
Physical and Comparative Mapping of Distal Mouse Chromosome 16
Abstract
Distal mouse Chromosome 16 (Chr. 16) includes a region of conserved linkage with human Chromosome 21 (Chr. 21). Mouse models of Down syndrome based on trisomy of distal Chr. 16 have several phenotypes similar to those seen in human patients and have proven useful for correlating dosage imbalance of specific genes with specific developmental anomalies. The degree to which such findings can be related to Down syndrome depends on how well the conserved synteny is maintained. Twenty-four genes have been mapped in both species and there are no discordancies, but the region could carry hundreds of genes. Comparative sequence represents the ultimate comparative map and will aid in identification of genes and their regulatory sequences. A physical map of the distal 4.5 Mb of Chr. 16 has been assembled as an essential step toward a map of sequence-ready templates. The map consists of 51 YACs and 15 BACs and includes 18 transcripts, 9 of which are mapped for the first time in mouse, and 3 of which are, for the first time, described in either species. YAC fragmentation was used to precisely localize the 49 markers on the map. Comparison of this physical map with that of the corresponding region on Chr. 21 shows conservation not only of gene order but of size in the 3 Mb fromCbr1 to Ets2; distal to Ets2, the human map is expanded.
[The sequence data described in this paper have been submitted to GenBank. See Tables 1 and 2 for accession nos.]
Distal mouse Chromosome 16 (Chr. 16) includes a region of conserved linkage with much of human Chromosome 21 (Chr. 21). This region extends from 21q11.1 to 21q22.3 and is bounded bySTCH (Kao et al. 1994; Reeves et al. 1998), the proximal-most known gene on Chr. 21, and MX, the distal-most known gene on Chr. 16 (Reeves et al. 1997). Twenty-four genes define this conserved linkage with no discordancies in the presence or order of genes in this region. Genes from the 3 Mb of the distal end of Chr. 21 are found in mouse on Chr. 17 and Chr. 10.
The conserved linkage between Chr. 21 and Chr. 16 provides the basis for mouse models for Down syndrome (DS) involving trisomy of all or part of Chr. 16. Animal models are important for the study of a developmental problem that cannot be examined easily in humans. The availability of inbred mice has the added advantage that the effects of gene dosage imbalance can be studied on uniform genetic backgrounds. The Ts65Dn mouse model for DS was generated by creating a translocation of distal Chr. 16 to the centromeric region of mouse Chr. 17 (Davisson et al. 1990). Animals carrying the marker chromosome and two copies of normal Chr. 16 (Ts65Dn mice) are at dosage imbalance for the region from proximal of App to Mx, which includes most of the known genes on Chr. 21. Analysis of such mice has demonstrated a behavioral deficit in the Morris swim maze, a test of learning involving integration of visiospatial cues (Escorihuela et al. 1995;Reeves et al. 1995; Coussons-Read and Crnic 1996). Craniofacial and neuroanatomical anomalies similar to those seen in DS individuals are also observed in Ts65Dn mice (J.T. Richtsmeier, L.L. Baxter, and R.H. Reeves, in prep.). The degree to which the genetic mechanisms producing these phenotypes reflect those seen in human depends on how well conserved syntenies are maintained between the two species.
The YAC contig of distal Chr. 16 will enhance comparative mapping efforts and functional studies using DS models. Gene discovery efforts in this region have focused on human; the resulting trapped exons and 3′ UTRs isolated by direct selection are frequently suboptimal probes for recombinational mapping in mouse but can be mapped on lower complexity targets such as YACs. This physical map provides higher resolution than recombinational maps and allows the first accurate comparison of gene spacing in this region. The YAC map is a useful step in the generation of sequence-ready templates covering distal Chr. 16. Comparative sequence will provide the ultimate comparative map, allowing analysis across species not only of gene coding regions but also gene spacing and regulatory elements.
RESULTS
Contig Construction
A 4.5-Mb contig was assembled from 51 YACs and 15 BACs and provides average depth of coverage of 4.4 YACs per marker, with a range of 1–12 YACs per marker. Twenty-four YAC clones are required to determine the marker order shown in Figure 1. The entire set of Chr. 16 YACs, their sizes, and STS content are available at http://physiology.med.jhu.edu/roger/roger.html. Seventeen YACs are from the large-insert Whitehead Center for Genome Research (WCGR) library (Haldi et al. 1996), 3 are from the St. Mary’s library (Chartier et al. 1992), 9 are from the Imperial Cancer Research Fund (ICRF) library (Larin et al. 1991), and the remaining 22 are from the original WCGR mouse library (Kusumi et al. 1993). On the basis of end-clone recoveries and remapping, the chimerism rate for YACs is ∼50%. Several large YACs containing only one or two Chr. 16 markers are presumed to be chimeric and were not analyzed extensively (data available at http://physiology.med.jhu.edu/roger/roger.html). A minimal tiling path of 11 YACs and 1 BAC (Shizuya et al. 1992) is shown in Figure 1. The contig contains 49 markers over the 4.5 Mb, for an average density of 1 marker per 92 kb.
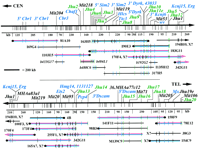
YAC contig spanning 4.5 Mb of distal mouse Chr. 16 from Cbr1to Mx. Twenty-four YACs and four BACs necessary for determination of marker order are shown. Markers are shown at thetop, with transcripts in blue, YAC/BAC end clones in green, and STS markers in black. Vertical lines beneath the marker map indicate fragmentation sites with physical distances in kilobases for selected intervals shown below. Minimal tiling path YACs are shown in red, and fragmented YACs are in blue. BAC clones names are in italics and prefixed by b. ( ) indicates that a marker is missing from a YAC and (‖) indicates that a marker is deleted. Known chimeric YACs have the symbol X at the chimeric end; those likely to be chimeric are indicated by X?. Where YAC orientation is known, centromeric ends are indicated (•). YACs continued from thetop half to the bottom are indicated by arrows. The complete set of 51 YACs and 15 BACs spanning this region are available for viewing at http://physiology.med.jhu.edu/roger/roger.html.
Transcripts
Eighteen of the markers are genes or transcribed sequences, of which 9 were mapped previously on distal Chr. 16 (Fig. 1; Table 2, below). Of the remaining nine markers, Cbr3 (Korenberg et al. 1997), Hlcs (Blouin et al. 1996a), Ttc3 (Ohira et al. 1996), Kcnj15 (Gosset et al. 1997; Ohira et al. 1997), andDscam (Yamakawa et al. 1998) were predicted to map to distal Chr. 16 based on their localization on human Chr. 21. Chaf1was shown to be syntenic to Chr. 16 by in situ hybridization (Blouin et al. 1996b) and is finely mapped here to a position corresponding to that found in human (Blouin et al. 1996b). Dscam was mapped using the mouse cDNA. Hlcs was mapped by PCR (Table1), and the remaining genes were mapped using ESTs matching the human genes as probes (Table 2).
Expressed Sequences
Primers
Three new genes have been identified on the contig. Two are human ESTs that are not known genes. Gene 1131127 matches human genomic sequence at the NotI site LL42SP and, thus, is well-localized on the human map (Ichikawa et al. 1993). In mouse, it lies within 50 kb of Hmg14. ESTs that overlap with1131127 can be used to build a transcript of 1.97 kb in silico, but this transcript has no open reading frame (ORF). This may be due to errors in single-pass sequencing of ESTs.
Another human EST, 43033, is contained in the 3′-noncoding region of human DYRK. Using the coding sequence ofDYRK as probe, a message of 6.1 kb has been observed in numerous human fetal tissues including brain (Guimera et al. 1996). Two transcripts, 6.1 and 3.1 kb, were found in adult mouse poly(A)+ RNA isolated from a number of tissues, including brain, lung, and heart. In situ hybridization found expression in mouse olfactory bulb, cortex, and regions of the hippocampus and hypothalamus (Guimera et al. 1996). Gene 43033 hybridizes to a 1.5-kb message in mouse cerebellum, cortex, liver, lung and heart, and in human cerebellar RNA (Fig.2). In contrast to the 6.1-kb DYRKtranscript, which is only detected in poly(A)+ RNA, 43033is readily detected in blots of total RNA. The longest ORF starting from a methionine codon in 43033 is 57 amino acids. Although the 3′-noncoding region of the mouse Dyrk gene has not been published, 43033 localizes to the same 10-kbEcoRI fragment as a 3′ Dyrk probe. Gene43033 matches numerous mouse ESTs and is also homologous to an EST from the Tamar wallaby, Macropus eugenii, so is most likely present in all mammals.
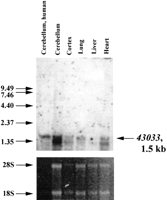
Northern blot analysis of EST 43033. 43033 was used as a probe against total RNA from human cerebellum (Clontech) and several mouse tissues. Sizes of the RNA ladder (GIBCO/BRL) are shown on theleft. The ethidium stained gel is shown below as a control for RNA loading.
The third new gene was identified from sequence of an STS from YAC 154C9, D16Jhu19e (Table 3), which matches six human ESTs and human genomic sequence from the MX1 region. This putative transcript is similar to the 2-19 protein precursor found on the human X chromosome (Bione et al. 1993). Further characterization of the gene on Chr. 21 is ongoing. The Tiam1 gene, which is nonrecombinant with Cbr1 and Dyrk on a commonly used backcross mapping panel (Song et al. 1997), was not found on the contig and hence must lie proximal to Cbr1.
D16JhuMarkers
Additional New Markers
Eighteen new markers on the contig are derived from YAC- and BAC-end clones (Table 3; Fig. 1, green). One of the 18 end clones contains a transcript; the acentric end of 182A2 matches part of the 3′ end of Kcnj6. The final new marker on the contig isD16Jhu1, an STS marker derived from YAC 342G11 (Table 3). The remaining 12 markers on the contig are STSs from WCGR. AllD16Mit markers on the contig are nonrecombinant on the WCGR genetic map (Dietrich et al. 1996). Two markers, D16Mit70 andD16Mit120, map to this region on a low-resolution intercross but are not found on this contig. This supports a previous report that they map far proximal to this region based on a higher resolution genetic map (Reeves et al. 1997).
The distal-most clones from the Mx region were hybridized with a (TTAGGG)n concatemer probe (Ijdo et al. 1991) to detect mouse subtelomeric repeats, but all were negative (data not shown). Although this contig includes the distal-most known gene on Chr. 16, either coverage does not yet extend to the telomeric region of the chromosome or these sequences are deleted from the YACs in the contig.
Physical Mapping by YAC Fragmentation
The distance between markers on minimal tiling path YACs was determined by YAC fragmentation (Fig. 1). More than 300 fragmentation derivatives were analyzed to identify 125 sites providing an average spacing of 1 site every 36 kb with a range from 10 to 250 kb. YACs were aligned by assessing common markers in the smallest fragmentation intervals on overlapping YACs. Markers were generally placed in the center of the fragmentation intervals to which they mapped except when clone size constraints gave a better localization within an interval. End clones were most useful in aligning YACs because they define the ends of precisely sized intervals. The only gap in YAC coverage falls between YACs I103E5 and 183G11, and is crossed by BAC 484L1 completing a physical map across the entire 4.5-Mb contig. Both ends of this BAC were localized on fragmentation derivatives of these YACs, demonstrating that the gap between them is <20 kb. The largest interval in the fragmentation map is that containing D16Jhu16and D16Jhu17, a distance of 250 kb. Fragmentation derivatives differing in length by ∼10 kb were visualized under the PFGE conditions utilized. All markers that are nonrecombinant on the genetic map are resolved on the physical map by YAC fragmentation, STS content mapping, or both (Fig. 3).
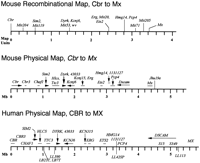
Comparison of mouse and human physical maps. A mouse recombinational map based on 1154 meioses (Reeves et al. 1997) is shown (top), spanning 3.5 map units with 16 markers at nine loci. Mouse and human physical maps are shown with the positions of transcripts indicated by a horizontal line beneath the marker name. Where gene orientation has been determined, an arrowhead indicates the direction of transcription. The consensus human physical map was compiled from several sources (Burmeister et al. 1991; Chumakov et al. 1992; Ichikawa et al. 1993;Dufresne-Zacharia et al. 1994; Patil et al. 1994; Gardiner et al. 1995;Gosset et al. 1995; Korenberg et al. 1995; Cabin et al. 1996; Osoegawa et al. 1996; Hubert et al. 1997; Ohira et al. 1997). With the exception of D21S15 and D21S349, the markers shown above the scale bar are known transcripts. Five NotI sites are shown beneath the map; LB23T and LB7T are separated by ∼30 kb. Gene 1131127 matches sequence from LL42SP.Where coding sequences on the human map are well localized, a horizontal bar is shown beneath the marker name. Less well-localized markers are placed in their approximate position without a horizontal bar.
The region between Ets2 and Pcp4 appears to be unstable in yeast. Several YACs used in the minimal tiling path in this area are known to be chimeric by end recovery or presumed to be chimeric based on YAC size, marker content, and marker localization by fragmentation analysis (e.g., 255F1 and 165A7). Given the problems with deleting YACs, the interval sizes in this region must be taken as minimal distances.
DISCUSSION
A YAC STS content map covering the distal 4.5 Mb of distal mouse Chr. 16 was assembled on the scaffold provided by a high-resolution recombinational map (Reeves et al. 1997). Gaps were crossed by isolation of end clones and screening of YAC libraries with these new markers. Known transcripts on the corresponding region of human Chr. 21 have been tested on this physical map. There are no discordancies in gene content or order. Where gene orientation is known in both, it too is the same (Fig. 3).
The STS content map was refined by YAC fragmentation to measure marker spacing. YAC fragmentation is a faster method for determining physical distances than restriction enzyme (RE) mapping with PFGE and provides a substrate for analysis both by PCR and hybridization. The fragmentation map gives an average resolution of 36 ± 10 kb, the limit of PFGE resolution under the conditions utilized. The fragmentation map is susceptible to errors because of undetected deletions in YACs. In this study redundant coverage and marker density were used to assess YAC integrity. The region around Ets2 and Pcp4 was found to be unstable in yeast. Coverage in this region depends on chimeric YACs. The chimeric portions of these YACs may stabilize the portions derived from Chr. 16. The corresponding region of Chr. 21 is also unstable in yeast. The basis for this instability may be revealed by comparative sequencing.
A sex-averaged composite map of Chr. 16 that shares nine loci and 16 markers with this contig covers 3.5 map units (Reeves et al. 1997; see Fig. 3). As the physical map of the corresponding region is 4.5 Mb, the measured 1.3 Mb per map unit is somewhat less than the genome-wide estimate of 1.8 Mb/cM in the mouse.
Assembly of a physical map across the entire mouse genome has begun at the WCGR (Haldi et al. 1996). Five YACs from this effort are part of the minimal tiling path presented here. On distal Chr. 16, two doubly linked contigs, WC-1804 and WC-902, cover part of the region described here. WC-902 spans at least 1.8 Mb, as measured here by fragmentation, but the six markers in common with this contig are in a different order. Nineteen YACs in WC-902 carry more than one marker, and the WCGR marker order requires 29 internal deletions in these YACs. When the marker order is changed to that presented here, 28 of the 29 gaps are eliminated, and the final gap is reduced from three missing markers to one.
Distal Chr. 16 is of special interest because of its conserved linkage with part of human Chr. 21. Comparative mapping has relied on markers (usually genes) that cross-hybridize between species. The generation of physical maps of the mouse allows comparison with human physical maps at the level of gene spacing, size, and orientation. Generation of an accurately measured physical map of distal Chr. 16 provides a basis for comparison with the corresponding region of Chr. 21.
Human and mouse maps are aligned at CBR1 based onNotI mapping and RE mapping of YACs and smaller clones (Fig.3). An accurate human physical map is available for the region between human CBR and ETS2 (Ichikawa et al. 1993;Dufresne-Zacharia et al. 1994; Patil et al. 1994; Gosset et al. 1995;Osoegawa et al. 1996; Ohira et al. 1997). Distances on the human and mouse physical maps are well-conserved between CBR1 andETS2. Both span ∼3 Mb and include 12 transcripts, for an average of 1 transcript/250 kb. Based on average gene density of 80,000 genes in a 3-Gb genome, 80 genes would be found in an “average” 3-Mb interval, so further gene discovery may be expected in this region. The overall conservation of distance could indicate some evolutionary pressure not only to maintain gene order but also spacing in the 80–100 million years since mouse and human shared a common ancestor. As more than one-third of the human and mouse genomes consist of repetitive elements (Smit 1996) and such elements and their locations differ between mouse and human, gene spacing and overall size may be important in the CBR1–ETS2 region. Comparative sequencing will be necessary to determine precise gene sizes and structures, identify conserved regulatory regions, and search for chromosomal structural elements important in maintaining the 3-Mb distance in both species.
In the ETS2–MX region, fewer common markers have been mapped in both species. EST 1131127 matches human genomic sequence around the NotI site LL42SP. Marker spacing is similar between the species up to this point. Distal to1131127, the mouse map is compressed compared to human. The distance from 1131127 to Mx is ∼1.3 Mb in mouse, whereas the corresponding human segment is >2 Mb. Undetected deletions in the mouse YACs around Pcp4 could have contributed to an underestimate of distance, but deletions on the order of 700 kb would likely have been detected given the marker density in this region. Although this region of Chr. 21 is covered by YAC maps (Chumakov et al. 1992; Gardiner et al. 1995; Korenberg et al. 1995) and almost completely converted to sequence-ready template (Soeda et al. 1995; Hubert et al. 1997), marker spacing in human has not been measured accurately. It will be interesting to determine the basis for differences in the physical map at the sequence level in areas where there is no discrepancy in gene content or order.
Comparison of genomic sequence between human and mouse can assist significantly in identification of gene regulatory sequences (Galili et al. 1997; Hardison et al. 1997). Mouse genomic sequences from YAC-end clones provide tantalizing hints of what may be revealed by comparative sequencing of the whole region. End clones D16Jhu4 andD16Jhu6, both just proximal to Sim2, have significant homology to human genomic DNA. Jhu6 matches aSim2-containing cosmid, clone A39D1-F2, at a point 1.7 kb upstream from exon 1. The match is 107/128 bases (83%), forP = 4.2 e −27. Jhu4 has 2 matches on the same cosmid, ∼9.5 kb proximal to the first exon ofSim2. These matches are 27/32 (84%) and 45/74 (69%) bases,P = 0.13. Although the P values for the short sequences are not strong, the highly conserved location of these hits in mouse and human argues against coincidence. These matches may identify regulatory regions for Sim2. An STS from YAC 154C9, transcribed sequence Jhu19e, matches human ESTs and genomic sequence around MX. Orientation of this new gene with respect to human MX is not known, but it is distal in mouse and thus the distal-most gene known on Chr. 16.
Two strong sequence matches involve recently released human genomic sequences from Chr. 21. The distal end clone of 100F3,D16Jhu14, hits human genomic sequence 15 kb distal toPCP4, its approximate location in mouse, atP = 2.8 e −25. An STS from YAC 342G1, D16Jhu1, matches human sequence 130 kb proximal ofKCNJ15 at P = 1.6 e −54. Neither of these mouse sequences matches a known gene or EST, and neither has an ORF longer than 60 amino acids; therefore, both are candidates for gene regulatory regions.
This high-resolution, fragmentation-based physical map of distal Chr. 16 provides the first opportunity to compare genomic regions as large as 4.5 Mb in human and mouse at a level beyond that of gene order. Whereas comparative sequence provides the ideal comparative map, resources for sequencing the mouse genome will lag behind human, despite the utility of comparative sequencing for identification of genes and regulatory elements. High-resolution physical maps produced quickly by YAC fragmentation provide an interim means for comparison and may be a useful way to identify regions of particular interest for comparative sequence analysis.
METHODS
YACs/BACs
YACs from WCGR mouse libraries (Kusumi et al. 1993; Haldi et al. 1996) were obtained from Research Genetics, Inc. (Huntsville, AL). YACs from the St. Mary’s (Chartier et al. 1992) and ICRF mouse libraries (Larin et al. 1991) were obtained from the Baylor University YAC screening core. YAC addresses were obtained by sequential screening of DNA pools, except those from the large-insert WCGR library, which were obtained from the Whitehead/MIT Mouse Physical Mapping database (http://www-genome.wi.mit.edu/cgi-bin/mouse/index). BACs were identified by hybridization of single-gene probes to filter arrays purchased from Research Genetics, Inc., and through the courtesy of Dr. Bruce Birren at WCGR.
Markers
D16Mit primer pairs were obtained from Research Genetics, Inc., and used according to WCGR conditions. PCR markers developed in this study are listed in Table 1, along with DNA sequence accession numbers from which primers were developed and their reaction conditions. New primers were developed for three WCGR markers:41.MHAa75d5, MHAa83a1, and 36.MHAa77c12.IMAGE clone ESTs were obtained from Research Genetics, Inc. (Table 2). Genes mapped previously on Chr. 16 have been described (Reeves et al. 1997; Mouse Genome Database athttp://www.informatics.jax.org/mgd.html).
YAC Manipulations
YACs were typed for markers by hybridization and/or PCR using glass bead prep yeast DNAs (Hoffman and Winston 1987). YACs in AB1380 were transferred to a his3Δ200 genetic background by karyogamy transfer to yPH925 (Spencer et al. 1994). YAC-end clones were recovered by the pICL/pLUS retrofitting procedure (Hermanson et al. 1991), except for YACs from the WCGR library constructed in pRML1 and pRML2, where end recovery by dilute ligation and electroporation does not require vector retrofitting (Spencer et al. 1993). End clones were sequenced from the T7 or T3 primer sites in the pICL and pLUS vectors or from the pYAC4 vector side using URAYAC-end or CENYAC-end primers (Table 1). YAC fragmentations were performed as described (Pavan et al. 1990), using mouse B1 repeat-containing vectors pWJ522 and pWJ528 (Heard et al. 1994). Fragmentation derivatives were separated by PFGE and sized after hybridization with a centric vector end probe.
Acknowledgments
We thank Jennifer S. Lee and Bruce Birren for BACs containingD16Mit204 and D16Mit205, and Udaya DeSilva and Eric Green for the end sequencing of BAC 484L1. This work was supported by U.S. Public Health Service awards from National Institute of Child Human Development and National Heart, Lung, and Blood Institute (J.R.K.) and U.S. Public Health Service awards HG00405 and HD24605 (R.H.R.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
Present addresses: 3Bloedel Hearing Research Center, University of Washington, Seattle, Washington 98195-7923 USA; 4Center for Molecular and Structural Biology, Medical University of South Carolina, Charleston, South Carolina 29425-2213 USA.
-
↵5 Corresponding author.
-
E-MAIL rreeves{at}welchlink.welch.jhu.edu; FAX (410) 955-0461.
-
- Received May 14, 1998.
- Accepted July 14, 1998.
- Cold Spring Harbor Laboratory Press








