
Identification and Characterization of a Homozygous Deletion Found in Ovarian Ascites by Representational Difference Analysis
Abstract
We have performed representational difference analysis (RDA) on DNA from tumor cells and normal fibroblasts isolated from the ascites of a patient with ovarian cancer. Five of six products of the RDA were homozygously deleted from the tumor DNA. One of these products has been characterized and identifies a homozygous deletion of ∼6.9 Mb at chromosome 9p21 in the original ovarian tumor material. This deletion encompasses CDKN2A (p16), CDKN2B (p15), and IFN-α. PCR analysis of other tumor cell lines using the novel STS based on the RDA product has shown it to lie between IFN-α and p16, and to identify the distal extent of a homozygous deletion in another ovarian cancer cell line. These data provide further evidence for a tumor suppressor locus distinct from, but mapping close to, p16 on 9p21. Cytogenetic analysis using comparative genomic hybridization (CGH) performed on the same primary tumor confirmed a loss of material from chromosome 9p. However, the CGH technique had neither the resolution nor the sensitivity to define a subregion of homozygous loss.
[The GenBank accession no. for this sequence is AF113912.]
Ovarian cancer is the fifth most common cause of death from cancer among women. It kills 80% of those 5000 diagnosed in the United Kingdom every year, primarily because of its late presentation. The majority of patients present with stage III and IV disease, for which the 5-year survival rates are 17% and 5%, respectively (Chang et al. 1994). It is clear that much more needs to be understood of the biology of this disease before significant advances in current diagnosis and treatment can be made. At the genetic level, several oncogenes have been shown to be activated, amplified, or overexpressed in ovarian cancer, including c-erbB2(HER2/neu) (Slamon et al. 1989), Ki-ras, H-ras, c-myc (Yokota et al. 1986), and c-fms(Bast et al. 1993; for reviews, see Chuaqui et al. 1997; Steel and Gabra 1997). However, relatively few recessively acting genes, or so-called tumor suppressor genes, have been directly implicated in the molecular pathology of ovarian cancer. Mutation of p53 has been observed in ∼52% of tumors (Teneriello et al. 1993), nm23expression has been lost or changed in 21.7% of tumors (Mandai et al. 1995), and E-cadherin (CDH1) is mutated in small numbers of ovarian tumors (Risinger et al. 1994). In familial cases of ovarian cancer, mutation of BRCA1 has been seen in only 10% of tumors (Merajver et al. 1995; Takahashi et al. 1995) and BRCA2 in even fewer (Takahashi et al. 1996).
Loss of both copies of a gene involved in regulation or control of cell growth or proliferation is detectable through loss of heterozygosity (LOH) studies. LOH analyses of ovarian blood/tumor pairs has revealed many regions of loss in ovarian tumors (for review, see Black et al. 1998). However, none of these studies has led directly to the identification of novel tumor suppressor genes. This is in part due to the ambiguous nature of the results of LOH analyses of ovarian tumors. Contaminating stromal tissue and contradictory or low-significance results can make it difficult to define a restricted region in which a novel tumor suppressor gene is located.
For other tumor types, regions of homozygous loss have proved very valuable for the identification of tumor suppressor genes, as the deletions are discrete and provide a limited and precise chromosomal interval for positional cloning. However, to date, the majority of such genomic lesions described both in ovarian and other cancers have been in the DNA of tumor cell lines, which is itself subject to in vitro karyotypic evolution. Techniques such as fluorescence in situ hybridization (FISH), Southern analysis, and dense microsatellite mapping can be used to detect homozygous deletions in primary tissue containing contaminating normal tissue (Cairns et al 1995). However, these methods require a high density of markers within a limited genomic region.
The technique of representational difference analysis (RDA) enables the whole genome of a tumor sample to be compared to its normal counterpart and, for regions of loss or amplification, to be isolated directly. First described by Lisitsyn (1993), the method uses PCR-generated “representations” of the genomic DNA starting materials to facilitate kinetic enrichment of sequences present in only one of the DNA sources.
We have applied the method of RDA to purified populations of tumor and normal fibroblast cells derived from a specimen of ascitic fluid from a patient with ovarian cancer, to identify regions of homozygous loss directly from patient material. Malignant ovarian ascites have been shown to share many of the histological and antigenic properties of the primary tumor (Hamilton et al. 1983; Provencher et. al 1993) and should contain genomic rearrangements and deletions associated with a poorly differentiated, highly malignant cellular phenotype. In most cases reported previously, RDA has been applied to tumor cell lines and their normal counterpart. By using primary material, we avoided cloning sequences that may have been lost through in vitro karyotypic evolution.
After performing RDA we have isolated and cloned several novel sequences that were homozygously deleted from malignant ascitic cells but were present in normal fibroblasts. One of the clones maps to 9p21, and we show that it is derived from a large deletion that encompasses not only the previously described tumor suppressor genesCDKN2A/p16 and CDKN2B/p15 but also other potential tumor suppressor genes.
We have also analyzed the primary material by CGH to identify gross rearrangements and deletions within the tumor and to provide a comparison with results obtained from RDA.
RESULTS
Representational difference analysis of malignant and fibroblast cells from a patient with ovarian cancer was carried out. Three rounds of kinetic enrichment were performed with tumor DNA in 100-fold excess. The products were subcloned and sequenced (Fig. 1). Six unique products were examined in more detail; five were shown by PCR and Southern blot analysis (data not shown) to be absent from the tumor while being present in the normal genomic counterpart (Fig.2), and therefore lay within a homozygous deletion specific to the tumor.
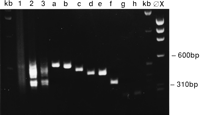
RDA results: difference products and cloned products. (Lanes1–3) Difference products after first, second, and third rounds of enrichment. (Lanes a–h) Results of PCR amplification of inserts of cloned RDA products.
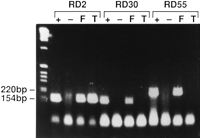
Analysis of RDA products: PCR of original material. PCR using primers derived from RDA products RD2, RD30, and RD55 on original fibroblast DNA (F); original tumor material (T); or genomic DNA from another individual (+); (-) Control reaction with no template DNA.
PCR on a monochromosome hybrid panel with primers derived from one of the products, RD55, showed that it mapped to chromosome 9. PCR with RD55 on cell lines derived from the original ascites specimen (PEO1, PEO4, PEO6, PEO1CDDP; see Methods) confirmed it to be homozygously deleted. PCR with RD55 on a panel of DNA samples from different human tumor cell lines showed it to be homozygously deleted in 2 of 17 cases. Mapping on a radiation hybrid (RH) panel of chromosome 9 placed the RDA product between α-interferon (IFN-α) and CDKN2 (p16) with odds of 1000:1 (Bouzyk et al. 1997).
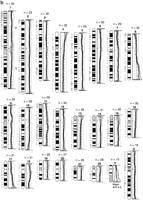
STS markers from 9p21.3 to pter were used to characterize the deletion in the original primary malignant ovarian material; the data are summarized in Figure 3. The same markers were also used to define the extent of the deletions in the tumor cell lines (Table 1).

Chromosome 9 map. Map of part of human chromosome 9p showing the status of DNA markers and gene sequences in PEO4 tumor DNA. The extent of the homozygous deletion in the ovarian tumor is indicated in bold.
PCR Analysis of Markers on Chromosome 9p in Original Tumor and Fibroblast DNA and in Cancer Cell Lines
The original tumor material and the cell lines derived from it (PEO1, PEO4, PEO6, PEO1CDDP) all had a deletion that encompassed IFN-α, p16, and p15. This deletion is flanked by the markers D9S162 and D9S171 and, from the RH map described by Bouzyk et al. (1997), could be as large as 6.9 Mb.
In addition to those cell lines derived from the original ascites specimen, 5 of 17 of the independent tumor cell lines characterized were homozygously deleted for p16 exon 2. Deletions of this region in some of these cell lines have been reported previously (Chenevix-Trench et al. 1994; Shih et al. 1997), and it confirms these results. In two of these cell lines, MCF7 and 59M, the deletion is confined by RD55 distally and D9S1748 proximally, and does not include p15 (CDKN2B). This distance is estimated to be <400 kb (Weaver-Feldhaus et al. 1994). Cell line SKOV3 was deleted for both p15 and p16, subtended by the markers D9S966 and RD55. These markers are all contained within a single CEPH mega-YAC with an estimated insert size of 1200 kb (Bouzyk et al. 1997). OVCAR5 was shown to be deleted for both p15 and p16 but not IFN-α. It is deleted for RD55, which places the RDA product proximal to IFN-α. Toward the centromere, the deletion in this cell line extends as far as, and includes D9S966. The next closest marker mapped is D9S171, which is retained but lies the other side of a gap in the RH map. This deletion could therefore be as large as 4.6 Mb. The deletion in MDA.MB.231 was shown to be even larger than that identified in the original tumor used in the RDA experiment and extends as far as D9S265 proximally, giving a maximum size estimate of ∼9 Mb.
Cytogenetic Analysis
Comparative genomic hybridization analysis of the original tumor material was carried out (see Methods). The ratios of fluorescent signal of tumor to normal DNA revealed multiple copy number abnormalities in this tumor (Fig. 4a,b). These are summarized in Table 2. The large homozygous deletion identified through RDA could be seen as a loss of material from 9pter to 9p12.
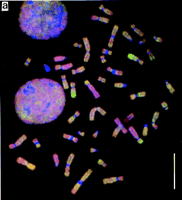
CGH results for PEO4 tumor DNA. (a) A digital image of a typical metaphase. Regions that are over-represented in the tumor are visualized as predominantly green, whereas regions that are under-represented or deleted from the tumor are seen as predominantly red. Bar, 10 μm. (b) The quantitative digital image analysis of fluorescence intensity ratios next to a diagram of the chromosome. The mean ratio (thick line) and ±1 s.e. (thin lines) of measurements from n metaphases for each chromosome are shown from pter to qter (top to bottom). The baseline value (1.0) representing the mean green-to-red ratio for the entire metaphase is shown as a broken line, and ratios 0.5 and 1.5 as dotted lines. Regions approaching a ratio of 0.5 were taken as losses; regions having a ratio >1.5 were taken as gains.
Summary of Chromosomal Gains and Losses Observed by CGH on PEO4 Tumor DNA
DISCUSSION
In 1994, Kamb et al. reported the localization of a novel tumor suppressor gene to chromosome 9p21 through mapping of homozygous deletions in melanoma cell lines. The gene,p16/CDKN2A, was shown to be homozygously deleted at high frequency in many other cell lines derived from different tumor types (Nobori et al. 1994). It was shown to be a member of a family of cyclin-dependent kinase (CDK) inhibitors that are important in normal cell-cycle regulation (Serrano et al. 1993).
Conflicting views have been presented however, as to the importance of p16 as a multitissue tumor suppressor gene. For example, although 74% of melanoma cell lines analyzed had homozygous deletions or intragenic mutations within p16, a frequency of only 19% was observed in primary melanomas, suggesting that deletions in p16 are necessary for growth of melanoma cells in culture and may not be so important in carcinogenesis in vivo (Walker et al 1998).
Despite the reported high levels of LOH at 9p21 in many different tumors (Caldas et al. 1994; Puig et al. 1995; Williamson et al. 1995), the inactivation of the second allele of p16 through microdeletion or point mutation as predicted by Knudson’s two-hit hypothesis has only been found in a limited number of tumor types, predominantly those of the pancreas (Caldas et al. 1994). However, homozygous deletions have been found in a growing number of primary tumors of different tumor types in the 9p21 region that harbors this gene (Cairns et al. 1995), suggesting that this is the predominant mechanism for gene loss. There is also evidence for inactivation of the second copy of the gene by methylation in some common cancers (Herman et al. 1995; Merlo et al. 1995), although this does not include ovarian carcinoma.
Although relatively high levels of homozygous deletion have been reported in ovarian tumor cell lines—4/8 (Rodabaugh et al. 1995) and 5/11 (Shih et al. 1997)—the frequency of homozygous deletions of this region reported in primary ovarian cancers is low—2/88 (Shih et al. 1997) and 1/67 (Campbell et al. 1995). Some groups claim higher frequencies of homozygous loss in primary tumors detected through multiplex PCR analysis (Ichikawa et al. 1996). However, LOH for this region is relatively high—48% (Campbell et al. 1995) and 45% (Chenevix-Trench et al. 1997)—but the number of gene-inactivating point mutations found in p16 in tumors with LOH is very small (Rodabaugh et al. 1995; Schultz et al. 1995). In addition no studies so far have detected inactivation of p16 by methylation in ovarian tumors (Ichikawa et al. 1996; Shih et al. 1997). Taken together, these data imply the involvement of another locus located around 9p21 in the genesis of ovarian cancer. A failure to find inactivation of either p16 or p15 in cutaneous malignant melanoma in which LOH frequencies are high also suggests that there may be another potential tumor suppressor gene in the region (Puig et al. 1995).
Our results from the analysis of ovarian cell lines and primary malignant ovarian tumor cells agree with a hypothesis for an additional tumor suppressor gene at 9p21. In several of the cell lines characterized (OVCAR5, PEOs), the homozygous deletions extend for a large distance beyond the defined tumor suppressor genes p16and p15. Interestingly the deletion detected in primary malignant ascites cells by RDA also extended across the same region, strengthening the case for the involvement of this additional gene in carcinogenesis in vivo.
The possible candidate gene IFN-α is excluded, as IFN-α is retained in OVCAR5 and the distal extent of the deletion is defined by RD55. This suggests that a novel tumor suppressor may lie either within the 400-kb region distal top16 limited by the novel RDA product RD55, or within 2.8 Mb proximal to p16, defined by the marker D16S171. Loss of function of this novel gene may account for the high frequency of LOH ob served at 9p21 in ovarian tumors that do not show inactivation of p16 by deletion, mutation, or methylation.
CGH analysis of DNA from the malignant ascites of tumor PEO4 reveals multiple regions of amplification and loss. A total of 20 copy number abnormalities (CNAs) were identified (see Table 2). There is a clear reduction in the ratio of green to red signal on 9p, from 9pter up to and including 9p21. This region covers the homozygous deletion detected by RDA at 9p21.3, but the loss of both copies of the chromosome at this point cannot be distinguished from the immediate proximal and distal regions, which we know to be conserved within the tumor from STS mapping (see Fig. 3). The size of the deletion on 9p21, detected by RDA at ∼7 Mb, is outside of the limits of resolution of CGH. This result highlights the limitations of CGH in the detection of biologically significant chromosomal deletions.
To our knowledge, this is the first report of CGH analysis of malignant ovarian ascites. The degree of CNA in this tumor demonstrated by CGH analysis is not unusual for an advanced stage ovarian epithelial carcinoma. Iwabuchi et al. (1995) report CNAs of between 0 and 30 in 44 different ovarian tumors, with high-grade tumors exhibiting, on average, more abnormalities than lower grade tumors. Twenty-three of the 24 well or moderately differentiated epithelial ovarian carcinomas analyzed by Tapper et al. (1997) show CNA ranging from 1 to 17, andWolff et al. (1997) report CNAs of between 1 and 31 in their panel of 24 ovarian carcinomas of different subtypes. Of the 17 distinct CNAs we observed in PEO4, most have been observed previously, including gains at 8q23–8q24, 11q13, and 17q22–qter and losses at 4q22–qter, 5p14, 6q16–qter, 9pter–p21, 13q, 16q22–qter, 17p, 17q12, 18q22–qter, and X. The following gains and losses of material had not been reported previously: +4p15, −9q21, −9q22.3–33, −11p15–pter, and −22q13–qter.
RDA has been used previously in the identification of homozygous deletions that have led toward the cloning of several novel tumor suppressor genes including BRCA2 (Schutte et al. 1995; Wooster et al. 1995), FHIT (Ohta et al. 1996), and MMAC/PTEN(Li et al. 1997; Steck et al. 1997). When we performed RDA on primary ovarian tumor DNA, five distinct products were isolated that were shown to be homozygously deleted from the tumor but retained within the fibroblast DNA from the same patient. One of these products has been shown to be part of a large homozygous deletion of chromosome 9p21, which encompasses IFN-α, p16, andp15 and possibly one or more additional unidentified tumor suppressor genes. We hope that characterization of the other four RDA products will lead to the identification of other novel loci that house tumor suppressor genes involved in ovarian cancer.
METHODS
Clinical Material
An ascites specimen was taken from a patient with advanced serous epithelial ovarian cancer. The cell line PEO1 was derived from the malignant cells in this sample (Wolf et al. 1987). Ascites taken from the patient after relapse some 10 months later was used to derive the cell lines PEO4, PEO6, and PEO1CDDP as described previously (Wolf et al. 1987; Langdon et al. 1988). An additional sample of ascites was cultured and trypsin/versene added until the fibroblast cells just became detached. These fibroblast cells could then be removed selectively and cultured separately. Several rounds of differential trypsinization were performed until distinct and pure fibroblast and tumor cell populations were obtained. DNA was isolated from the two cell types (Nucleon DNA extraction kit, ScotLab), and it was from these samples that representations were derived for the difference analysis procedure.
Cell lines
PEO14, PEO16, PEO23, PEA1, and PEA2 are all cell lines derived from epithelial ovarian tumors (Wolf et al. 1987; Langdon et al. 1988); PEA1 and PEA2 are derived from the same patient. OVCAR3, OVCAR4, OVCAR5, A2780, 59M, OAW42, OAW28, and SKOV3 are all human ovarian cancer cell lines obtained from the American Tissue Culture Collection (ATCC). HeLa Ohio was a gift from A. Cranston (Queen’s University, Belfast, UK); HCT116, a colorectal cancer cell line, was a gift from S. Farrington (MRC–HGU), ZR75.1, MCF7, and MDA.MB.231 are breast cancer cell lines available from the ATCC.
RDA
RDA was performed exactly as described by Lisitsyn et al. 1993 (a detailed protocol was supplied by N. Lisitsyn and has now been published in Lisitsyn and Wigler 1995).
Briefly, 2 μg of DNA from the two cell populations was digested with BglII and then ligated to the first primer pair set (R Bgl 24 and R Bgl 12) that had been annealed previously. R Bgl 24 was used as a primer for PCR on 40 ng of the catch-linkered templates, to generate tester (fibroblast) and driver (tumor) amplicons. The first set of catch linkers was removed from the amplicons by restriction endonuclease digestion with BglII. The tester amplicon was gel-purified and the DNA re-extracted from the agarose using a Qiaquick gel extraction kit (Qiagen Ltd., Crawley, UK). One microgram of purified tester amplicon was ligated to annealed primer pair set 2 (J Bgl 24 and J Bgl 12). Subtractive hybridization was performed using 40 μg of the prepared driver amplicons and 400 ng of the prepared tester. Selective amplification of self-reannealed tester fragments was carried out by performing 10 cycles of PCR using J Bgl 24 as a primer. This produced the first-round difference product. Remaining single-stranded DNA (ss-DNA) molecules were removed by digestion with mung bean nuclease (New England Biolabs), before performing a further 20 cycles of PCR. Linkers were removed from the first-round difference product again by digestion with BglII. The third primer pair set of annealed oligonucleotides (N Bgl 24 and N Bgl 12) was ligated to the digested difference product. A further two rounds of subtractive hybridization and selective amplification were carried out, using primer pair set 2 as catch linkers on the second-round product to generate the third round difference product.
Cloning of RDA Products
Products of the final round of PCR were digested withBglII and shotgun-cloned into BglII-digested, dephosphorylated pBS plasmid vector (Stratagene), a gift from Tony Brookes (MRC–HGU). Cloned products were sequenced using T3 and T7 as primers in either a dideoxy sequencing reaction (Sequenase) with35S incorporation, or they were cycle-sequenced usingTaq FS fluorescent dideoxy termination mix (ABI), and then analyzed on a 373 automated ABI sequencing apparatus.
PCR Analysis
Specific PCR primers were derived from the sequences. For RDA product RD55, these were 5′-TACAACAGGATCAAGAAGGC-3′ and 5′-AGAGAAACCGAGAAGAAACC-3′. PCR conditions were as follows: 50 ng of template, 300 ng of primer, 50 mm KCl, 10 mmTris (pH 9), 0.1% Triton X-100, 1.5 mm MgCl2, 200 μm dNTPs, and 1 unit of Taq Polymerase (ICRF). Cycling conditions were as follows: 93°C for 1 min ×1; 70°C for 30 sec, 72°C for 30 sec, 91°C for 30 sec ×2; 68°C for 30 sec, 72°C for 30 sec, 91°C for 30 sec ×2; 66°C for 30 sec, 72°C for 30 sec, 91°C for 30 sec ×2; 64°C for 30 sec, 72°C for 30 sec, 91°C for 30 sec ×2; 62°C for 30 sec, 72°C for 30 sec, 91°C for 30 sec ×2; 60°C for 30 sec, 72°C for 30 sec, 91°C for 30 sec ×20; 72°C for 5 min.
Human monochromosome somatic cell hybrid panel was from the HGMP Resource Centre (Hinxton, Cambridge, UK). STS markers p16 exon 2, p15 exon 2, and c.1b were a kind gift from Kathy Williamson (MRC HGU) and are described in Kamb et al. (1994). Primers for IFN-α and TYRP1 were a gift from Cathy Abbot (University of Edinburgh, UK) (sequence is held by GenBank). Primers for other STS markers—D9S288, D9S178, D9S285, D9S156, D9S157, D9S162, D9S1749, D9S1748, D9S1752, D9S942, D9S171, D9S265, D9S272, D9S176, D9S1748, D9S1747, D9S942, and D9S1752—were synthesized by ICRF support services according to the sequences held in GenBank.
CGH
CGH analysis was carried out on metaphase spreads prepared from an unsynchronized culture of fresh phytohemaglutinin-stimulated lymphocytes from a healthy male (46XY) using standard procedures of hypotonic treatment and methanol/acetic acid fixation (3:1). Total genomic tumor DNA and normal total genomic DNA were labeled by nick translation with fluorochrome-conjugated nucleotides FITC11–dUTP (Amersham, UK) and Texas Red–5-dUTP (Dupont, Boston, MA), respectively. Another sample of total genomic DNA from a normal female was labeled with FITC-11–dUTP as a control. The size of the resultant labeled DNA fragments was checked as being in the range of 600–1700 bp.
Hybridization experiments were carried out as described previously (Kallioniemi et al. 1994) with minor modifications. Briefly, normal lymphocyte metaphase chromosomes were prepared by incubation at 37°C in 100 μg/ml RNase, 2× SSC for 1 hr. They were then dehydrated through an ethanol series (70%, 90%, 100%) and denatured in 70% formamide/2× SSC at 70°C for 3 min. After quenching in cold 70% ethanol, the dehydration was completed by passing slides through 90% and 100% ethanol.
Equal amounts of previously labeled tumor and normal DNA (400 ng), together with 5 μg of unlabeled Cot1 DNA (GIBCO), was precipitated, dried, and resuspended in 10 μl of hybridization mix (50% formamide, 10% dextran sulfate, 2× SSC, 0.1% Tween 20). This was then denatured at 70°C for 5 min and applied immediately to the lymphocyte metaphase preparations. After hybridization at 37°C for 48 hr the slides were washed four times for 3 min in each of 50% formamide/2× SSC, and 2× SSC at 45°C and in 0.1× SSC at 60°C. The slides were then transferred to 4× SSC/0.1% Tween 20 and counterstained with 1.5 μg/ml DAPI in Vectashield antifade solution (Vector Labs, Peterborough, UK).
The slides were analyzed using a Zeiss Axioplan microscope equipped with a CCD camera and a filter system. Excitation of the individual fluorochromes was done by means of a single band pass excitation filter in a computer-controlled wheel. The information was processed using a Sun Sparc10 workstation and the red-to-green ratios were calculated using X-woolz software developed by J. Piper (Piper et al. 1995). Twenty-five to 30 metaphases were captured, and the fluorescence ratios for each chromosome type were derived from the three color images. The chromosomes were digitally segmented and the interchromosomal background removed from the chromosomal fluorescence. The fluorescence intensities along the length of all the chromosomes in each metaphase were then calculated and normalized so that the overall green-to-red ratio for each metaphase was 1.0. Thereafter, a drop in standard deviation below 1.0 was defined as a loss in DNA copy number and a rise above 1.0 as a gain in that particular region of the tumor genome. Only images that showed a uniform fluorescence in both green and red signal across the metaphase were analyzed.
Acknowledgments
We thank Nick Hastie for his ideas and advice, the MRC HGU Reprographics department for figures, K. Williamson and C. Abbott for primers, A. Cranston and S. Farrington for cell lines, S. Langdon and A.J. Brookes for materials and advice, and Georgia Chenevix-Trench for her comments. This work was funded by the ICRF and the UK MRC.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL vivienne{at}hgu.mrc.ac.uk; FAX 44-131-343-2620.
-
- Received October 13, 1998.
- Accepted December 10, 1998.
- Cold Spring Harbor Laboratory Press








