
A key aspect of research in genetics is associating sequence variations with heritable phenotypes. The most common variations are single nucleotide polymorphisms (SNPs), which occur approximately once every 500–1000 bases in a large sample of aligned human sequence. Because SNPs are expected to facilitate large-scale association genetics studies, there has recently been great interest in SNP discovery and detection. In collaboration with the National Human Genome Research Institute (NHGRI), the National Center for Biotechnology Information (NCBI) has established the dbSNP database (http://www.ncbi.nlm.nih.gov/SNP) to serve as a central repository for molecular variation. Designed to serve as a general catalog of molecular variation to supplement GenBank (Benson et al. 1999) database submissions can include a broad range of molecular polymorphisms: single base nucleotide substitutions, short deletion and insertion polymorphisms, microsatellite markers, and polymorphic insertion elements such as retrotransposons.
Although the name dbSNP is a slight misnomer given the variations represented, SNP polymorphisms are the largest class of variation in the database, and the name dbSNP, selected at the request of NHGRI, reflects this fact. For the sake of brevity, we elected to use the term SNP as a shorthand for “variation” in the database notation and documentation (http://www.ncbi.nlm.nih.gov/SNP/get_html.cgi?whichHtml=how_to_submit). Thus terms used in the documentation like “submitted SNP” or “reference SNP” refer to all classes of variation in the database and should be regarded as meaning “a submitted report of variation” and “a reference report of variation.” Furthermore, it should be noted that in serving its role as the variation complement to GenBank, dbSNP does not restrict submissions to only neutral polymorphisms. Submissions are welcome on all classes of simple molecular variation, including those that cause rare clinical phenotypes.
Submissions to dbSNP come from a variety of sources including individual laboratories, collaborative polymorphism discovery efforts, large-scale genome sequencing centers, and private industry. The data collected range from the tightly focused characterization of particular genes to broadly sampled levels of variation from random genomic sequence. The distribution of reported marker density across the genome is thus expected to be mixed, with an expected minimum density of 1/3000 bases in regions of random genomic sequence, and local regions of higher density around well-characterized genes. Each variation submitted to dbSNP must have an identifier provided by the submitter (called a “local” identifier by dbSNP), and each is issued a unique identifier, formatted as an integer prefixed with ss (forsubmitted SNP), for example, ss334. An ss number is thus permanently associated with the submitter’s identifier, and it can be treated as a formal accession number by the scientific publishing community.
For Each Variation, dbSNP Includes Links to Populations, Specific Locations within Molecular Sequences, and Assay Methods
Linking to Genomic Location
The sequence location permits us to specify the specific base(s) altered, and although obtained in several ways, it is always pinpointed within flanking sequence in the dbSNP submission. Simultaneous submission of either STS data documenting how to isolate the marker with PCR techniques, explicit linking to a GenBank accession number, and postsubmission computational analysis of the polymorphism and flanking sequence can all be used to align the flanking sequence to other sequence records in the NCBI databases. These alignments are analyzed to localize the variation and its flanking sequence within the genome. The quality and accuracy of this localization is determined by the quality and nature of the sequence; variations in segments of low-complexity sequence will be more difficult to localize than variations reported from segments of complex, unique sequence.
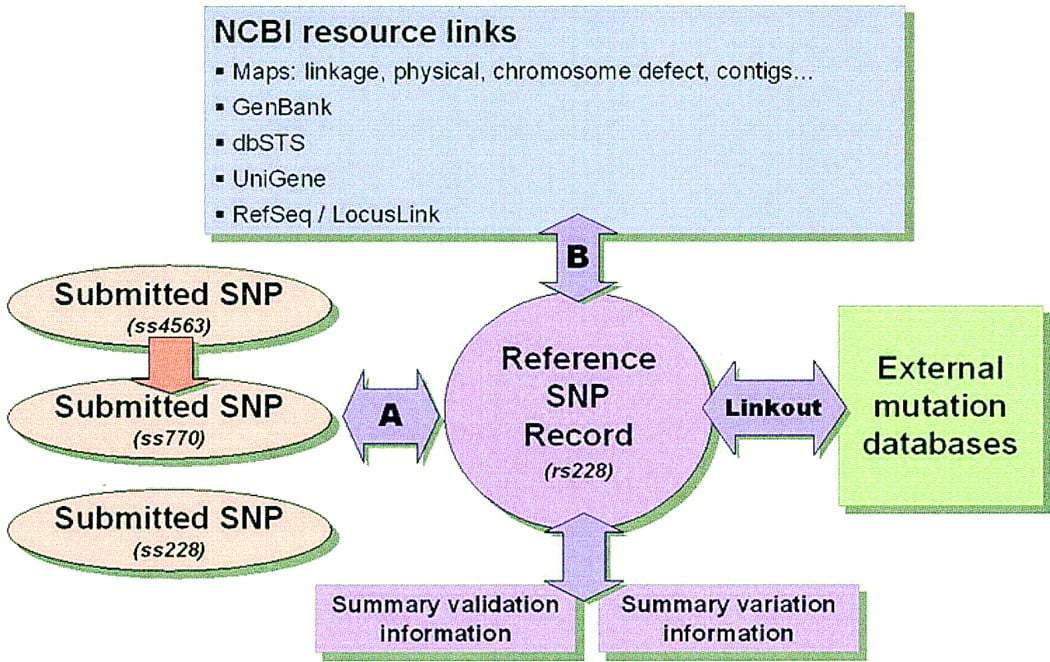
To represent multiple submissions of variation at the same genome location, dbSNP maintains two types of records: ss records of each original submission, and reference SNP (rs) records that are constructed (and periodically reconstructed) by the algorithm discussed below. As of this writing there are 4790 submissions to dbSNP, which have been resolved to a set of 4713 unique variations. These cases are recognized by a postsubmission computational analysis consisting of several steps (see Box Figure B1.).
The NCBI dbSNP data model. Individual submissions to dbSNP are represented by the three ovals on the left. Postsubmission computations identify sets of identical submissions and cluster them into single reference SNP records (step A). Submissions that are determined to be unique at the time of analysis also have reference SNP records constructed at this time as well. All reference SNP records are analyzed by a second round of BLAST analysis in an attempt to annotate them onto other NCBI resources (step B) as discussed in the text. Illustrated here is a real case in the database in which two groups independently reported a variation (ss228, ss770), and a third group (ss4563) submitted additional frequency data on the published variation ss770. All three records were grouped into the single reference record, rs228. Links are established between individual submissions and external databases or servers, such as locus-specific mutation databases or submitter web sites by using the LINKOUT line type. These links will direct users to additional information on a particular variation.
Construction of Reference SNP Summary Records
During the submission process, all of the flanking sequences of dbSNP submissions are compared, pairwise, with the BLAST algorithm (Altschul et al. 1990) to identify cases of independent discovery and reporting. Independent discovery is not a rare event, as many of the groups involved with SNP discovery are working from a large but common set of initial reagents, e.g., sequences from dbEST (Boguski et al. 1993), clones from the genome sequencing pipeline, and submissions from dbSTS (Olson et al. 1989). In addition, new submissions to dbSNP can consist exclusively of additional frequency or genotype data on previously submitted variations. In such cases, the submission of a variation’s flanking sequence can be substituted with a reference to the database record for which the new data apply by using the SNP_LINK line type in the SNPASSAY section. Sets of two or more identical submissions are identified by a stepwise algorithm that first checks flanking sequence for probable identity and then checks the set of STS or GenBank accession numbers that are submitted with the records to ensure that their best representatives have been identified as high-scoring pairs (HSP), in the NCBI BLAST database (Fig. 1, step A). Our current acceptance criteria have been optimized through heuristic analysis of the BLAST output returned for each pair of markers and their flanking sequence. These criteria are as follows: (1) The variable sites must be at the same position in the sequence alignment returned by ungapped BLAST; (2) there must be a maximum of five partial matches in the aligned sequence [defined as an exact match between two International Union of Pure and Applied Chemistry (IUPAC) ambiguous nucleotide codes (e.g., R-R) or a match between a nucleotide and an ambiguity code of which it is a set member (e.g., A-R)]; (3) there must be a maximum of one mismatch in the BLAST alignment (e.g., A−T, A−G, or A−C); and (4) there must be a percent identity score, P ≥ 0.89, where P = (I + M)/min(qlen,slen), Iis the number of identical matches in the alignment, M is the number of partial matches in the alignment, and min(qlen,slen) is the minimum length of the two sequences (query and subject) returned from the BLAST alignment. Criterion 4 is necessary to eliminate false matches produced by very short, but highly significant, sequence alignments returned by the BLAST algorithm. Pairs of successful matches as defined by the above criteria are then evaluated for sequence similarity in their submitter-associated accession numbers. This extra level of validation is performed when possible to ensure that our presumed pairs occur within the context of a larger region of identical sequence. Sequence similarity is currently checked by selecting the longest sequence from the set of accession numbers (STS or GenBank) for each marker, and querying the NCBI HSP database to ensure that the pair have been externally neighbored for NCBI BLAST analysis or Entrez (http://www.ncbi.nlm.nih.gov/Entrez/) retrieval. By working through all pairs of submitted records in such a fashion, sets of two or more identical records can be collected into a single reference SNP cluster. These reference SNP records are numbered sequentially and are prefixed with rs to distinguish them from individual submission records. The annotation of reference SNP records onto other NCBI resources is accomplished by a second round of ungapped BLAST analysis of the flanking sequences against the GenBank divisions NR, EST, STS, GSS, and HTGS (Fig. 1, step B). The current acceptance criteria for this process are (1) a maximum of five partial matches; (2) a maximum of two mismatches; and (3) an identity score of P ≥ 0.95. These high-stringency criteria were adopted to reduce the false-positive hit rate in our initial pass, and they may be modified as we continue our heuristic optimization of the algorithm.
Reference SNP records contain summary information for the variation such as the longest extent of flanking sequence considering all of the submissions for this variation, the list of submitted records in the set, and summary allele frequencies. They serve as the data object that will be anchored to other NCBI resources such as reference sequences1 and the human genome contig assemblies. NCBI is currently working with NHGRI to establish a convention for reporting validation information for the variations in dbSNP. When these standards are adopted, both submitted SNP and reference SNP records will be classified and searchable by their degree of validation.
Linking Populations
Submitters describe the populations containing the variations using free text fields to classify their sample as specifically as possible. Submissions also require a specification of the sample size specified as the number of chromosomes that were examined in the course of discovery of the variation. Certain population samples that are publicly available have standardized descriptions and can be used by any submitter with data from those resources. Examples include the National Institutes of Health Polymorphism Discovery Resource (NIHPDR) (Collins et al. 1998) and the CEPH family collection (http://www.cephb.fr/). Of particular importance is a description of the sample in which the variation was first discovered and reported. Many subsequent analyses of the frequency data in dbSNP will require knowledge of these ascertainment conditions (sample size and sample composition) to remove any bias effects (e.g., Sherry et al. 1997).
Allele frequencies may be reported by population for each allele of a particular variation. Additionally, individual genotypes may be submitted for any variation. Genotype data will be useful for further analysis in genome association studies (e.g., Lai et al. 1998) or testing and refining haplotype reconstruction algorithms (e.g.,Peterson et al. 1999), especially for samples from the sets of public reagents discussed above. It is important for submitters to describe the population from which individuals whose genotypes are submitted were sampled. This is especially important in cases where the samples are not publicly available. It is the hopeful intention of NCBI that future data on allele frequencies, genotypes, or multilocus haplotypes will be submitted to dbSNP as supplemental data about previously reported variations.
Future Directions
dbSNP is in the early stages of a maturing database. In addition to obtaining variation data by original submissions, dbSNP is developing data exchange protocols with other public variation and mutation databases, such as HGBASE (human genicbiallelic sequences) (http://hgbase.interactiva.de) and The SNP Consortium (TSC) public database (Masood 1999). The same algorithm (see Box Figure B1.) for mapping to reference SNPs will be applied to this variation data. Other issues under development are an extension of the database to support haplotype data objects, expanded integration of dbSNP records to other NCBI resources such as UniGene,2 expanded query facilities and graphical user interfaces to permit structured queries and batch retrieval of results, and online web submission tools to complement the established batch submission process.
Notes
[1] Corresponding author.
Notes
[2] E-MAIL [email protected]; FAX (301) 435-7794.
[3] The NCBI Reference Sequence project (RefSeq) provides sequence standards for the naturally occurring molecules of the central dogma, from chromosomes to mRNAs to proteins. RefSeq standards provide a foundation for the functional annotaion of the human genome and a stable reference point for mutation analysis, gene expression studies, and polymorphism discovery. (http://www.ncbi.nlm.nih.gov/LocusLink/refseq.html)
[4] UniGene is an experimental system for automatically partitioning GenBank sequences into a nonredundant set of gene-oriented clusters. Each UniGene cluster contains sequences that represent a unique gene, as well as related information such as the tissue types in which the gene has been expressed and map location. (http://www.ncbi.nlm.nih.gov/UniGene/index.html)
REFERENCES
- ↵S.F. AltshulW. GishW. MillerE.W. MyersD.J. Lipman(1990) J. Mol. Biol. 5:403–410.
- ↵D.A. BensonM.S. BoguskiD.J. LipmanJ. OstellB.F. OuelletteB.A. RappD.L. Wheeler(1999) Nucleic Acids Res. 27:12–17.
- ↵M.S. BoguskiT.M. LoweC.M. Tolstoshev(1993) Nat. Genet. 4:332–333.
- ↵F.S. CollinsL.D. BrooksA. Chakravarti(1998) Genome Res. 8:1229–1231.
- ↵E. LaiJ. RileyI. PurvisA. Roses(1998) Genomics 54:31–38.
- ↵E. Masood(1999) Nature. 398:545–546.
- ↵M. OlsonL. HoodC. CantorD Botstein(1989) Science 245:1434–1435.
- ↵R.J. PetersonD. GoldmanJ.C. Long(1999) Hum. Genet. 104:177–187.
- ↵S.T. SherryH.C. HarpendingM.A. BatzerM. Stoneking(1997) Genetics 147:1977–1982.