cladogram was constructed by the neighbor-joining method using ClustalX and visualized with TreeView. Bootstrap values on 1000 replications are shown next to the nodes. Arrows indicate on the host phylogeny the evolutionary period corresponding to the exceptional event leading to the apparition of a new type of LTR-retrotransposon. A schematic representation of the evolution of the structural features of the LTR-retrotransposons is shown below the name of the retrotransposon types. LTRs are represented by boxes containing a black triangle, ORF1 by white boxes and ORF2 by gray boxes. The tRNA genes complementary to PBS are indicated above ORF1. The star in Tca2-like elements represents the stop codon separating ORF1 and ORF2.
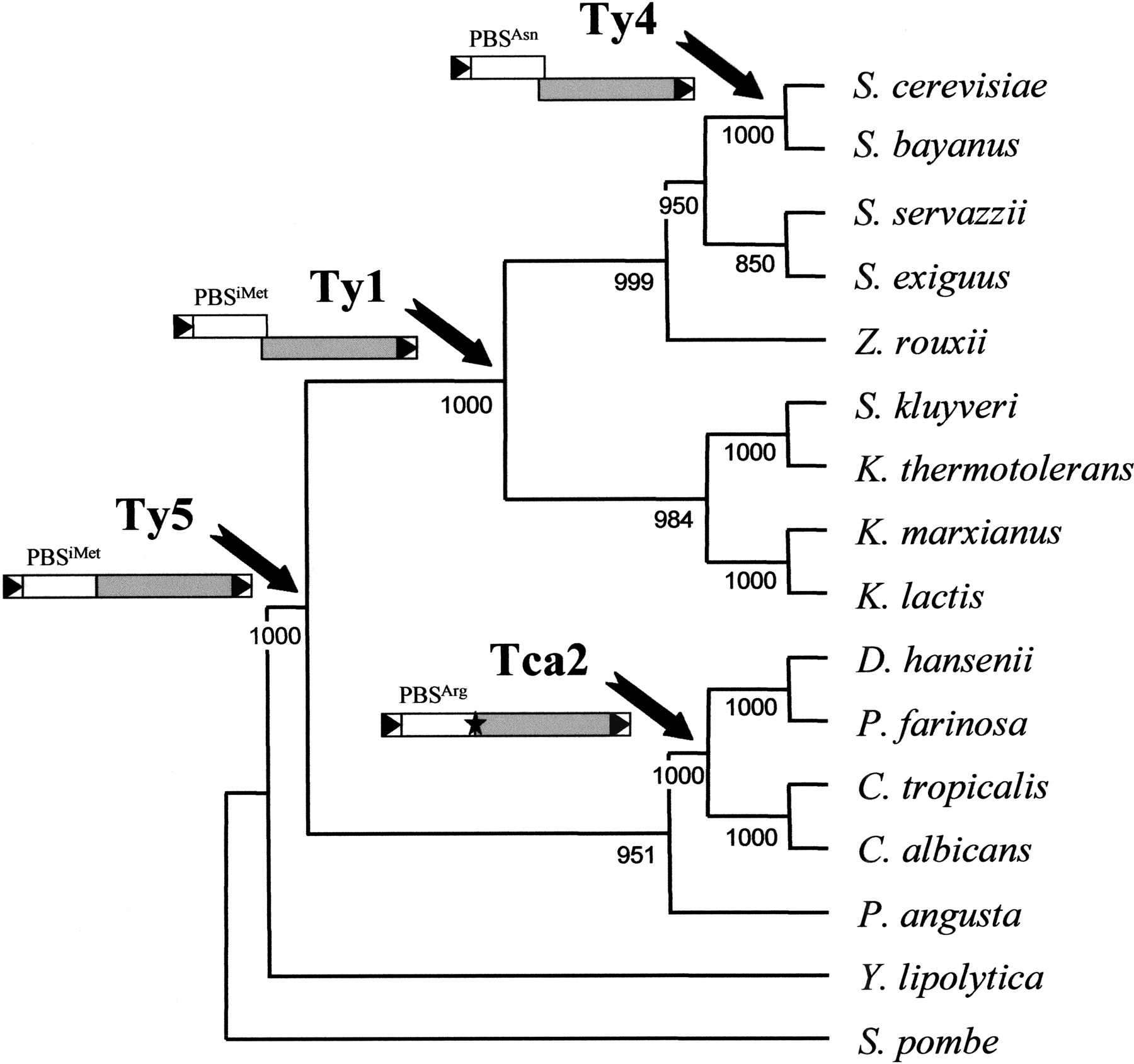
Ty5-like elements are probably the most ancient group of Ty1/copiaelements in hemiascomycetous yeasts. Although their genetic structure has been conserved, with all members encoding a single ORF corresponding to a gag-pol gene fusion, they present a low level of sequence conservation (up to 47% aa identity inpol). Thus, these elements might be an old component of hemiascomycetous yeasts that probably arose after the speciation ofY. lipolytica (Fig. 6). This would also explain why most hemiascomycetous species (9 of 14 in Fig. 6) have lost this type of element. The presence of a single ORF, compared with two in the other Ty1/copia elements, is reminiscent of the scenario for the evolution of non-LTR retrotransposons, in which the most ancient element has only one ORF, and the most recent types have two (Malik et al. 1999).
We propose that the first exceptional event that arose during Ty1/copia evolution in hemiascomycetous yeasts was the acquisition of different strategies for regulating the stoichiometry ofgag and pol. Thus, the acquisition of a stop-codon separating ORF1 and ORF2 took place in the lineage leading to the Tca2 clade. Because only C. albicans and D. hanseniipossess modern members of this retrotransposon type, we suggest that this exceptional event arose after the speciation of P. angusta, just before the differentiation of D. hanseniiand P. farinosa on the one hand and C. tropicalis andC. albicans on the other hand (Fig. 6). Another strategy consisting of a +1 frameshift mechanism was acquired by the Ty1-like ancestor and has been retained during evolution by Ty4-like elements (Fig. 6). Because the six families of full-length Ty1-like elements (Ty1, Ty2, Tse1, Tsk1, Tkl1, and Tkm1) that form this very homogeneous group with highly conserved sequences (up to 79.9% aa identity inpol) and a phylogeny congruent with the phylogeny of the host species are all contained into species of the Saccharomycesand Kluyveromyces genera, we propose that the event leading to the appearance of the Ty1 ancestor arose before the generaSaccharomyces and Kluyveromyces differentiated (Fig.6). Similarly, we propose that the acquisition of a different PBS gave the Ty4-like ancestor a selective advantage. Given that Ty4-like elements (Ty4 and Tsu4) share highly conserved sequences (68% identity on the 1461 aa of pol) and are only present in S. cerevisiae and S. bayanus, we suggest that Ty4-like elements are relatively recent and that they arose before the speciation of the sensu stricto group (Fig. 6).
LTR retrotransposons are constantly evolving, so new types will probably arise. Jordan and McDonald (1999b) showed that there is a high level of genomic turnover of Ty elements in S. cerevisiae. However, we do not know how the new types appear. We suggest several different possibilities. The first entails the accumulation of point mutations that allowed the new element to escape the regulatory repression of the host. As no major evolutionary events differentiate Ty1 from Ty2, this is a prominent example of elements separated from each other by nucleotide divergence only. This implies that Ty1 and Ty2 evolved independently as a result of functional constraints, either in the same genome or in two different genomes that crossed after differentiation of the retrotransposons by allotetraploidization (Seoighe and Wolfe 1999) or by a cross between two local races that mixed recently. We do not have sufficient evidence to differentiate between these different hypotheses concerning the origin of the modernS. cerevisiae genome.
The second possibility is that internal elements recombined with the host's DNA, which led to the acquisition of new PBS, for example (contributing to coevolution between host and transposon), or by interelement recombination as reported by Jordan and McDonald (1998). The third alternative is that a partial or complete retrotransposon or an invading retrovirus recombined with an endogenous LTR retrotransposon via horizontal transfer. Unlike in insect retrotransposons (Jordan et al. 1999), there is no evidence that horizontal transfer is involved in the acquisition of LTR retrotransposons in yeasts. The only example is Tsk1 from S. kluyveri, which is closer to Ty1 than is Ty2 (79.9% identity for 1322 aa in pol between Ty1 and Tkl1 vs. 75.6% for 1351 aa between Ty1 and Ty2); whereas host genomes are more distantly related. However, this example needs further documentation.
CONCLUSIONS
We used an exceptionally large set of data on hemiascomycetous yeasts to analyze the LTR retrotransposons identified in these species in detail. Our results help to elucidate the evolution of these elements and, in particular, provide an insight into the genomic evolution of yeasts. We have developed a model for the evolution of elements from the Ty1/copia group. However, it was not as easy to establish a model for the evolution of the Ty3/gypsy group because there is little sequence information on this type of element. The large-scale genome sequencing project that is underway (P.F. Cliften, pers. comm.; GDR Génolevures) should confirm the evolutionary model proposed for Ty1/copia elements and provide accurate information that can be extended to Ty3/gypsyelements.
METHODS
Strains
The strains used in this study were the same as those used in the Génolevures project (Souciet et al. 2000): S. bayanus syn.S. uvarum (CLIB533), S. exiguus (CBS379), S. servazzii (CBS4311), Z. rouxii (CBS732), S. kluyveri(CBS3082), K. thermotolerans (CBS6340), K. lactis(CLIB210), K. marxianus var. marxianus (CBS712), P. angusta (CBS4732), D. hansenii var. hansenii (CBS767),P. farinosa (CBS7064), C. tropicalis (CBS94), andY. lipolytica (CLIB89).
Accession Numbers
The EMBL and GenBank accession numbers of the sequences of the elements used in this study and of the new transposable elements are listed in Table 4.
Retrotransposons Studied
| Organism | Element | Accession |
| Mammalians | ||
| Homo sapiens | L1Hs(LINE) | M22333 |
| Plants | ||
| Arabidopsis thaliana | Ta1-3 | X13291 |
| Nicotiana tabacum | Tnt1 | X13777 |
| Insects | ||
| Drosophila melanogaster | gypsy | AF033821 |
| Drosophila melanogaster | copia | P04146 |
| Drosophila melanogaster | 1731 | X07656 |
| Fungi | ||
| Cladosporium fulvum | CfT-1 | Z11866 |
| Magnaporthe grisea | MAGGY | L35053 |
| Hemiascomycetous yeasts | ||
| Candida albicans | Tca2 | AF05215 |
| Candida albicans | Tca3 | AF061575 |
| Candida albicans | Tca4 | AF078809 |
| Candida albicans | Tca5 | AF65434 |
| Candida albicans | Tca8 | AF142436 |
| Candida tropicalis | Tct3[ii] | AL440929, AL438891, AL441434 |
| Debaryomyces hansenii | Tdh2 | AJ439551 |
| Debaryomyces hansenii | Tdh3[i] [ii] | AL436369 |
| Debaryomyces hansenii | Tdh5 | AJ439552 |
| Kluyveromyces lactis | Tkl1.1[i] | AJ439548 |
| Kluyveromyces lactis | Tkl1.2[i] [ii] | AJ439549 |
| Kluyveromyces marxianus | Tkm1 | AJ439546 |
| Kluyveromyces thermololerans | LTRkt1[iii] | AJ439556 |
| Kluyveromyces thermotolerans | LTRkt2[iii] | AJ439557 |
| Pichia angusta | Tpa5 | AJ439553 |
| Pichia angusta | LTRpa1[iii] | AJ439558 |
| Saccharomyces bayanus | Tsu4 | AJ439550 |
| Saccharomyces bayanus | LTRsu1[iii] | AJ316069 |
| Saccharomyces exiguus | Tse1 | AJ439547 |
| Saccharomyces exiguus | Tse3 | AJ439555 |
| Saccharomyces exiguus | Tse5.1[i] | AJ439554 |
| Saccharomyces exiguus | Tse5.2[i] [ii] | AJ439560 |
| Saccharomyces kluyveri | Tsk1 | AF492702 |
| Saccharomyces kluyveri | Tsk3[i] [ii] | AL406386 |
| Saccharomyces paradoxus | Ty5 | U19263 |
| Saccharomyces servazzii | Tss1[i] [ii] | AL404444 |
| Saccharomyces servazzii | Tss3[i] [ii] | AL404175, AL403291, AL402542 |
| Yarrowia lipolytica | Ylt1 | AJ310725 |
| Yarrowia lipolytica | Tyl3[i] [ii] | AL414488, AL414575 |
| Yarrowia lipolytica | LTRyl1[iii] | AJ439559 |
| Zygosaccharomyces rouxii | Tzr3[i] [ii] | AL392764, AL395629, AL394114, AL392218, AL394501 |
| Other yeasts than Hemiascomycetes | ||
| Cryptococcus neoformans | Tcn1 | Retrobase |
| Cryptococcus neoformans | Tcn2 | Retrobase |
| Cryptococcus neoformans | Tcn3 | Retrobase |
| Cryptococcus neoformans | Tcn4 | Retrobase |
| Cryptococcus neoformans | Tcn5 | Retrobase |
| Cryptococcus neoformans | Tcn6 | Retrobase |
| Cryptococcus neoformans | Tcn9 | Retrobase |
| Cryptococcus neoformans | RF3 | Retrobase |
| Cryptococcus neoformans | RF5 | Retrobase |
| Schizosaccharomyces pombe | Tf1 | A36373 |
[i] Defective element.
[ii] Partial sequence.
[iii] LTR.
Sequence Screening and Element Assembly
Sequence data for the 13 hemiascomycetous yeasts are available on the Génolevures website athttp://cbi.labri.u-bordeaux.fr/Genolevures/Genolevures.php3. In addition to the Ty5 sequence of S. paradoxus, we used a database of S. cerevisiae Ty elements (Horst Feldmann, unpubl.). We compared the amino acid sequences of the RSTs from the 13 yeast species with those of the Ty elements with theBLASTX version 2.0.8 program with the blosum62 substitution matrix. We used the Staden programs of the Madison Institute Genetics Computer Group (GCG) sequence analysis package (Devereux et al. 1984) to assemble the sequences. We assembled the complete elements by sequencing the clones containing the RST of interest and, if part of the element was missing from the plasmid libraries, by PCR amplification of genomic DNA fragments. Sequencing data were obtained from overlapping reads on both strands.
Multiple Sequence Alignment and Phylogenetic Analysis
Pairwise comparisons were performed using the FASTA orBestfit programs from the GCG package. We aligned multiple amino acid sequences with the PILEUP program of the GCG package and ClustalW (Thompson et al. 1994). Alignments were adjusted manually with GeneDoc (K.B. Nicholas and H.B. Nicholas, http://www.psc.edu/biomed/genedoc/) so that our alignments were consistent with previously published multiple alignments of protease, IN, RT, and RNAseH domains (Xiong and Eickbush 1990; Springer and Britten 1993; Jordan and McDonald 1999b). The GCG program Distances was used to calculate distance matrixes with the Kimura correction method, and the GCG programGrowTree was used to construct and to view UPGMA trees. For bootstrap analyses, neighbor-joining trees were constructed usingClustalX (Thompson et al. 1997) and viewed withTreeView (Page 1996).
Southern Hybridization
Genomic DNA was prepared in Seakem GTG agarose (FMC, USA) plugs as described previously (Vezinhet et al. 1990). The DNA was digested and subjected to electrophoresis in a CHEF Mapper apparatus (Bio-Rad Laboratories) in 1% Pulsed Field Certified Agarose (Bio-Rad Laboratories) gels in 0.5× TBE buffer at 12°C. Genomic DNA was digested with EcoRI, BamHI, HindIII, orPstI and then was separated by field inversion gel electrophoresis (FIGE) for 20 hr, 39 min with forward and reverse voltages of 9 V/cm and 6 V/cm, respectively. Initial and final pulses were 0.11 sec and 0.67 sec, respectively. DNA was transferred onto GeneScreen nylon membranes (Dupont de Nemours NEN, USA) as described previously (Zimmermann and Fournier 1996). DNA/DNA hybridizations were performed as described previously (Sambrook et al 1989) with DNA probes labeled with (α−32P) dCTP using the Megaprime labeling kit (Amersham Life Science, UK). Probes were obtained by PCR amplification under the following conditions: 4 min at 94°C, 30 cycles of 30 sec at 94°C, followed by 30 sec at the Tm of the primers, 1 min per kb at 72°C with 2.5 units of Taq DNA polymerase (Appligene Oncor, France). Long-range PCR amplifications of genomic DNA were run in a Perkin-Elmer 2400 thermocycler using the Expand high-fidelity PCR system (Boehringer Mannheim, Germany).
WEB SITE REFERENCES
http://bioc111.otago.ac.nz:591/retrobase/home.htm; Retrobase site.
http://cbi.labri.u-bordeaux.fr/Genolevures/Genolevures.php3; Génolevures project.
http://www.inra.fr/clib/english/genolevu.htm; Collection of yeasts of biotechnological interest.
http://www.psc.edu/biomed/genedoc/; GeneDoc.
http://www-sequence.stanford.edu/group/candida; Sequencing ofCandida albicans at the Stanford Genome Technology Center.
http://www-sequence.stanford.edu/group/C.neoformans/index.html; Stanford Genome Technology Center, Cryptococcus neoformansgenome project.
E.B. was supported by the EEC scientific research grant QLRI-1999-01333. This work was supported by INRA, CNRS, the GDR/CNRS 2354 “GénolevuresII,” and by two BRG grants (Ressources Génétiques des Microorganismes n°11–0926–99 and Gestion des Collections de Levures de Fromage).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Notes
[19] Corresponding author.
Notes
[20] E-MAIL [email protected]; FAX 33-1-30-81-54-57.
[21] Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.219202.
REFERENCES
- ↵G. BlandinB. LlorenteA. MalpertuyP. WinckerF. ArtiguenaveB. Dujon(2000a) Genomic exploration of the hemiascomycetous yeasts: 13. Pichia angusta. FEBS Lett. 487:76–81.
- ↵G. BlandinO. Ozier-KalogeropoulosP. WinckerF. ArtiguenaveB. Dujon(2000b) Genomic exploration of the hemiascomycetous yeasts: 16. Candida tropicalis. FEBS Lett. 487:91–94.
- ↵J.D. Boeke(1989) Transposable elements in Saccharomyces cerevisiae. in Mobile DNA, eds D.E. BergM.M. Howe(American Society for Microbiology, Washington, DC.) pp 335–374.
- ↵M. Bolotin-FukuharaC. Toffano-NiocheF. ArtiguenaveG. Duchateau-NguyenM. LemaireR. MarmeisseR. MontrocherC. RobertM. TermierP. WinckerM. Wesolowski-Louvel(2000) Genomic exploration of the hemiascomycetous yeasts: 11. Kluyveromyces lactis. FEBS Lett. 487:66–70.
- ↵E. BonC. NeuvégliseS. CasaregolaF. ArtiguenaveP. WinckerM. AigleP. Durrens(2000a) Genomic exploration of the hemiascomycetous yeasts: 5. Saccharomyces bayanus var. uvarum. FEBS Lett. 487:37–41.
- ↵E. BonC. NeuvégliseA. LépingleP. WinckerF. ArtiguenaveC. GaillardinS. Casaregola(2000b) Genomic exploration of the hemiascomycetous yeasts: 6. Saccharomyces exiguus. FEBS Lett. 487:42–46.
- ↵E.B. CambareriB.C. JensenE. SchabtachE.U. Selker(1989) Repeat-induced G-C to A-T mutations in Neurospora. Science 244:1571–1575.
- ↵S. CasaregolaC. NeuvégliseA. LépingleE. BonC. FeynerolF. ArtiguenaveP. WinckerC. Gaillardin(2000) Genomic exploration of the hemiascomycetous yeasts: 17. Yarrowia lipolytica. FEBS Lett. 487:95–100.
- ↵D.L. ChalkerS.B. Sandmeyer(1992) Ty3 integrates within the region of RNA polymerase III transcription initiation. Genes Dev. 6:117–128.
- ↵J. ClareP. Farabaugh(1985) Nucleotide sequence of a yeast Ty element: Evidence for an unusual mechanism of gene expression. Proc. Natl. Acad. Sci. 82:2829–2833.
- ↵J. de MontignyC. SpehnerJ. SoucietF. TekaiaB. DujonP. WinckerF. ArtiguenaveS. Potier(2000) Genomic exploration of the hemiascomycetous yeasts: 15. Pichia sorbitophila. FEBS Lett. 487:87–90.
- ↵J. DevereuxP. HaeberliO. Smithies(1984) A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 12:387–395.
- ↵A. EigelH. Feldmann(1982) Ty1 and delta elements occur adjacent to several tRNA genes in yeast. EMBO J. 1:1245–1250.
- ↵G. FischerC. NeuvégliseP. DurrensC. GaillardinB. Dujon(2001) Evolution of gene order in the genomes of two related yeast species. Genome Res. 11:2009–2019.
- ↵Génolevures(2000) Genomic exploration of the hemiascomycetous yeasts. FEBS Lett. 487:3–149.
- ↵A. GoffeauB.G. BarrellH. BusseyR.W. DavisB. DujonH. FeldmannF. GalibertJ.D. HoheiselC. JacqM. Johnston(1996) Life with 6000 genes. Science 274:546, , 563–567..
- ↵T.J. GoodwinR.T. Poulter(2000) Multiple LTR-retrotransposon families in the asexual yeast Candida albicans. Genome Res. 10:174–191.
- ↵(2001) The diversity of retrotransposons in the yeast Cryptococcus neoformans. Yeast 18:865–880, ibid.
- ↵T.J. GoodwinJ.E. OrmandyR.T. Poulter(2001) L1-like non-LTR retrotransposons in the yeast Candida albicans. Curr. Genet. 39:83–91.
- ↵C. GoyonG. Faugeron(1989) Targeted transformation of Ascobolus immersus and de novo methylation of the resulting duplicated DNA sequences. Mol. Cell. Biol. 9:2818–2827.
- ↵I.K. JordanJ.F. McDonald(1998) Evidence for the role of recombination in the regulatory evolution of Saccharomyces cerevisiae Ty elements. J. Mol. Evol. 47:14–20.
- ↵(1999a) The role of interelement selection in Saccharomyces cerevisiae Ty element evolution. J. Mol. Evol. 49:352–357, ibid.
- ↵(1999b) Tempo and mode of Ty element evolution in Saccharomyces cerevisiae. Genetics 151:1341–1351, ibid.
- ↵I.K. JordanL.V. MatyuninaJ.F. McDonald(1999) Evidence for the recent horizontal transfer of long terminal repeat retrotransposon. Proc. Natl. Acad. Sci. 96:12621–12625.
- ↵J.M. KimS. VanguriJ.D. BoekeA. GabrielD.F. Voytas(1998) Transposable elements and genome organization: A comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 8:464–478.
- ↵C.P. KurtzmanC.J. Robnett(1998) Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie van Leeuwenhoek 73:331–371.
- ↵A. LépingleS. CasaregolaC. NeuvégliseE. BonH. NguyenF. ArtiguenaveP. WinckerC. Gaillardin(2000) Genomic exploration of the hemiascomycetous yeasts: 14. Debaryomyces hansenii var. hansenii. FEBS Lett. 487:82–86.
- ↵B. LlorenteP. DurrensA. MalpertuyM. AigleF. ArtiguenaveG. BlandinM. Bolotin-FukuharaE. BonP. BrottierS. Casaregola(2000a) Genomic exploration of the hemiascomycetous yeasts: 20. Evolution of gene redundancy compared to Saccharomyces cerevisiae. FEBS Lett. 487:122–133.
- ↵B. LlorenteA. MalpertuyG. BlandinF. ArtiguenaveP. WinckerB. Dujon(2000b) Genomic exploration of the hemiascomycetous yeasts: 12. Kluyveromyces marxianus var. marxianus. FEBS Lett. 487:71–75.
- ↵H.S. MalikT.H. Eickbush(2001) Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res. 11:1187–1197.
- ↵H.S. MalikW.D. BurkeT.H. Eickbush(1999) The age and evolution of non-LTR retrotransposable elements. Mol. Biol. Evol. 16:793–805.
- ↵G.D. MatthewsT.J. GoodwinM.I. ButlerT.A. BerrymanR.T. Poulter(1997) pCal, a highly unusual Ty1/copia retrotransposon from the pathogenic yeast Candida albicans. J. Bacteriol. 179:7118–7128.
- ↵H. NakayashikiK. IkedaY. HashimotoY. TosaS. Mayama(2001) Methylation is not the main force repressing the retrotransposon MAGGY in Magnaporthe grisea. Nucleic Acids Res. 29:1278–1284.
- ↵C. NeuvégliseE. BonA. LépingleP. WinckerF. ArtiguenaveC. GaillardinS. Casaregola(2000) Genomic exploration of the hemiascomycetous yeasts: 9. Saccharomyces kluyveri. FEBS Lett. 487:56–60.
- ↵R.D.M. Page(1996) TREEVIEW: An application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12:357–358.
- ↵E.P. PlantT.J. GoodwinR.T. Poulter(2000) Tca5, a Ty5-like retrotransposon from Candida albicans. Yeast 16:1509–1518.
- ↵J. SambrookE.F. FritschT. Maniatis(1989) Molecular cloning: A laboratory manual (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY), 2nd ed..
- ↵N. Schmid-BergerB. SchmidG. Barth(1994) Ylt1, a highly repetitive retrotransposon in the genome of the dimorphic fungus Yarrowia lipolytica. J. Bacteriol. 176:2477–2482.
- ↵C. SeoigheK.H. Wolfe(1999) Yeast genome evolution in the post-genome era. Curr. Opin. Microbiol. 2:548–554.
- ↵J. SoucietM. AigleF. ArtiguenaveG. BlandinM. Bolotin-FukuharaE. BonP. BrottierS. CasaregolaJ. de MontignyB. Dujon(2000) Genomic exploration of the hemiascomycetous yeasts: 1. A set of yeast species for molecular evolution studies (1). FEBS Lett. 487:3–12.
- ↵M.S. SpringerR.J. Britten(1993) Phylogenetic relationships of reverse transcriptase and RNase H sequences and aspects of genome structure in the gypsy group of retrotransposons. Mol. Biol. Evol. 10:1370–1379.
- ↵R. StuckaC. SchwarzloseH. LochmullerU. HackerH. Feldmann(1992) Molecular analysis of the yeast Ty4 element: Homology with Ty1, copia, and plant retrotransposons. Gene 122:119–128.
- ↵J.D. ThompsonD.G. HigginsT.J. Gibson(1994) CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680.
- ↵J.D. ThompsonT.J. GibsonF. PlewniakF. JeanmouginD.G. Higgins(1997) The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882.
- ↵F. VezinhetB. BlondinJ.-N. Hallet(1990) Chromosomal DNA patterns and mitochondrial DNA polymorphism as tools for identification of enological strains of Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 32:568–571.
- ↵J. VolffC. KortingM. Schartl(2001) Ty3/gypsy retrotransposon fossils in mammalian genomes: Did they evolve into new cellular functions? Mol. Biol. Evol. 18:266–270.
- ↵D.F. VoytasJ.D. Boeke(1992) Yeast retrotransposon revealed [letter]. Nature 358:717.
- ↵(1993) Yeast retrotransposons and tRNAs. Trends Genet. 9:421–427, ibid.
- ↵Y. XiongT.H. Eickbush(1990) Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 9:3353–3362.
- ↵Y. YoshinakaI. KatohT.D. CopelandS. Oroszlan(1985) Murine leukemia virus protease is encoded by the gag-pol gene and is synthesized through suppression of an amber termination codon. Proc. Natl. Acad. Sci. 82:1618–1622.
- ↵M. ZimmermannP. Fournier(1996) Electrophoretic karyotyping of yeasts. in Nonconventional yeasts in biotechnology, ed K. Wolf(Springer-Verlag, Berlin, Heidelberg, New York), pp 101–116.
- ↵M.E. Zolan(1995) Chromosome-length polymorphism in fungi. Microbiol. Rev. 59:686–698.
- ↵S. ZouN. KeJ.M. KimD.F. Voytas(1996) The Saccharomyces retrotransposon Ty5 integrates preferentially into regions of silent chromatin at the telomeres and mating loci. Genes & Dev. 10:634–645.