
A Resource of Mapped Human Bacterial Artificial Chromosome Clones
- Vivian G. Cheung1,
- Heather L. Dalrymple1,
- Sandya Narasimhan1,
- Jason Watts1,
- Gregory Schuler2,
- Anton K. Raap3,
- Michael Morley1, and
- Alan Bruzel1
- 1Department of Pediatrics, The Children's Hospital of Philadelphia, University of Pennsylvania, Philadelphia, Pennsylvania 19104 USA; 2National Center for Biotechnology Information, National Institutes of Health, Bethesda, Maryland 20894 USA; 3Department of Molecular Cell Biology, Leiden University Medical Center, Leiden, The Netherlands
Abstract
To date, despite the increasing number of genomic tools, there is no repository of ordered human BAC clones that covers entire chromosomes. This project presents a resource of mapped large DNA fragments that span eight human chromosomes at ∼1-Mb resolution. These DNA fragments are bacterial artificial chromosome (BAC) clones anchored to sequence tagged site (STS) markers. This clone collection, which currently contains 759 mapped clones, is useful in a wide range of applications from microarray-based gene mapping to identification of chromosomal mutations. In addition to the clones themselves, we describe a database, GenMapDB (http://genomics.med.upenn.edu/genmapdb), that contains information about each clone in our collection.
The goal of this project is to provide a collection of large-insert clones that are anchored to STS markers. The great success of the human genome project has provided high-quality BAC libraries, dense maps of genetic markers, genome sequences, and high throughput technologies such as DNA microarrays. The human genome project has also provided well-characterized cDNA clones that are used in constructing cDNA microarrays for gene expression profiling. However, well-characterized clones for genomic microarrays, which would be the tool of choice for gene mapping (Cheung et al. 1998) and identification of chromosomal mutations (Pinkel et al. 1998), are not available. Genome centers are concentrating efforts to make available highly accurate runs of genomic sequences; it is not their goal to provide characterized genomic clone sets (Collins et al. 1998).
This paper describes ordered sets of genomic clones from eight human chromosomes. These mapped genomic clones are available as characterized glycerol stocks. Information about each clone is publicly available on the World Wide Web in our database, GenMapDB (http://genomics.med.upenn.edu/genmapdb). The combination of the clones and the database will provide resources for construction of genomic microarrays, physical mapping, comparative genomics, and as probes for fluorescent in situ hybridization.
RESULTS
Identification of STS-Linked Human BAC Clones
Our BAC clones are anchored to mapped STS markers that span eight human chromosomes at average intermarker distances of ∼1 Mb. We use the Roswell Park Cancer Institute (RPCI) human male BAC library as starting material (Oseogawa et al. 1998) (http://bacpac.med.buffalo.edu/human_bac.html). The STS markers used for selecting BAC clones are chosen from the GeneBridge4 (GB4) radiation hybrid (RH) panel (Gyapay et al. 1996) and from the physical map of chromosome X (Nagaraja et al. 1997). The probes used to identify corresponding BAC clones are PCR amplicons from these STS markers. Because the goal of our project is to create a resource of mapped BAC clones, we use very stringent selection criteria in placing clones on the map.
The mapping of the BAC clones to STS markers was a four-step procedure consisting of two rounds of hybridization (steps 1 and 2) and two rounds of PCR verification (steps 3 and 4) (Fig. 1). All the clones in our map passed these steps. Clones identified by hybridization, but failing PCR verification, were not included in our map. This selection criterion minimizes false-positive clones that can result from screening methods relying solely on either DNA–DNA hybridization or PCR.
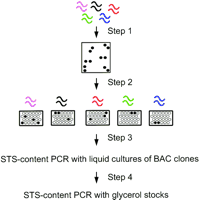
Schematic of the procedure for STS-based BAC mapping. (Step 1) Radiolabeled STS-probes are multiplexed and hybridized onto RPCI-11 segment 2 human male BAC library high-density filters. Liquid cultures of single colonies from positive clones are used to make dot blots. (Step 2) Individual probes are then hybridized onto the dot blots for verification and matching of clones to STS probes. (Steps 3 and 4) The mapped clones are then verified by two separate rounds of STS-content PCR, one round with liquid cultures of BAC clones (step 3) and another round with glycerol stocks of the mapped BAC clones (step 4).
In the first step, a set of radiolabeled STS probes (up to10 probes per set) were multiplexed and hybridized onto the RPCI high-density BAC filters. Positive clones were identified from strong signals on the autoradiograms. Single-colony isolates of the positive clones were grown in liquid cultures that were then spotted onto nylon membranes. In the second step, these dot blots were hybridized with the individual radiolabeled STS probes used in the first step. This step allowed us to verify the identity of the clones from the mixture used in the first step and to match the clones to their corresponding STS probes. About 70% of the clones that were positive in step 1 were also positive in step 2. Because the high-density BAC filters were used repeatedly (up to seven times), some false-positive clones might be positive signals that were carried over from previous hybridizations. A small portion of false-positive clones were adjacent to true positive clones, these might be the result of contamination in the filter preparation process.
The clones that mapped to each STS by hybridization were then verified in steps 3 and 4 by two separate rounds of PCR amplifications. In step 3, STS-content PCR reactions were performed with the liquid cultures of the BAC clones as templates. Step 4 was another round of PCR amplification, but the templates used were the glycerol stocks prepared from the liquid cultures. This step ensured that there was no bookkeeping error in preparing the glycerol stocks. This is critical because the glycerol stocks are the reagents for long-term storage of the mapped clones and for their distribution. About 80% of the clones that were positive in the hybridization steps were positive in the PCR-verification steps. For each PCR, the sizes of the amplicons from the clone-based PCR were compared with the sizes of genomic DNA-based PCR to detect and minimize spurious amplifications. In addition, the sizes of the amplicons were checked against the published sizes of the STSs. Glycerol stocks of the mapped clones are available through Research Genetics, Inc. (Huntsville, AL; www.resgen.com).
Fluorescent In Situ Hybridization
A small number of the mapped BAC clones were also verified by fluorescent in situ hybridization (FISH). Figure 2shows three BAC clones hybridizing to three consecutive cytogenetic bands on chromosome X as predicted by their map locations.
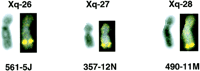
FISH hybridization showing three BAC clones that mapped to Chromosome X. The three clones are 561-7J that mapped to DXS7274 (124 Mb from Xpter, Xq26 signal), 357-12N that mapped to DXS7503 (132 Mb from Xpter, Xq27 signal), and 490-11M that mapped to DXS7378 (151 Mb from Xpter, Xq28 signal).
Mapped Human BAC Clones
The resource described here is composed of clones from eight human chromosomes, chromosomes 17, 18, 19, 20, 21q, 22q, X, and Y. For the acrocentric chromosomes, clones were not mapped to the p arms. There are a total of 408 STS markers on the map with 759 mapped BAC clones. Of the 408 STS markers, 137 are known genes and many of the remaining markers are expressed sequence tags. Table 1 lists the distribution of clones for each chromosome. For chromosomes 17, 18, 19, 20, 21q, 22q, and X, we achieved an average intermarker distance of 1.1–1.7 Mb (see Table 1). However, for chromosome Y, the average intermarker distance is 5.2 Mb. In the total map, there are eight gaps that are >5 Mb; three are on chromosome Y, one is found on each of the chromosomes 17, 18, 19, 20, and 21q. Most of the gaps are in genomic regions that lack appropriate RH markers (markers with high lod scores, free of repetitive sequences). Selecting anchoring markers for chromosome Y was difficult because there is no RH map for chromosome Y and there are large regions of repetitive sequences on chromosome Y. As more information becomes available on the genomic architecture of chromosome Y, we will be able to fill in the map to achieve a denser coverage.
Distribution of Mapped BAC Clones for Each Chromosome
Database
Detailed information about our BAC map, including the STS-markers and their corresponding BAC clones is publicly available on the World Wide Web from our database, GenMapDB (http://genomics.med.upenn.edu/genmapdb). An example of a search query with GenMapDB is shown in Figure 3.
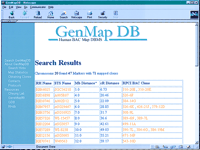
Physical mapping information for chromosome 20. Shown is a part of a table in our database GenMapDB (http://genomics.med.upenn.edu/genmapdb), which includes STS markers, their physical addresses, and BAC clones corresponding to each STS-marker.
GenMapDB is a relational database, designed to serve as a resource of the mapped BAC clones. It allows users to retrieve clones that map to an entire chromosome or a region of a chromosome. The output for each query includes information about clones mapped to the queried genomic interval along with the STS content of these clones. We have made additional information about the STS markers available through hyperlinks to genome databases such as Rhdb (http://www.ebi.ac.uk/RHdb) and GeneMap99 (http://www.ncbi.nlm.nih.gov/genemap99/) (Deloukas et al. 1998).
Accuracy of the Mapped Clones
The accuracy of our map depends on factors such as the accuracy of the map position of the markers used as anchors for the BAC clones, the precision of the techniques for STS-based BAC mapping, and the quality of the BAC clones. Whenever possible, we used RH markers that have a high lod score (lod >3.0). Physical distances between markers were used with the understanding that they are based on interpolations from distances of framework markers in the RH map. For chromosome X, instead of RH markers, we used mostly STS markers on the YAC-based map, hence the map location of the markers should be quite accurate. As for the precision of the BAC mapping method, we are confident that the clone positions are accurate. Each clone is anchored to an STS marker after two rounds of hybridization with the marker as probe and two separate rounds of amplification with the clone as template under stringent PCR conditions. Lastly, the quality of this BAC map also depends on the quality of the clones. In this project, the second round of hybridization and the two PCR verification steps were performed with single colony cultures to avoid polyclonality. The BAC clones should have a fairly low rate of chimerism as compared with YAC clones. FISH analysis of a large set of these clones will establish the chimerism of the BACs used.
DISCUSSION
Here, we describe a 1-Mb BAC map for eight human chromosomes and the database that stores the mapping data. This project is ongoing in our laboratory. As clones are mapped, we will deposit them in the database, GenMapDB (http://genomics.med.upenn.edu/genmapdb), until we achieve a 1-Mb map for the entire genome. The entire map will include at least 3500 clones. The average insert size of the RPCI BAC clones is 168 kb (http://bacpac.med.buffalo.edu/human_bac.html). Thus, the DNA in the clones of our final map will represent ∼600 Mb or 20% of the entire human genome.
Next, we will further characterize the mapped clones by providing molecular fingerprints (HindIII) and end sequences of the clones. As we perform HindIII digests on the clones, the set of restriction fragments corresponding to each clone will be made available on our website. The HindIII fingerprints will serve as a reliable marker for users of the clones to verify the identity and integrity of the clones. In addition, the clones will be FISH mapped to confirm their genomic addresses and to ensure that each clone maps only to one genomic location.
The mapped BAC clones will be useful in a variety of projects such as gene mapping, cytogenetic studies, and comparative genomics. In addition, this resource allows the determination of the genomic addresses of DNA fragments isolated by the gene mapping technique known as direct identical-by-descent (IBD) mapping (Cheung et al. 1998). For direct IBD mapping, the mapped clones will be placed in map order onto a DNA microarray. Then, the DNA fragments isolated with genomic mismatch scanning (Nelson et al. 1993; Cheung and Nelson 1998; Cheung et al. 1998; McAllister et al. 1998) will be labeled and hybridized onto the genomic microarrays.
When extended to all chromosomes, our genomic DNA microarray can also be used to map the genomic addresses of any DNA fragments and to determine chromosomal mutations similar to traditional comparative genomic hybridization (CGH). Compared with traditional CGH, BAC-based microarray hybridizations improve the mapping resolution from 5 Mb to 1 Mb (Pinkel et al. 1998). In addition, microarray-based hybridizations can be automated, whereas traditional CGH requires highly skilled personnel to prepare chromosome spreads. A whole genome microarray will be an important tool in analyzing chromosomal changes in cancer cells and other cells in which aberrations such as chromosomal deletions and amplifications are common. The preparation of the BAC clones and the database described in this paper are the first steps in making this microarray a reality.
METHODS
Selection of STSs
All of the STS markers used to construct the maps for chromosome 17–22, and part of chromosome X were obtained from markers collected and mapped by the International RH Consortium. In most cases, markers were selected only from the high confidence RH bins (1000:1 bins). Markers not obtained from RH maps were obtained from Genome Database (GDB, http://www.gdb.org) and the physical map of human chromosome X (Nagaraja et al. 1997).
The STS markers were selected on the basis of several criteria: (1) they are repeat-free, as determined with RepeatMasker (http://ftp.genome.washington.edu/cgi-bin/RepeatMasker); (2) they are greater than 125 bp to ensure efficient radiolabeling by random priming, and (3) the confidence of their genome address is high as determined by high lod scoring (lod>3) in RH maps or prior physical mapping data.
Generation of Radiolabeled STS Probes
Probes were generated by PCR amplifications with oligonucleotide primer sequences obtained from RHdb (http://www.ebi.ac.uk/RHdb/) with primers synthesized by Research Genetics, Inc. (Huntsville, AL). The PCR mixture was composed of genomic DNA (100 ng), 0.4 mmprimers, 200 μm dNTP, 2.5 units of Taq DNA polymerase (Promega), 2.5 mm MgCl2, with 1× reaction buffer (Promega) in a final volume of 50 μl. Amplifications were generally carried out by an initial denaturation at 96°C for 5 min, followed by 30 cycles of 94°C for 45 sec, 55°C, or 57°C for 45 sec, 72°C for 45 sec, and a final extension of 72°C for 5 min. Some amplifications required substantially different PCR conditions (refer to GenMapDB for details). All amplifications were carried out in PTC-100 thermal cyclers (MJ Research). An aliquot of each amplicon was checked by gel electrophoresis and its size compared with the published size of the STS marker. Unincorporated primers and nucleotides were removed with a PCR purification kit (Qiagen).
The purified DNA samples were radiolabeled by random priming. The reaction mixture was composed of 25 ng of denatured STS amplicon, 1× hexanucleotide mix (Boehringer Mannheim), 25 μm dATP, 25 μm dTTP, 25 μm dGTP, 50 μCi [α32P]dCTP and 2 units of Klenow enzyme (Promega) in a total volume of 20 μl. The reaction mixture was then incubated at 37°C for 30 min. The labeled amplicons were passed through a Sephadex G-50 column to remove unincorporated dNTPs. Counts per minute of 32P were measured.
Hybridization
Up to 10 labeled probes (each probe with a specific activity of ∼0.5 μCi/ng) were pooled and hybridized onto one set of RPCI-11 segment 2 Human Male BAC library high density filters. Prior to hybridization, probes were separately preannealed with human Cot-1 DNA (GIBCO-BRL), while the filters were prehybridized with 200 μg/ml salmon sperm DNA in 10% SDS, 7% PEG solution at 65°C for 1 hr. The pool of probes was hybridized onto the filters with fresh 200 μg/ml salmon sperm DNA in 10% SDS, 7% PEG hybridization solution at 65°C overnight.
After hybridization, the filters were washed and allowed to expose films overnight. Signals were then read from the autoradiographs. Because the BAC clones were printed onto the filters in duplicates with eight different duplicate pair orientations, the clones that were positive in one of the eight duplicate pair orientations were scored as positives.
BAC clones corresponding to positive signals were grown overnight on LB plates containing 170 μg/ml chloramphenicol. Single colonies from each plate were then grown in LB liquid cultures with chloramphenicol. From each liquid culture, 2-μl aliquots were spotted onto a nylon membrane to make a dot blot containing positive clones from the previous round of hybridization. Multiple copies of these membranes were made so that each membrane could be hybridized to one probe from the pool of probes used in the first round of hybridization. The hybridization conditions used for these dot blots were identical to those used for the high-density BAC filters. The positive signals from each dot blot allowed matching of the BAC clones to each STS marker.
PCR Verification
The BAC clones that were mapped by two steps of hybridization were verified by PCR amplification of the STS from the clones. PCR conditions used were identical to those described under generation of radiolabeled STS probes.
Glycerol stocks of verified clones were prepared. A second round of PCR was performed using the glycerol stocks as templates to ensure that their STS contents were correct.
FISH Analysis
FISH was performed following standard protocols with biotinylated BAC DNAs hybridized to metaphase chromosomes of a normal male. Unlabeled Cot-1 DNA was used to suppress hybridization to repetitive DNA. Biotin was detected with Streptavidin-Fluorescein and counterstained with 4,6′-diamidino-2-phenylindole.
Acknowledgments
We thank Eric Geelen for performing the FISH experiments. This work was supported by grants from the Merck Genome Institute (V.G.C), National Institutes of Health grants DC00154 and HG01880 (V.G.C).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Note Added in Proof
Since the writing of this manuscript, we have mapped clones for three additional chromosomes, chromosomes 14, 15, and 16. For details, please refer to our database, GenMapDB.
Footnotes
-
4 Corresponding author.
-
E-MAIL vcheung{at}mail.med.upenn.edu; FAX (215) 590-3709.
-
- Received June 10, 1999.
- Accepted August 12, 1999.
- Cold Spring Harbor Laboratory Press








