Abstract
All animals live in tight association with complex microbial communities, yet studying the effects of individual bacteria remains challenging. Bacterial feeding nematodes are powerful systems to study host-microbe interactions as worms can be grown on monoxenic cultures. Here, we present three different types of resources that may assist future research of cross-species interactions in the nematode Pristionchus pacificus and also in other organisms. First, by sequencing the genomes of 84 Pristionchus-associated bacteria, we establish a genomic basis to study host-microbe interactions, and we demonstrate its utility in identifying candidate pathways in the bacteria affecting chemotaxis behavior and survival in the nematodes. Second, we generate nematode transcriptomes of P. pacificus nematodes on 38 bacterial diets and characterize 60 coexpression modules with differential responses to environmental microbiota. Third, we link the microbial genome and host transcriptome data by predicting a global map of more than 2800 metabolic interactions. These interactions represent statistical associations between variation in bacterial metabolic potential and differential transcriptomic responses of coexpression modules in the nematode. Analysis of the interactome identifies several intestinal modules as the primary response layer to diverse microbiota and reveals a number of broadly conserved metabolic interactions. In summary, our study establishes a multiomic framework for future mechanistic studies in P. pacificus, and may also be conceptually transferred and reimplemented in other organisms in order to investigate the evolution of the host–microbe interactomes.
The human gut microbiome exemplifies the intricate associations between organisms and bacteria, highlighting the complexity of cross-kingdom interactions and their impact on host development and disease. These interactions are influenced by the heterogeneity of the gut microbiome, as well as variations in host diet and genetic background, making them challenging to study. In recent years, the nematodes Caenorhabditis elegans and Pristionchus pacificus have emerged as a powerful model for investigating host–microbiota interactions. These models allow for detailed examination of individual interactions through the use of monoxenic bacterial cultures, where worms are grown on single bacterial strains (Zhang et al. 2017; Akduman et al. 2020). Both nematodes engage with bacteria in multiple ways: they are bacterial feeders, typically consuming the Escherichia coli OP50 strain in laboratory settings, and the bacterial diet significantly influences its development, as exemplified by the vitamin B12 production by Comamonas that impacts C. elegans development (Watson et al. 2014). Moreover, both nematodes harbor complex microbiomes in their natural habitat, with certain bacteria enhancing the host's fitness under stress (Dirksen et al. 2016; Meyer et al. 2017; Slowinski et al. 2020; Lo et al. 2022). Whereas many of the previous studies focused on understanding individual interactions at a mechanistic level (Iatsenko et al. 2014; Watson et al. 2014; Akduman et al. 2020), we lack a broad overview of the global landscape of interactions between nematodes and bacteria. Recently, we have started to characterize transcriptomic profiles of P. pacificus nematodes in response to diverse microbiota and used them to test the hypothesis that the adaptation to novel microbiota might be facilitated by the evolution of novel genes (Athanasouli et al. 2023). This revealed evidence that novel genes might be preferentially integrated into specific environmentally responsive networks. However, we have very little understanding about what environmental signals these networks are sensing.
In this study, we characterize bacterial metabolic variation as well as variation in nematode gene networks that respond to these bacteria. Applying a systems biology approach, we compile the first cross-species interactome of P. pacificus by statistically linking bacterial genomic and nematode transcriptomic data.
Results
Sequencing of 84 bacteria establishes the genomic basis to study host–microbe interactions
In order to establish a phylogenomic framework that could be used to study differential effects of bacteria on P. pacificus nematodes, we sequenced 84 strains from a large bacterial collection isolated previously from Pristionchus-associated environments (Akduman et al. 2018). This collection mostly comprised Proteobacteria as well as some members of the phyla Firmicutes, Actinobacteria, and Bacteroidetes, which are among the most abundant bacteria found in Pristionchus nematodes and their beetle hosts (Meyer et al. 2017). Eighty-four bacterial strains were selected for whole-genome sequencing based on criteria such as fast growth, ease of DNA extractions, and low propensity to contamination. (Supplemental Table S1). The resulting genome assemblies were highly complete as indicated by a median single-copy BUSCO completeness of 99% with an interquartile range (IQR) of 98%–100%. The largest parts of the bacterial genomes could be assembled into contigs spanning more than 100 kb. Specifically, the N50 value, which indicates the minimum contig length among the largest contigs that account for at least half of the genome, was 324 kb (IQR = 107–498 kb) and median genome assembly size was 4.9 Mb (IQR = 4.4–5.6 Mb). Gene annotation yielded a median of 4562 gene models per assembly (IQR = 4184–5225) with a median single-copy BUSCO completeness level of 99% (IQR = 98%–100%). A complete overview of these different quality measures can be found in Supplemental Table S1. This set of bacterial genomes builds the basis for the current and future studies investigating host–microbe interactions in the Pristionchus system.
Pristionchus-associated microbiota harbor previously uncharacterized bacterial strains
The predicted proteomes of all 84 genomes were taken to reconstruct a bacterial phylogeny (see Methods; Fig. 1). The different lineages were mostly consistent with previous genus assignments based on 16S sequencing (Akduman et al. 2018). To more accurately classify the strains, we performed homology searches against the nonredundant version of NCBI and determined the nomenclature of our bacteria by assigning each strain to the bacterial genus with the majority of best hits (Supplemental Table S2). During this process, we observed a low median percentage of identity (<90%) for the strains Sphingobacterium L2, Pseudomonas L74, and Erwinia V71 to their respective best hits in NCBI. In addition, we were unable to definitively determine the genus for the closely related strains V69 and V91, despite a median identity with available Phytobacter genomes of 90.6% and 96%, respectively. We hypothesized that these bacterial strains could either represent novel isolates or have no associated whole-genome sequencing data in the NCBI database. Therefore, we searched the literature for recent, comprehensive phylogenies of these genera (Girard et al. 2021; Kakumanu et al. 2021; Brady et al. 2022; Smits et al. 2022) and recreated species phylogenies including our strains and their best hits in the NCBI database. The reconstructed phylogenies for the three bacterial strains with low median identity (L2, L74, V71) revealed that our strains are outgroups to the clusters containing their best hits in their respective phylogenetic trees but are still more closely related to them compared to any other genus (Supplemental Fig. S1). The reconstructed phylogeny including the two strains V69 and V91 exhibited the same patterns and indicated that both bacteria should be classified as Phytobacter strains (Supplemental Fig. S1). These findings support that Pristionchus-associated microbiota constitute a previously undersampled environment that harbors novel bacterial strains and species.
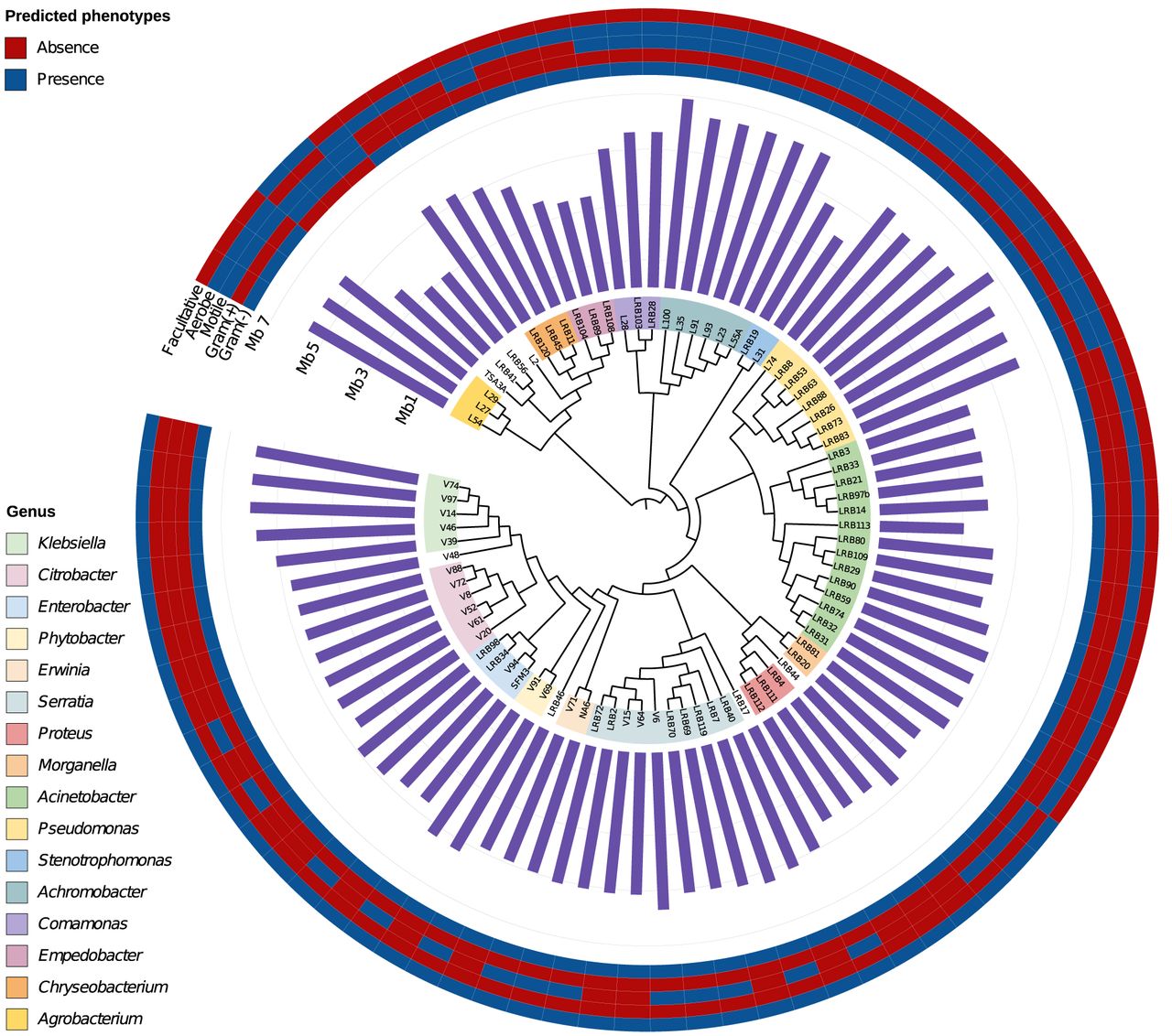
Phylogenomics of 84 bacterial genomes. The tree depicts the bacterial phylogeny acquired using RAxML after whole-genome sequencing, genome assembly, and orthogroup detection. Phenotype prediction with Traitar3 revealed that the overwhelming majority of bacterial strains sequenced are aerobic, with the exception of TSA3A and LRB41, which are facultative anaerobic. Our collection contains only three gram-positive strains, and the rest are gram-negative. Genome sizes may vary even within a genus, as is the case with Acinetobacter, where we observe genomes ranging between 3 and 5 Mb.
More than 1000 metabolic pathways vary across the bacterial collection
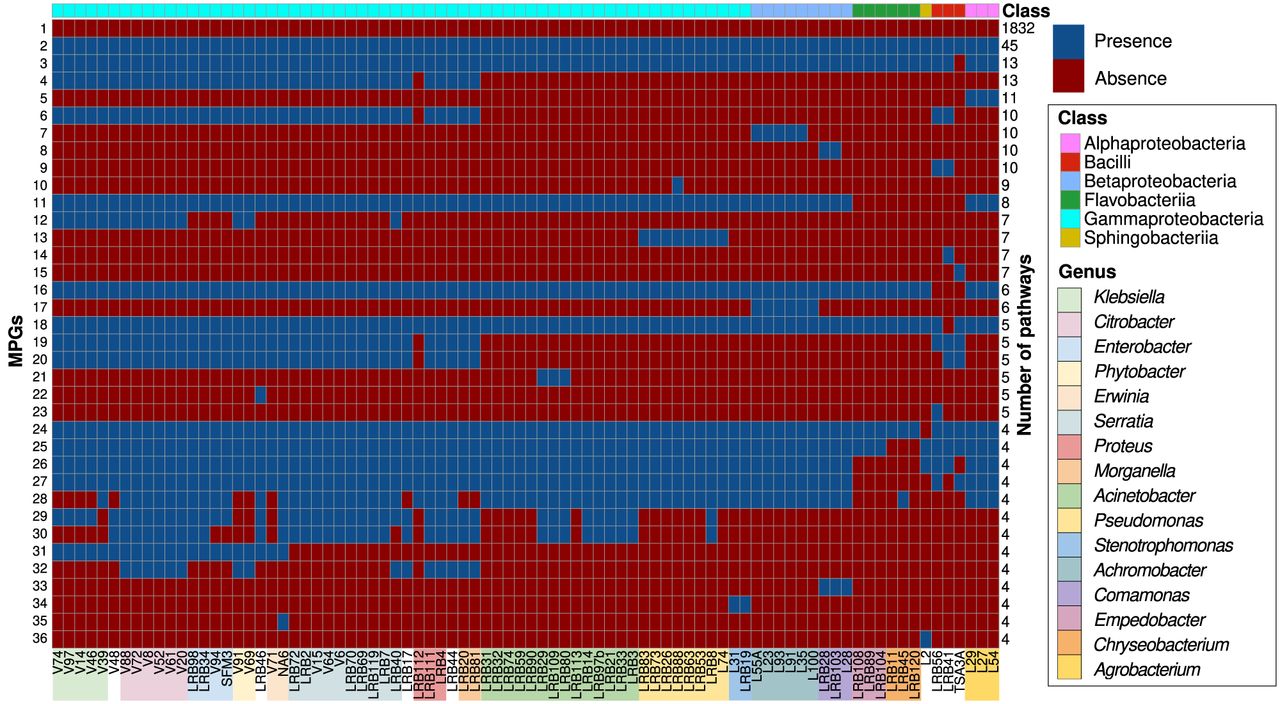
In order to characterize the variation of metabolic potential among members of the Pristionchus-associated microbiota, we reconstructed metabolic networks for the 84 bacterial genomes with the gapseq approach using existing pathway information from the MetaCyc database (Caspi et al. 2012; Zimmermann et al. 2021). MetaCyc was preferred to KEGG due to the higher detail it offers with regard to the available pathways. The gapseq pipeline considers a pathway to be present if at least 80% of the reaction or at least two thirds of the key reactions can be found (Zimmermann et al. 2021). This can lead to false negative calls in cases of incompletely annotated pathways. Similarly, relying on genomic data can lead to false positive calls in cases where a pathway is present but not active under the tested condition. However, both effects should be mitigated by the high number of bacterial genomes in our collection. We then investigated the absence/presence patterns of metabolic pathways among the 84 bacteria and defined metabolic pathways groups (MPGs) based on identical patterns (Fig. 2). This reduced the number of total metabolic pathways from 2902 (Supplemental Table S3) to 715 MPGs. The two most abundant MPGs denote 1832 and 45 individual pathways that are either absent or present in all strains (Fig. 2), respectively. Whereas most pathways that were not detected in our data correspond to pathways that are known only from eukaryotes, examples of the core metabolic pathways that were annotated in all strains relate to the TCA cycle, biosynthesis of several amino acids as well as fatty acid biosynthesis and elongation pathways, pointing toward necessary pathways for the survival of bacterial cells (Supplemental Table S3). The third most abundant pathway group MPG 3 denotes 13 pathways that were not found in Enterococcus casseliflavus strain TSA3A which is one of only three gram-positive strains in our collection (Fig. 1). Its genome assembly has good contiguity (N50 = 234 kb) and is highly complete (BUSCO completeness = 98%), which suggests that this pattern is unlikely to be due to a lower quality of this particular genome. Other MPGs show strong phylogenetic signatures such as MPG 7 and 13 that are restricted to the genera Achromobacter and Pseudomonas, respectively. We would therefore conclude that most of the variation in metabolic potential reflects genome evolution rather than technical differences. To explore the amount of metabolic variation across the bacterial phylogeny, we counted the number of pathway differences for pairwise comparison within and across genera. This showed significantly fewer differences in the metabolic potential for strains of the same genus (Supplemental Fig. S2). However, even strains of the same genus can differ in dozens of metabolic pathways, which can potentially explain the previously observed variability in the transcriptomic profiles and other phenotypes of P. pacificus nematodes in response to different bacteria from the same genus (Akduman et al. 2018; Athanasouli et al. 2023). Overall, this analysis revealed 1025 metabolic pathways that exhibit absence/presence variation in our bacterial collection.
Variation in bacterial metabolic potential. The heat map shows the predicted presence or absence of the most abundant MPGs across our genome collection (grouped according to the bacterial phylogeny). This revealed the metabolic potential of the individual strains. The majority of MPGs detected were absent from all strains, with a core of 45 bacterial pathways predicted always present. The rest of the MPGs exhibit distinct patterns even within the same genus.
Interspecies association studies identify candidate pathways affecting nematode survival and chemotaxis behavior
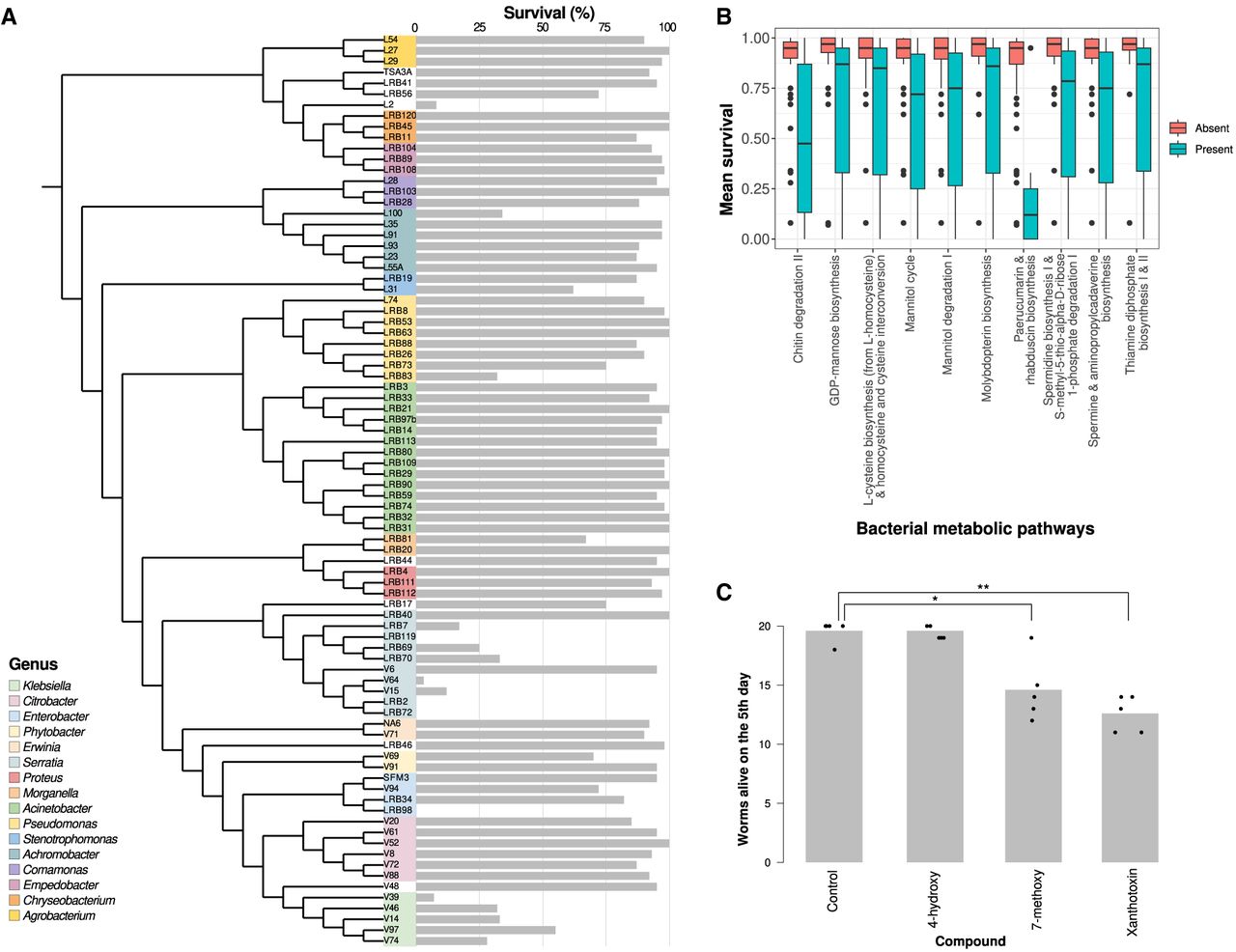
During the initial culture-based characterization of Pristionchus-associated microbiota, Akduman et al. (2018) performed assays for chemotaxis behavior and survival with over a hundred bacterial strains, many of which are part of our collection (Fig. 3A). In a pilot experiment, we employed our phylogenomic data to screen for candidate pathways, and their associated metabolic products, that could possibly impact nematode survival and chemotaxis behavior. To this end, we tested for associations between the presence or absence of an MPG with the behavioral and survival phenotypic data mentioned above. With this strategy, we identified 24 MPGs exhibiting significant association with chemotaxis (P < 0.05, Wilcoxon test) (Supplemental Fig. S3; Supplemental Table S4) and 40 MPGs that are associated with survival (Fig. 3B) (Supplemental Fig. S3; Supplemental Table S5). Among these candidate pathways, chitin degradation represents one of the pathways that has previously been shown to contribute to pathogenicity of certain bacteria (Zhang et al. 2014; Chen et al. 2015). However, the most dramatic difference in survival is associated with the presence or absence of the paerucumarin and rhabduscin biosynthesis pathways. Both pathways are partially overlapping, and rhabduscin as well as coumarin derivatives have previously been reported to exhibit nematicidal activity (Molnar et al. 2017; Guo et al. 2018; Abebew et al. 2022; Ibrahim et al. 2025). However, to our knowledge, bacterial production of coumarin derivatives has never been linked to pathogenicity against nematodes (Kurz et al. 2003). To complement this analysis with an additional screen for other virulence factors, we combined the predicted proteomes of the 84 bacteria with the virulence factor database (VFDB) (Liu et al. 2022) and performed a similar association study. This yielded 15 orthologous clusters with significant association (P < 0.05, Wilcoxon test), among which members of the paerucumarin biosynthesis gene cluster showed the strongest and most consistent effect (Supplemental Fig. S4). Closer examination of the presence of these genes in our data set revealed that these orthologs are exclusively present in some strains of the genus Serratia. Because the paerucumarin gene cluster has been initially characterized in Pseudomonas (Clarke-Pearson and Brady 2008), it is not known if the homologs of those genes in Serratia will produce exactly the same compound. Thus, hypothesizing that paerucumarin, rhabduscin, or similar metabolites could contribute to the pathogenicity of Serratia strains, we tested three coumarin-derivatives—4-hydroxycoumarin, 7-methoxycoumarin, and Xanthotoxin—for their effect against P. pacificus. All three compounds are commercially available and have previously been shown to possess some nematicidal activity in other species (Molnar et al. 2017; Guo et al. 2018; Abebew et al. 2022). Among those, 7-methoxycoumarin and Xanthotoxin caused substantial lethality at 1 mM concentrations (P < 0.05, t-test) (Fig. 3C). This supports that some coumarin derivatives have nematicidal activity against P. pacificus and that such compounds could potentially contribute to the increased pathogenicity of Serratia strains. However, further experimental validation is needed to elucidate the underlying pathways and confirm causality.
Association of nematode survival with the bacterial pathways. (A) The tree shows the bacterial phylogeny together with previously determined survival data (Akduman et al. 2018). (B) An association study between the MPGs and mean survival data narrowed down the MPGs with a significant effect on the trait, from hundreds to 41, a subsample of which is shown here. (C) Supplementation tests with three types of coumarins showed that 7-methoxycoumarin and Xanthotoxin affect nematode survival (P < 0.05, t-test).
Transcriptomic profiles of P. pacificus worms on 38 bacteria establish a catalog of molecular phenotypes
Having demonstrated the utility of the bacterial genomes to uncover candidate pathways impacting nematode survival and behavior, we complement the existing genomic data with phenotypic data that reflects the regulatory and metabolic response networks in the worm. To this end, we generated nematode transcriptome profiles for a subset of 38 dietary bacteria that belong to the three classes, Alpha-, Beta- and Gammaproteobacteria, and represent 16 genera. These 38 bacteria were selected because they supported complete development from egg to adult for at least two generations, which was not the case for all the strains. For each bacterial strain, we generated two biological replicates by collecting F1 worms 72 h after egg-laying (see Methods). Although most animals should have reached adulthood at this time under the standard E. coli OP50 diet (Sun et al. 2021), some bacterial diets either accelerate or slow down development, leading to a shift of the transcriptomic profiles toward either older or younger developmental stages (Akduman et al. 2020; Athanasouli et al. 2023). We decided to sequence the transcriptomes of mixed-stage populations because we had previously observed that even when adult worms are manually picked from plates, a strong developmental signature is still visible in the resulting RNA-seq data. This is because worms will still develop at different speeds, and chronological age might not necessarily correspond to developmental age (Athanasouli et al. 2023). As a consequence, differences in stage composition likely explain the larger extent of transcriptomic variation in our study, that is, quite a number of samples showed correlation coefficients r < 0.9 (Supplemental Fig. S5), which was not the case in our previous study (Athanasouli et al. 2023). Generally, we observed that biological replicates of the same bacterial diet exhibit significantly less variation than transcriptomic profiles from different bacterial diets (Supplemental Fig. S5). With regard to the taxonomic grouping of the bacteria, we observe that bacterial strains of the same genus show more similar transcriptomic responses in the worms than members of different genera (Supplemental Fig. S5). In summary, our transcriptomic data represent molecular phenotypes of P. pacificus nematodes that exhibit considerable variation in response to diverse microbiota.
Specific network modules respond to signals from diverse bacteria
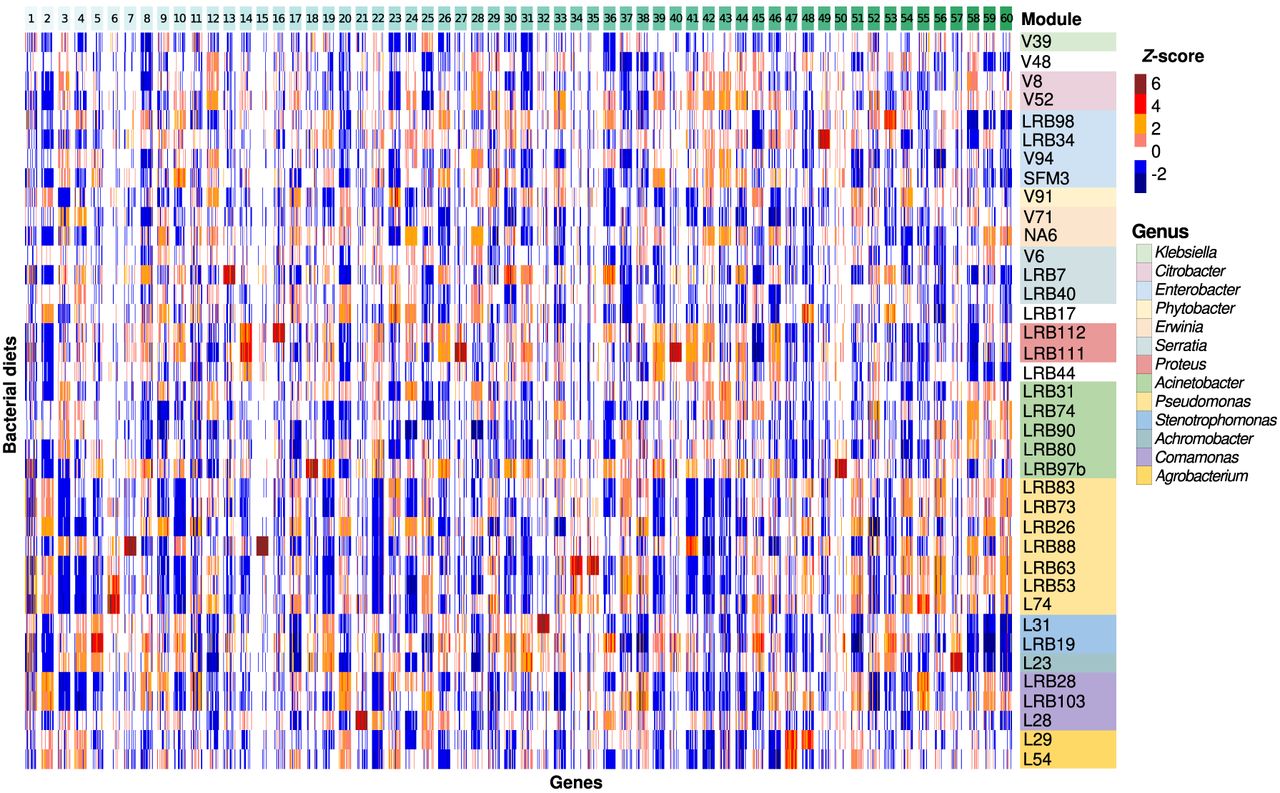
Whereas the above-mentioned transcriptomic analysis captures overall transcriptomic similarities, specific effects of individual bacteria might be overshadowed by global patterns. To investigate the effects of individual bacteria on smaller gene sets that may represent specific regulatory or metabolic network modules, we constructed a gene coexpression network from the newly generated RNA-seq data following our previously established protocol (see Methods). Overall, our coexpression network captures 24,375 (84%) of P. pacificus genes, of which 13,972 are found in the 60 largest modules with more than 20 genes (Supplemental Table S6). To test for the consistency between the current and the previously generated coexpression network (Athanasouli et al. 2023), we compared the gene set overlap between modules of both studies (Supplemental Fig. S6). Although the current study spans a wider developmental range than our previous study, the majority of modules from the current network match only a single module in the previous network. Specifically, the two largest modules (modules 1 and 2) of both networks appear to correspond to each other. This supports that the construction of coexpression networks is largely robust with regard to the underlying transcriptomic data set. Visualization of expression values in these modules across the different microbiota demonstrated that individual modules are induced by specific bacteria (Fig. 4). For example, the second largest module 2 shows high expression on most Pseudomonas strains, whereas module 7 exhibits highest expression only on the Pseudomonas protegens strain LRB88. Note that some of these patterns may represent indirect effects of altered development on diverse bacterial diets. This is shown by distinct expression signatures of specific coexpression modules when overlaying them with the developmental transcriptomic data of P. pacificus (Supplemental Fig. S7; Sun et al. 2021). For example, module 1 peaks late in development close to adulthood and is slightly preceded by a peak of module 2. However, no matter if the expression of those genes is a direct or indirect consequence, it can be interpreted as a specific response of P. pacificus nematodes to a distinct microbiota (Athanasouli et al. 2023). Thus, different bacterial diets induce specific transcriptomic responses of diverse modules.
Expression of modules across the different bacterial diets. The Z-score-normalized expression of the top 60 coexpression modules across the different diets was visualized as a heat map. The expression of the biological replicates was averaged in order to produce a single expression profile for each of the 38 diets. Modules with more than 50 genes were randomly downsampled to aid the visualization of smaller modules and the comparisons between them. Expression patterns were unique for each module and depended on the bacterial diet.
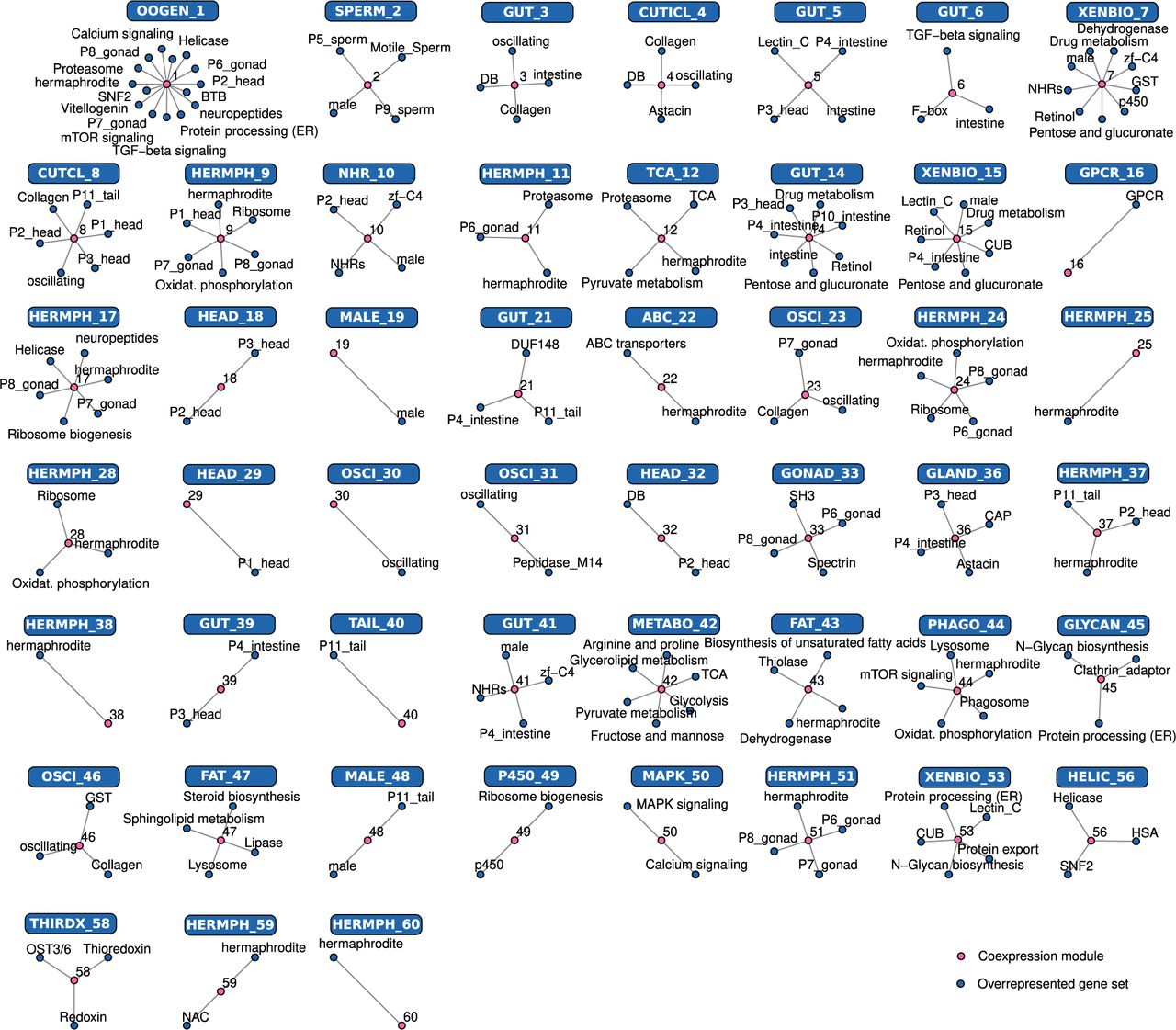
Most coexpression modules can be functionally labeled
The biological interpretation of coexpression modules requires their functional annotation. Given that roughly one third of P. pacificus genes have no detectable homologs in C. elegans (Athanasouli et al. 2023) and that detailed experimental studies have revealed distinct functions of highly conserved genes (Moreno et al. 2016; Theska and Sommer 2024), the ability to functionally annotate gene sets based on conservation alone is limited. Therefore, we integrated multiple data sets to functionally annotate the top 60 modules of the network by performing enrichment analyses. This included overrepresentation of protein domains (Mistry et al. 2021), regionally enriched genes as identified from spatial transcriptomics (Rödelsperger et al. 2021), KEGG pathways (Kanehisa and Goto 2000), and previous gene expression studies focusing on sex-biased genes (Rödelsperger et al. 2018), intestinal transcriptome (Lightfoot et al. 2016), and developmental oscillations (Supplemental Table S7; Sun et al. 2021). This allowed the labeling of 50 modules with biological terms based on the strongest enrichment patterns (Fig. 5; Supplemental Table S8). For example, the largest module 1 (OOGEN_1) is overrepresented in hermaphrodite-biased and gonad-enriched genes, which indicates that it largely represents oogenesis (Fig 5; Supplemental Table S8). This is further supported by the expression of germline markers like spo-11, dmc-1, hcp-4 (cenp-c), syp-4, and hop-1 (Rillo-Bohn et al. 2021). However, module 1 also contains neuropeptides and head-enriched genes, suggesting that also a portion of neuronal genes are captured in this module. The second largest module 2 (SPERM_2) seems to be more homogeneous, as all overrepresented terms point towards spermatogenesis (Fig. 6). The assignments of module 1 and 2 are also in line with the module comparison with our previous study (Supplemental Fig. S6) and the developmental signature of both modules (Supplemental Fig. S7), as sperm are generated during late larval development whereas oocytes are produced during adulthood (Rudel et al. 2005). Whereas many modules could only be associated with broad terms such as developmental oscillations or hermaphrodites, some individual modules can be assigned to very specific pathways or anatomical regions. For example, module 36 (GLAND_36) is strongly associated with gland cell-specific expression (Sieriebriennikov et al. 2020). We used the transcriptomic responses to different bacteria to arbitrarily label a handful of modules that did not show any enrichment in the above-mentioned data set (Supplemental Table S8). Thus, module 13 (LRB7_13) is strongly upregulated on Serratia quinivorans strain LRB7 and module 27 (LRB111_27) exhibits highest expression on Proteus terrae strain LRB111 (Fig. 4). Altogether, the characterization of the 60 largest coexpression modules functionally annotates a large portion of the P. pacificus gene set which includes many lineage-specific genes for which no homologs are known in C. elegans (Athanasouli et al. 2023; Lo et al. 2024). These data will be helpful to link specific transcriptomic responses to biological processes in the worm.
Enrichment analysis of the gene coexpression modules using existing transcriptomic data sets and functional annotation. The networks summarize the results of the overrepresentation analysis for specific coexpression modules (Supplemental Table S7). Based on the strongest enrichment patterns, we connected 50 of the processed 60 modules with biological processes and protein domains, allowing the functional labeling of the majority of the modules (Supplemental Table S8).
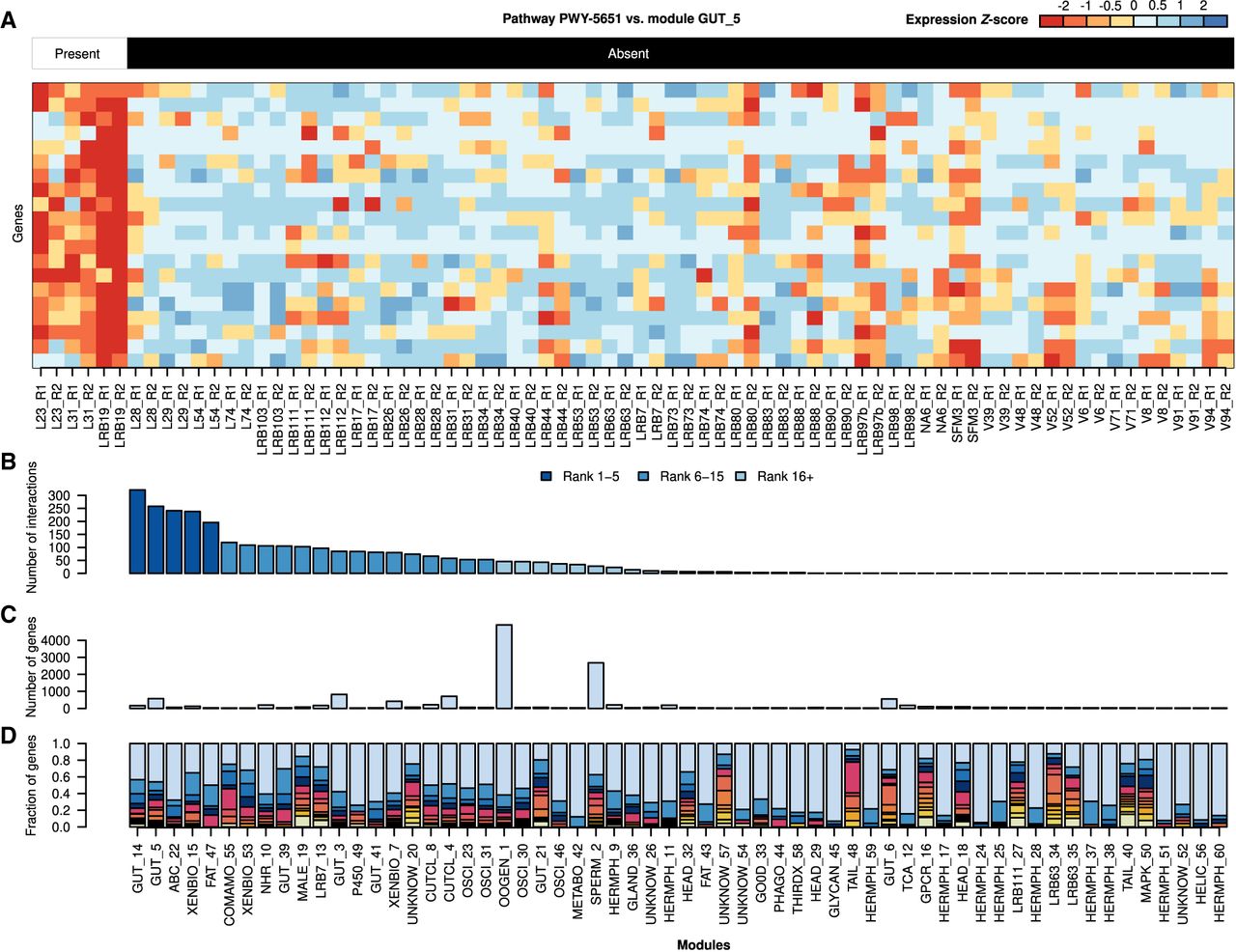
Interactions between bacterial pathways and coexpression modules. (A) The heat map shows a representative interaction between pathway PWY-5651 (L-tryptophan degradation to 2-amino-3carboxymuconate semialdehyde) and module GUT_5. Twenty randomly chosen genes from this module are shown together with their expression across different bacterial diets. These genes exhibit much higher expression on bacteria that have the PWY-5651 pathway. (B) The bars show the numbers of interactions per coexpression module. All of the top five and 11 out of the top 15 modules are associated with the intestine, detoxification, or specific metabolic pathways. The number of genes per module (C) and the phylostratigraphic distribution across modules (D) show that the five modules exhibiting the highest number of interactions are not the largest modules and are also not enriched in diplogastrid-specific orphan genes (phylostratum ≤ 9).
Linking bacterial pathways to coexpression modules predicts a global map of metabolic interactions
A main goal of this study was to test if we can trace the transcriptomic response of individual coexpression modules in P. pacificus to candidate metabolic pathways in the bacteria. Hereby, we define such links as statistical associations between variations in bacterial metabolic potential with differential responses of coexpression modules in the worms. To this end, we combined the bacterial MPGs and the nematode coexpression modules into a bipartite network and calculated the interaction of each MPG with a given coexpression module based on the significance of the overlap between the genes within a coexpression module and the genes that exhibit differential expression in response to the absence or presence of an MPG. This predicted a cross-species interactome of 2852 metabolic interactions (6.7% of all possible interactions) that involved 620 (86%) MPGs and 42 coexpression modules (Supplemental Table S9; Fig. 6A). Given that such a high number of MPGs are associated with a transcriptomic response of at least one coexpression module suggests that nematodes are highly sensitive to changes in dietary composition and that altered concentrations of most metabolites will have molecular consequences. The bacterial biosynthesis pathway that had the highest number of interactions produced phosphatidylcholine (PWY-6826), which was recently proposed as a key player regulating lipid homeostasis in C. elegans (Laranjeira et al. 2024). Within the interactome, we identified other metabolites and associated pathways that had already been implicated in regulating various traits in C. elegans and other nematodes. Thus, arginine and ornithine have been recently shown to affect reproductive fitness in C. elegans and parasitic nematodes (Venzon et al. 2022) and are also predicted to affect module OOGEN_1 (Supplemental Table S9). Similarly, intermediate metabolites of the pyrimidine pathways have been shown to affect reproduction and lifespan (Wan et al. 2019). Thus, although the complete cross-species interactome just represents predicted interactions that need to be validated using classical genetic approaches, it captured specific metabolites that have been previously shown to affect different biological processes in C. elegans and other nematodes. This suggests that the interactome displays a certain degree of conservation, making it a potentially valuable resource also outside the P. pacificus research community.
Host-microbe interactions may not be the dominant force driving the formation of novel genes in the P. pacificus genome
Whereas the detailed functional investigation of individual interactions is beyond the scope of our study, we further characterized global properties of the interactome. Using the number of predicted interactions as a proxy for the environmental responsiveness of a given coexpression module, we can identify the main modules that are responsible in sensing and processing bacterial metabolites. Notably, all of the top five modules and 11 out of the top 15 modules that interact with the highest number of bacterial pathways are associated with the intestine, detoxification, or specific metabolic pathways (Fig. 6B), which is consistent with the hypothesis that variation in metabolic potential of dietary bacteria should primarily affect the nematode gut. On the contrary, several hermaphrodite-enriched modules showed very low numbers of interactions, pointing toward a certain robustness of specific reproduction-associated processes. In summary, the inferred cross-species interactome identifies intestinal- and detoxification-related modules as components of the primary response layer to diverse microbiota. This further supports the general structure of our interactome as the module labels were inferred independently from the cross-species interactions. Furthermore, overlaying the number of interactions with the module size (Fig. 6C) and the distribution of phylostrata (gene age classes) (Athanasouli et al. 2023) across modules (Fig. 6D) shows that the most responsive modules do not contain the largest numbers of genes in total and are also not enriched in diplogastrid-specific orphan genes (Athanasouli et al. 2023). Notably, the largest module (SPERM_2), which exhibits an enrichment of diplogastrid-specific orphan genes, only has an intermediate number of interactions. These findings suggest that host-microbe interactions are unlikely the dominant force driving the emergence of novel genes in Pristionchus nematodes and point toward a major contribution of sexual conflict reflecting similar findings in flies (VanKuren et al. 2024).
Discussion
All animals live in tight associations with microbial communities, and bacteria can have a significant impact on the biology of their host. However, quantifying the effects of individual bacteria remains a major challenge in many study systems. In this study, we took advantage of bacterial feeding nematodes, where individual interactions can be studied in isolation (Zhang et al. 2017). This distinguishes our work from most metagenomic studies that investigate the aggregate effects of highly complex microbial communities on their hosts (Shalev et al. 2022; Lo et al. 2024; Salazar-Jaramillo et al. 2024). In addition, in contrast to most other studies focusing on the effect of single bacteria on specific nematode traits (Iatsenko et al. 2014; Watson et al. 2014; Akduman et al. 2020; O'Donnell et al. 2020; Lo et al. 2022), we undertake a systems-level approach to gain broad insights into the metabolic interactions between nematodes and their bacterial diets. We generated expression profiles of P. pacificus nematodes on 38 different bacterial diets. This represents, to our knowledge, the largest nematode transcriptomic study in response to different microbiota, and this data will be of substantial value for future studies in P. pacificus that explore the environmental responsiveness of specific genes or gene sets. By integrating these transcriptomic and additional phenotypic data with 84 newly sequenced bacterial genomes, our study illuminates both sides of this cross-kingdom interaction and allows us to associate phenotypic and transcriptomic variation in the nematode to the underlying metabolic variation in the bacteria. This is conceptually similar to the study of Zimmermann et al. (2020), who associated metabolic potential of members of the C. elegans microbiome with specific traits, but extends this idea by considering the response of specific coexpression networks in the worm as molecular phenotypes. This established a first global map of more than 2800 metabolic interactions between P. pacificus and its bacterial diets. Although the structure of this network is likely influenced by sampling of bacteria and correlated phylogenetic patterns across MPGs, it can be taken as a starting point for future studies investigating the effect of individual bacteria or metabolic pathways on specific nematode traits. Two main features of our interactome reinforce the assertion that it reflects true biological signals. First, given that the functional annotation of coexpression modules was completely independent from the association with bacterial metabolic pathways, it appears striking that most of the most responsive modules are associated with the intestine and detoxification processes. This is consistent with the assumption of the intestine as the primary response layer and thus supports the overall structure of the interactome. Second, the fact that many of the prominently highlighted metabolic pathways or metabolites, like chitin degradation (Chen et al. 2015), coumarins (Guo et al. 2018), phosphatidylcholine (Laranjeira et al. 2024), arginine, and ornithine (Venzon et al. 2022) have been shown to affect various biological processes in C. elegans or other nematodes points toward some degree of conservation of the map of metabolic interactions. The conserved nature of the metabolic interaction map suggests that our findings in P. pacificus may extrapolate to diverse host–microbe systems, informing mechanistic studies across taxa. Finally, to our knowledge, our work represents the first application of such a systems-level approach to predict a cross-species interactome in nematodes. Thus, this approach may be transferred to virtually any bacterial feeding nematode in order to establish species-specific maps of metabolic interactions and to explore the evolution of cross-species interactomes.
Methods
Bacterial culture conditions
The bacterial strains used in this study correspond to the strains isolated previously (Akduman et al. 2018). All strains were initially restreaked from a glycerol stock on Lysogeny Broth (LB) plates and grown overnight in LB. All bacterial isolates were grown at 30°C according to previous protocols (Akduman et al. 2018), whereas Escherichia coli OP50 was cultured at 37°C.
Nematode growth conditions
The wild-type strain of Pristionchus pacificus (PS312) was maintained at 20°C on nematode growth medium (NGM) seeded with 300 µL E. coli OP50 before use. Every 5 days, three adults were transferred to fresh NGM plates with a worm pick. Both NGM and E. coli OP50 were sourced from in-house stock.
Whole-genome sequencing of bacterial strains
All bacterial strains to be sequenced were grown overnight in LB in 15-mL Falcon tubes, and DNA samples were obtained using the Epicentre MasterPure Complete DNA and RNA Purification kit (Illumina). DNA libraries were prepared using the Illumina DNA Prep kit according to the manufacturer's guidelines. The libraries were quantified using both a Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and a Bioanalyzer (Agilent Technologies) and normalized to 2.5 nM. Samples were sequenced as 150-bp single-end reads on multiplexed lanes of an Illumina HiSeq 3000 in our in-house sequencing facility.
Genome assembly and metabolic potential identification of bacterial strains
The bacterial genomes were assembled from the raw reads using SPAdes (Prjibelski et al. 2020) (version 3.15.1, parameters: ‐‐careful -o -1 -2 -t 30 –cov-cutoff “auto”) and annotated with PROKKA (Seemann 2014) (version 1.14.6, parameters: ‐‐addgenes) and eggNOG-Mapper (Cantalapiedra et al. 2021) (version 2.1.0-1, eggnog DB version: 5.0.2). Genome assembly and annotation were evaluated using BUSCO (Manni et al. 2021) and QUAST (Gurevich et al. 2013). The predicted bacterial protein sequences were used as input to detect orthogroups and reconstruct the phylogeny with OrthoFinder (Emms and Kelly 2015). The bacterial genomes were used to predict the presence or absence of metabolic pathways with gapseq (Zimmermann et al. 2021) (version 1.2, parameters: find -p all, MetaCyc [Caspi et al. 2012] as the default database) as well as the presence of 67 bacterial phenotypes with the implementation of Traitar (Weimann et al. 2016) in Python (version 3.0.1, parameters: predict). The bacterial metabolic pathways were grouped (MPGs) based on their presence/absence patterns across the bacterial strains to reduce redundancy. This decreased the number of bacterial metabolic pathways to be assessed from 2902 to 712. The nomenclature of the bacterial strains was determined by applying DIAMOND (Buchfink et al. 2021) (version 2.1.4.158, parameters: makedb and blastp) to compare the predicted bacterial protein sequences against the nonredundant version of the NCBI database (August 2023 version).
Association of MPGs with chemotaxis and survival
In the initial study, the chemotaxis index ranges from −1 (repulsion) to 1 (attraction) and the survival index from 0 to 100, the latter reflecting the percentage of worms that were alive on the fifth day posttransfer (Akduman et al. 2018). For our analysis, we binarized the assay data by assigning the values below 40% survival and 0.2 chemotaxis as zero and the rest as one. The threshold for the binarization was determined based on the distribution of values. To test the association between each MPG and the nematode phenotypes, we divided the assay data into two vectors, representing the set of values when the nematode was maintained on bacterial strains with and without the MPG, and performed a Wilcoxon test, followed by Bonferroni correction (P < 0.05). To associate bacterial virulence factors with survival, we downloaded the 4236 core proteins from the Virulence Factor DataBase (Liu et al. 2022) (2024-07-31) and performed pairwise all-against-all BLASTP (Camacho et al. 2009) searches for all bacterial protein sets combined with the VFDB data (e-value < 10−5). The widely used clustering algorithm MCL was used to cluster the data into 1605 orthogroups. These orthogroups were then tested for association with survival using a Wilcoxon test with FDR correction (P < 0.1).
Supplementation experiments
To check the nematicidal effect of coumarin derivatives in vivo, we supplemented 4-hydroxycoumarin (Sigma-Aldrich, CAS: 1076-38-6, purity: >97.5%), 7-methoxycoumarin (Sigma-Aldrich, CAS: 531-59-9, purity: >98%), and Xanthotoxin (TCI Deutschland, CAS: 298-81-7, purity: >98%) to the nematode. The coumarins were added directly to liquid NGM to a final concentration of 1 mM, and the solution was subsequently poured into 6-cm plates (11 mL per plate). The plates were seeded with 300 µL of E. coli OP50 the next day and left to grow a bacterial lawn for 2 days. After that time, 20 young adult hermaphrodites were transferred on each plate, and their survival was tracked daily for 5 days. The surviving nematodes were transferred onto new plates containing the coumarins on the third day after the initial transfer to avoid food depletion and misidentification from their offspring. Mortality was determined by lack of response to prodding with the pick. For the control and each coumarin, we performed five replicates.
Dietary experiments
Eggs from P. pacificus adults were obtained and spotted on NGM plates seeded with 75 µL bacterial overnight cultures to produce the parental generation (P0) on each diet. Twenty-five gravid hermaphrodites from the P0 were transferred to a new plate seeded with the same bacterial strain and were left to lay eggs for 5 h, after which point they were removed. The F1 worms were collected 72 h after the initial transfer. Collection of the worms required washing the plate with autoclaved M9− buffer, centrifugation at 500g for a minute, and discarding the supernatant in order to remove as much of the collected bacterial lawn as possible. The worm pellets were flash-frozen in liquid N2 and were used for RNA extraction or stored at −80°C for later processing.
RNA sequencing
For total RNA extraction, the frozen worm pellets were treated with TRIzol followed by purification with the Zymo RNA Clean & Concentrator 25 kit according to the manufacturer's instructions. The extracted RNA was quantified and quality-assessed with a NanoDrop ND1000 spectrometer (PeqLab) and a Qubit 2.0 Fluorometer. The samples were shipped to Novogene for library preparation and messenger RNA (mRNA) sequencing. Libraries were sequenced as 150-bp paired-end reads on an Illumina NovoSeq 6000 platform.
RNA-seq and coexpression network analysis
For the coexpression analysis, we only used newly generated RNA-seq data because the samples from our previous study were obtained using different protocols and were only sequenced as single replicates. The raw reads were aligned to the reference Pristionchus pacificus genome (version El Paco) with STAR (Dobin et al. 2013) (version 2.7.1a) and quantified with featureCounts (Liao et al. 2014) from the Subread R package (version 2.0.1) based on the latest annotations (Athanasouli et al. 2020). After filtering the count matrix to remove genes with less than 10 reads total according to the reference manual, we reduced the number of P. pacificus genes to be assessed from 28,896 to 24,335. The read counts across different conditions and replicates were normalized with the DESeq2 (Love et al. 2014) counts function (option: normalized = TRUE) and were input into the Markov CLustering software (MCL) (van Dongen and Abreu-Goodger 2012) (version 22-282, r = 0.7, I = 2) to create a gene coexpression network. The network modules were tested for the enrichment of protein domain annotations (Pfam) (Mistry et al. 2021), metabolic pathways (KEGG) (Kanehisa and Goto 2000), and gene expression sets from previous studies (Athanasouli et al. 2023) with Fisher's exact test followed by Bonferroni correction (P < 0.05) in R (version 4.4.0) (R Core Team 2024). Phylostrata were defined as described previously (Athanasouli et al. 2023): in short, BLASTP searches (e-value < 0.001) were carried out against predicted proteomes of related nematodes (Howe et al. 2017; Prabh et al. 2018), and phylostrata were defined based on the presence of homologs in the most distantly related species. The network modules were assessed further for their interactions with the bacterial metabolic potential. Specifically, for each MPG, the normalized counts of the genes in each module were separated into two vectors based on the presence or absence of the MPG in the bacterial strain in order to represent the expression of a specific gene in a module in the presence or absence of an MPG. For each gene, we separated the expression values into two vectors based on the absence or presence of an MPG. These expression vectors were compared using a Wilcoxon test, and they were compared to calculate the fold change in expression. To determine the interaction of a module with an MPG, we retained the genes where the fold change was lower than 0.5 or higher than 2 and performed multiple hypothesis testing for the module genes that fulfilled the condition (Bonferroni < 0.1). The list of genes identified as interacting with the MPGs were tested for module enrichment using Fisher's exact test (Bonferroni-adjusted P-value < 0.05) to determine the interaction between the MPGs and the coexpression modules. The bipartite network was implemented using Python (version 3.10.12).
Data access
All sequencing data generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/) under accession number PRJEB80633.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Nicholas Youngblut, Boris Macek, Thorsten Langner, and all members of the Sommer lab for helpful discussions. This work was funded by the Max Planck Society.
Author contributions: Conceptualization: C.R. Investigation: M.A. and T.L. Data curation: M.A. Formal analysis: M.A. Visualization: M.A. and C.R. Writing—original draft: M.A. Writing—review and editing: C.R. Supervision: C.R.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280848.125.
References
- ↵Abebew D, Sayedain FS, Bode E, Bode HB. 2022. Uncovering nematicidal natural products from Xenorhabdus bacteria. J Agric Food Chem 70: 498–506. 10.1021/acs.jafc.1c05454
- ↵Akduman N, Rödelsperger C, Sommer RJ. 2018. Culture-based analysis of Pristionchus-associated microbiota from beetles and figs for studying nematode-bacterial interactions. PLoS One 13: e0198018. 10.1371/journal.pone.0198018
- ↵Akduman N, Lightfoot JW, Röseler W, Witte H, Lo W-S, Rödelsperger C, Sommer RJ. 2020. Bacterial vitamin B12 production enhances nematode predatory behavior. ISME J 14: 1494–1507. 10.1038/s41396-020-0626-2
- ↵Athanasouli M, Witte H, Weiler C, Loschko T, Eberhardt G, Sommer RJ, Rödelsperger C. 2020. Comparative genomics and community curation further improve gene annotations in the nematode Pristionchus pacificus. BMC Genomics 21: 708. 10.1186/s12864-020-07100-0
- ↵Athanasouli M, Akduman N, Röseler W, Theam P, Rödelsperger C. 2023. Thousands of Pristionchus pacificus orphan genes were integrated into developmental networks that respond to diverse environmental microbiota. PLoS Genet 19: e1010832. 10.1371/journal.pgen.1010832
- ↵Brady C, Kaur S, Crampton B, Maddock D, Arnold D, Denman S. 2022. Transfer of Erwinia toletana and Erwinia iniecta to a novel genus Winslowiella gen. nov. as Winslowiella toletana comb. nov. and Winslowiella iniecta comb. nov. and description of Winslowiella arboricola sp. nov., isolated from bleeding cankers on broadleaf hosts. Front Microbiol 13: 1063107. 10.3389/fmicb.2022.1063107
- ↵Buchfink B, Reuter K, Drost H-G. 2021. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 18: 366–368. 10.1038/s41592-021-01101-x
- ↵Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10: 421. 10.1186/1471-2105-10-421
- ↵Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J. 2021. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol 38: 5825–5829. 10.1093/molbev/msab293
- ↵Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, 2012. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 40: D742–D753. 10.1093/nar/gkr1014
- ↵Chen L, Jiang H, Cheng Q, Chen J, Wu G, Kumar A, Sun M, Liu Z. 2015. Enhanced nematicidal potential of the chitinase pachi from Pseudomonas aeruginosa in association with Cry21Aa. Sci Rep 5: 14395. 10.1038/srep14395
- ↵Clarke-Pearson MF, Brady SF. 2008. Paerucumarin, a new metabolite produced by the pvc gene cluster from Pseudomonas aeruginosa. J Bacteriol 190: 6927–6930. 10.1128/JB.00801-08
- ↵Dirksen P, Marsh SA, Braker I, Heitland N, Wagner S, Nakad R, Mader S, Petersen C, Kowallik V, Rosenstiel P, 2016. The native microbiome of the nematode Caenorhabditis elegans: gateway to a new host-microbiome model. BMC Biol 14: 38. 10.1186/s12915-016-0258-1
- ↵Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635
- ↵Emms DM, Kelly S. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16: 157. 10.1186/s13059-015-0721-2
- ↵Girard L, Lood C, Höfte M, Vandamme P, Rokni-Zadeh H, van Noort V, Lavigne R, De Mot R. 2021. The ever-expanding Pseudomonas genus: description of 43 new species and partition of the Pseudomonas putida group. Microorganisms 9: 1766. 10.3390/microorganisms9081766
- ↵Guo Q-Q, Du G-C, Li Y-X, Liang C-Y, Wang C, Zhang Y-N, Li R-G. 2018. Nematotoxic coumarins from Angelica pubescens Maxim. f. biserrata Shan et Yuan roots and their physiological effects on Bursaphelenchus xylophilus. J Nematol 50: 559–568. 10.21307/jofnem-2018-045
- ↵Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. 10.1093/bioinformatics/btt086
- ↵Howe KL, Bolt BJ, Shafie M, Kersey P, Berriman M. 2017. Wormbase ParaSite − a comprehensive resource for helminth genomics. Mol Biochem Parasitol 215: 2–10. 10.1016/j.molbiopara.2016.11.005
- ↵Iatsenko I, Yim JJ, Schroeder FC, Sommer RJ. 2014. B. subtilis GS67 protects C. elegans from gram-positive pathogens via fengycin-mediated microbial antagonism. Curr Biol 24: 2720–2727. 10.1016/j.cub.2014.09.055
- ↵Ibrahim H, Nchiozem-Ngnitedem V-A, Dandurand L-M, Popova I. 2025. Naturally-occurring nematicides of plant origin: two decades of novel chemistries. Pest Manag Sci 81: 540–571. 10.1002/ps.8504
- ↵Kakumanu ML, Marayati BF, Wada-Katsumata A, Wasserberg G, Schal C, Apperson CS, Ponnusamy L. 2021. Sphingobacterium phlebotomi sp. nov., a new member of family Sphingobacteriaceae isolated from sand fly rearing substrate. Int J Syst Evol Microbiol 71: 004809. 10.1099/ijsem.0.004809
- ↵Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28: 27–30. 10.1093/nar/28.1.27
- ↵Kurz CL, Chauvet S, Andrès E, Aurouze M, Vallet I, Michel GPF, Uh M, Celli J, Filloux A, De Bentzmann S, 2003. Virulence factors of the human opportunistic pathogen Serratia marcescens identified by in vivo screening. EMBO J 22: 1451–1460. 10.1093/emboj/cdg159
- ↵Laranjeira AC, Berger S, Kohlbrenner T, Greter NR, Hajnal A. 2024. Nutritional vitamin B12 regulates RAS/MAPK-mediated cell fate decisions through one-carbon metabolism. Nat Commun 15: 8178. 10.1038/s41467-024-52556-3
- ↵Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. 10.1093/bioinformatics/btt656
- ↵Lightfoot JW, Chauhan VM, Aylott JW, Rödelsperger C. 2016. Comparative transcriptomics of the nematode gut identifies global shifts in feeding mode and pathogen susceptibility. BMC Res Notes 9: 142. 10.1186/s13104-016-1886-9
- ↵Liu B, Zheng D, Zhou S, Chen L, Yang J. 2022. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res 50: D912–D917. 10.1093/nar/gkab1107
- ↵Lo W-S, Han Z, Witte H, Röseler W, Sommer RJ. 2022. Synergistic interaction of gut microbiota enhances the growth of nematode through neuroendocrine signaling. Curr Biol 32: 2037–2050.e4. 10.1016/j.cub.2022.03.056
- ↵Lo W-S, Sommer RJ, Han Z. 2024. Microbiota succession influences nematode physiology in a beetle microcosm ecosystem. Nat Commun 15: 5137. 10.1038/s41467-024-49513-5
- ↵Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. 10.1186/s13059-014-0550-8
- ↵Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. 2021. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol 38: 4647–4654. 10.1093/molbev/msab199
- ↵Meyer JM, Baskaran P, Quast C, Susoy V, Rödelsperger C, Glöckner FO, Sommer RJ. 2017. Succession and dynamics of Pristionchus nematodes and their microbiome during decomposition of Oryctes borbonicus on La Réunion Island. Environ Microbiol 19: 1476–1489. 10.1111/1462-2920.13697
- ↵Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, 2021. Pfam: the protein families database in 2021. Nucleic Acids Res 49: D412–D419. 10.1093/nar/gkaa913
- ↵Molnar M, Komar M, Brahmbhatt H, Babić J, Jokić S, Rastija V. 2017. Deep eutectic solvents as convenient media for synthesis of novel coumarinyl Schiff bases and their QSAR studies. Molecules 22: 1482. 10.3390/molecules22091482
- ↵Moreno E, McGaughran A, Rödelsperger C, Zimmer M, Sommer RJ. 2016. Oxygen-induced social behaviours in Pristionchus pacificus have a distinct evolutionary history and genetic regulation from Caenorhabditis elegans. Proc Biol Sci 283: 20152263. 10.1098/rspb.2015.2263
- ↵O'Donnell MP, Fox BW, Chao P-H, Schroeder FC, Sengupta P. 2020. A neurotransmitter produced by gut bacteria modulates host sensory behaviour. Nature 583: 415–420. 10.1038/s41586-020-2395-5
- ↵Prabh N, Roeseler W, Witte H, Eberhardt G, Sommer RJ, Rödelsperger C. 2018. Deep taxon sampling reveals the evolutionary dynamics of novel gene families in Pristionchus nematodes. Genome Res 28: 1664–1674. 10.1101/gr.234971.118
- ↵Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. 2020. Using SPAdes de novo assembler. Curr Protoc Bioinformatics 70: e102. 10.1002/cpbi.102
- ↵R Core Team. 2024. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/.
- ↵Rillo-Bohn R, Adilardi R, Mitros T, Avşaroğlu B, Stevens L, Köhler S, Bayes J, Wang C, Lin S, Baskevitch KA, 2021. Analysis of meiosis in Pristionchus pacificus reveals plasticity in homolog pairing and synapsis in the nematode lineage. eLife 10: e70990. 10.7554/eLife.70990
- ↵Rödelsperger C, Röseler W, Prabh N, Yoshida K, Weiler C, Herrmann M, Sommer RJ. 2018. Phylotranscriptomics of Pristionchus nematodes reveals parallel gene loss in six hermaphroditic lineages. Curr Biol 28: 3123–3127.e5. 10.1016/j.cub.2018.07.041
- ↵Rödelsperger C, Ebbing A, Sharma DR, Okumura M, Sommer RJ, Korswagen HC. 2021. Spatial transcriptomics of nematodes identifies sperm cells as a source of genomic novelty and rapid evolution. Mol Biol Evol 38: 229–243. 10.1093/molbev/msaa207
- ↵Rudel D, Riebesell M, Sommer RJ. 2005. Gonadogenesis in Pristionchus pacificus and organ evolution: development, adult morphology and cell-cell interactions in the hermaphrodite gonad. Dev Biol 277: 200–221. 10.1016/j.ydbio.2004.09.021
- ↵Salazar-Jaramillo L, de la Cuesta-Zuluaga J, Chica LA, Cadavid M, Ley RE, Reyes A, Escobar JS. 2024. Gut microbiome diversity within Clostridia is negatively associated with human obesity. mSystems 9: e0062724. 10.1128/msystems.00627-24
- ↵Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069. 10.1093/bioinformatics/btu153
- ↵Shalev O, Karasov TL, Lundberg DS, Ashkenazy H, Pramoj Na Ayutthaya P, Weigel D. 2022. Commensal Pseudomonas strains facilitate protective response against pathogens in the host plant. Nat Ecol Evol 6: 383–396. 10.1038/s41559-022-01673-7
- ↵Sieriebriennikov B, Sun S, Lightfoot JW, Witte H, Moreno E, Rödelsperger C, Sommer RJ. 2020. Conserved nuclear hormone receptors controlling a novel plastic trait target fast-evolving genes expressed in a single cell. PLoS Genet 16: e1008687. 10.1371/journal.pgen.1008687
- ↵Slowinski S, Ramirez I, Narayan V, Somayaji M, Para M, Pi S, Jadeja N, Karimzadegan S, Pees B, Shapira M. 2020. Interactions with a complex microbiota mediate a trade-off between the host development rate and heat stress resistance. Microorganisms 8: 1781. 10.3390/microorganisms8111781
- ↵Smits THM, Arend LNVS, Cardew S, Tång-Hallbäck E, Mira MT, Moore ERB, Sampaio JLM, Rezzonico F, Pillonetto M. 2022. Resolving taxonomic confusion: establishing the genus Phytobacter on the list of clinically relevant Enterobacteriaceae. Eur J Clin Microbiol Infect Dis 41: 547–558. 10.1007/s10096-022-04413-8
- ↵Sun S, Rödelsperger C, Sommer RJ. 2021. Single worm transcriptomics identifies a developmental core network of oscillating genes with deep conservation across nematodes. Genome Res 31: 1590–1601. 10.1101/gr.275303.121
- ↵Theska T, Sommer RJ. 2024. Feeding-structure morphogenesis in “rhabditid” and diplogastrid nematodes is not controlled by a conserved genetic module. Evol Dev 26: e12471. 10.1111/ede.12471
- ↵van Dongen S, Abreu-Goodger C. 2012. Using MCL to extract clusters from networks. Methods Mol Biol 804: 281–295. 10.1007/978-1-61779-361-5_15
- ↵VanKuren NW, Chen J, Long M. 2024. Sexual conflict drive in the rapid evolution of new gametogenesis genes. Semin Cell Dev Biol 159-160: 27–37. 10.1016/j.semcdb.2024.01.005
- ↵Venzon M, Das R, Luciano DJ, Burnett J, Park HS, Devlin JC, Kool ET, Belasco JG, Hubbard EJA, Cadwell K. 2022. Microbial byproducts determine reproductive fitness of free-living and parasitic nematodes. Cell Host Microbe 30: 786–797.e8. 10.1016/j.chom.2022.03.015
- ↵Wan Q-L, Meng X, Fu X, Chen B, Yang J, Yang H, Zhou Q. 2019. Intermediate metabolites of the pyrimidine metabolism pathway extend the lifespan of C. elegans through regulating reproductive signals. Aging (Albany NY) 11: 3993–4010. 10.18632/aging.102033
- ↵Watson E, MacNeil LT, Ritter AD, Yilmaz LS, Rosebrock AP, Caudy AA, Walhout AJM. 2014. Interspecies systems biology uncovers metabolites affecting C. elegans gene expression and life history traits. Cell 156: 759–770. 10.1016/j.cell.2014.01.047
- ↵Weimann A, Mooren K, Frank J, Pope PB, Bremges A, McHardy AC. 2016. From genomes to phenotypes: Traitar, the microbial trait analyzer. mSystems 1: e00101-16. 10.1128/mSystems.00101-16
- ↵Zhang L, Yu J, Xie Y, Lin H, Huang Z, Xu L, Gelbič I, Guan X. 2014. Biological activity of Bacillus thuringiensis (Bacillales: Bacillaceae) chitinase against Caenorhabditis elegans (Rhabditida: Rhabditidae). J Econ Entomol 107: 551–558. 10.1603/EC13201
- ↵Zhang J, Holdorf AD, Walhout AJ. 2017. C. elegans and its bacterial diet as a model for systems-level understanding of host-microbiota interactions. Curr Opin Biotechnol 46: 74–80. 10.1016/j.copbio.2017.01.008
- ↵Zimmermann J, Obeng N, Yang W, Pees B, Petersen C, Waschina S, Kissoyan KA, Aidley J, Hoeppner MP, Bunk B, 2020. The functional repertoire contained within the native microbiota of the model nematode Caenorhabditis elegans. ISME J 14: 26–38. 10.1038/s41396-019-0504-y
- ↵Zimmermann J, Kaleta C, Waschina S. 2021. gapseq: informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. Genome Biol 22: 81. 10.1186/s13059-021-02295-1