
SeqHelp: A Program to Analyze Molecular Sequences Utilizing Common Computational Resources
Abstract
Here we descibe a tool to analyze molecular sequences utilizing the internet and existing computational resources for molecular biology. The computer program SeqHelp organizes information from database searches, gene structure prediction, and other information to generate multiply aligned, hypertext-linked reports to allow for fast analysis of molecular sequences. The efficient and economical strategy in this program can be employed to study molecular sequences for gene cloning, mutation analysis, and identical sequence search projects.
Computational tools are important components in generating and understanding novel genetic sequences. A gene identification project typically includes the following components: (1) generation and assembly of DNA sequences from a genetic region of interest; (2) database searches to find similar or homologous sequences; (3) construction of the genomic structure of the putative gene; (4) if searching for disease susceptibility genes, screening for mutations in candidate genes; (5) multiple sequence comparison and other analyses. Computer programs have dramatically improved the efficiency of these analyses. Some well-known examples of these computational tools include PHRED (Ewing et al. 1998; Ewing and Green 1998) and PHRAP [http://bozeman.mbt.washington.edu/phrap.docs/phrap.html (P. Green, unpubl.)] for sequence generation and assembly, the BLAST family of programs (Altschul et al. 1990), FASTA and FASTP (Pearson 1990) for database searches, and GRAIL (Xu et al. 1994) and Genefinder (C. Wilson and P. Green, unpubl.) for gene structure prediction.
Although these and many other computer programs are excellent tools in specific areas of analysis, they often do not provide an easy interface for experimental biologists to analyze information simultaneously from multiple resources. A tool to integrate a variety of information to provide the ability to visually analyze the overall structure as well as details of information for the underlying sequence is highly desirable for the experimental biologist. Display of a data sequence multiply aligned with related sequences, along with immediate access to relevant information during sequence analysis, would greatly expedite gene identification studies. Programs such as Genotator (Harris 1997) and DrawMap (T. Smith, unpubl.) provide graphical display of genomic structure including predicted exons, selected database search results, and other information. These programs generally provide a high-level display of genetic information, but detailed display of sequence information is limited and access to data via the internet is not provided. In part, inspired by these programs, the present work is designed to exploit some commonly available computational resources to provide a simple, yet efficient, tool for visually studying DNA sequences in gene hunting and other molecular research projects.
RESULTS AND DISCUSSION
Overview
The present work utilizes a set of readily available software, which are among the best in their respective fields of application, and can be applied to DNA sequences in a plain text file or generated from electrophoresis image files (chromatograms). For each data sequence, the program SeqHelp will, at the user’s option, call other programs for gene prediction, masking of repeat elements, and database searches, and gather the information from these programs into a visual display of integrated, hypertext-linked information for genomic analysis. The general approach is schematically given in Figure 1, and the programs used in specific components are described in Methods.
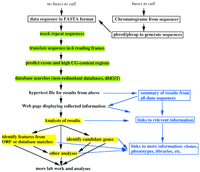
Schematic for sequence analysis utilizing multiple information sources.
For automatically sequenced data, chromatograms from the ABI sequencer are first transferred to a UNIX-based computer workstation. The program PHRED (Ewing et al. 1998, Ewing and Green 1998a,b) is then used to call the bases and translate them into DNA sequences. After screening off vector sequences, the program PHRAP [http://bozeman.mbt.washington.edu/phrap.docs/phrap.html (P. Green, unpubl.)] is used to analyze the sequences and assemble them into contiguous DNA sequences (contigs) where overlapping sequences are identified. SeqHelp is then applied to the resulting data for analysis.
Information Presentation
SeqHelp organizes the database search results into an HTML file, in which the data sequence is aligned with all constituent local sequences, if the data sequence is a contig, and with genomic, EST, cDNA, or amino acid sequences identified from database searches. Repeat elements, predicted exons, and predicted CpG islands are also shown for each sequence. For each sequence identified from the database search, hypertext links point to database search results and their relevant records in the remote databases. Discrepancies in the alignments are highlighted with a different color to alert the investigator. The six ORFs are displayed over the DNA sequence, with ORFs corresponding to predicted exons highlighted in color. Predicted CpG islands are highlighted by color on the data sequence. A summary report with hypertext links is also generated for all data sequences (Fig.2). Any computer program capable of browsing hypertext files can then be used to visualize and study the data as web pages.

A truncated summary report.
The summary information page can be used to manage sequence data for a sequencing project with a hypertext browser. The investigator can quickly browse this page to monitor the information on the individual sequences and the progress of the overall project. Information for the individual sequences can be used to identify candidate genes and other features by comparing sequence similarities, predicted exons, and studying relevant information that can be readily accessed via the internet. A genomic sequence will typically contain individual exons separated by introns. Intron/exon boundaries are identified by alignment of individual exons to ESTs and amino acid sequences. DNA sequences matching ESTs or amino acid sequences can be selected as candidate genes for further analysis. Multiple local sequences matching a contig can be used to study the consistency of the constituent sequences.
Applications
Our goals in genomic research are to (1) translate the electrophoregrams into molecular sequences; (2) identify candidate genes through database searches and gene prediction methods; (3) monitor the progress of sequencing projects; (4) provide instant access to relevant genomic information; and (5) compare multiple sequences, inside or away from the laboratory. SeqHelp has been applied to our gene cloning and analysis efforts and successfully met our goals. For illustration, the partial results for analyzing a sequence containing the human DFNA1 gene (Lynch et al. 1997) are displayed in Figure 3. The predicted exons, cDNAs, amino acid sequences from the public databases, repeat elements, as well as the constituent sequences from the local database for the sequencing project, are appropriately displayed. Clicking on the right-hand links leads to the database search results in BLAST output format, from which appropriate database entries can be accessed by clicking on the respective links. Candidate genes are identified from examination of such annotated sequences and links to relevant databases.

A truncated example of the analysis of a contig containing the humanDFNA1 gene, a homolog of the Drosophila diaphanous gene. Numbers at left between the first three horizontal lines are for illustration purposes only and are not in the actual display. Lines 1–6 are translations of the six reading frames of the sequence segment, displayed above the data sequence (line 7). Lines 8–11 are results from BLAST searches against public and local databases shown in alignment with the data sequence. Line8 is the result from searches against nonredundant amino acid databases, line 9 against nonredundant nucleic acid databases, and lines 10 and 11 against local database (sequencing reads). Sequences, as in lines 10 and 11,are the constituent sequences used to construct the consensus data sequence on line 7. Discrepancies between data sequence and database sequences are indicated in magenta. Line 7 displays part of a predicted CpG island displayed in orange. Lines 3and 5 contain predicted exons and are displayed in red. Other lines can be interpreted similarly. Additional database search results have been omitted. Clicking on the right-hand link will lead to the web page for relevant database search results in BLAST output format for details of the matches. From this web page, further links can lead to complete entries of the relevant data in remote databases. (No repeat elements or ESTs have been found in the displayed segment).
Among its other applications, SeqHelp has been used to annotate sequence data in preparation for submission to public databases, to monitor the progress of sequencing projects, and to compare multiple sequences. Interestingly, when constructing the genomic sequence of a specific gene, aligning its known cDNA sequence (or its homolog) against local sequences in the relevant sequencing project can reveal the boundaries of exons. A complete display of a 117-kb genomic sequence containing the human BRCA1 gene (GenBank accession no. L78836; Smith et al. 1996) and other examples can be accessed viahttp://polaris.mbt.washington.edu.
Design Issues
Dissemination of genomic information encompasses the study of the data sequence relative to the existing information of known genetic sequences. Such information is now readily available on the Internet, which provides unprecedented accessibility to information of virtually any kind. Computation can now be carried out with commercially or publicly available internet browsing programs, and many programs now allow the analyses of genetic data over the Internet. Furthermore, the HTML form of the database search results by the BLAST suite of programs (Altschul et al. 1990) and Entrez (Schuler et al. 1996) provide links to multiple genomic databases, from which further links to other relevant information are possible. SeqHelp facilitates immediate linkage to such information in the novel sequence for fast analysis. Furthermore, because SeqHelp organizes information for analysis on hypertext files, the results can be studied using any computer capable of the most basic hypertext browsing via the Internet.
The choice of computer programs to be employed naturally should consider their merits. Because every existing computer program has superior performance in special cases, our choice of programs was based on their general ability to solve problems in their respective areas of application. The BLAST programs have been highly regarded and widely accepted for database searches, although their sensitivity in database searches is sometimes compromised; RepeatMasker is based on the most up-to-date databases of repeat sequences and is highly effective in masking known repeat elements; PHRED has the highest success rate of translation for electrophoregrams from an automatic sequencer, and PHRAP provides an efficient way of assembling individual sequences into contiguous sequences of practically any size; Genefinder has been very successful for gene prediction in Caenorhabditis elegans,although its ability to predict genes in humans is not as successful, like any other program for such purpose. In addition, these programs can be adapted easily for batch processing, which is highly desirable in large-scale sequencing projects. One design philosophy of SeqHelp is to quickly employ existing, high-quality technology in genomic research. These programs meet these criteria and provide the fastest, most economical means for an integrated approach to meet our requirements in sequence analysis. Additional programs and databases can be incorporated as additions to SeqHelp, but their inclusion should be based on their purposes and ease of interface.
The selection criteria for database matches has to be a compromise between including too many low similarity sequences and dismissing potentially homologous but distantly related sequences. In positional cloning practices, the selection of database search results can vary widely, depending on the evolutionary distance between genes reported in the databases and a homolog in the novel sequence. In a gene-search project, the investigator is interested in genomic, cDNA, or amino acid sequences that show similarity to a novel sequence of interest. Closely related genomic and cDNA sequences generally show a higher level of similarity, whereas distant members of a gene family may show weak homologies. If an EST or a cDNA segment were part of a gene in the novel sequence, the similarity is very high. On the other hand, an amino acid sequence may display only weak homology to a distant relative in the novel sequence. Using only a high similarity requirement could exclude potentially important new genes. Thus, the investigator must decide on the level of stringency for the selection criteria. In our research, although selection criteria do vary, we have normally included database matches for nucleic, cDNA, EST, and local genomic sequences with at least a 70% similarity and <1% probability of being a random match, and amino acid matches with at least 50% similarity. These selection criteria seem to have included the appropriate search results for our analyses.
Alternative Programs
Other programs are available that serve a similar purpose as SeqHelp, and each provides certain, but distinct, advantages. Obviously, these programs are alternative choices in genomic analysis. A brief comparison of SeqHelp to some of these programs is provided in the ensuing paragraphs.
As mentioned before, SeqHelp was motivated in part by Genotator (Harris 1997), which is an excellent tool for sequence annotation and visual analysis. It provides a graphical display of high-level information from database searches and gene structure prediction by multiple programs, an interactive mechanism for user-defined characteristics, and indication of some other miscellaneous information. It does not, however, provide hypertext links to information, and its display of low-level similarity sequence data, particularly multiply aligned sequences, is limited.
Another program, PowerBlast (Zhang and Madden 1997), provides a set of powerful tools, including a graphical display of the structure of the sequence being studied, various forms of reports for database search results, as well as hypertext links to entries in the results. However, it presents only a selection from the database search results, and these are identified using rather stringent matching criteria. It also provides direct links to the remote databases but without first allowing the user to examine the database search results.
SeqHelp shares the same purposes as Genotator, PowerBlast, and other sequence annotation and display software, but its own features will serve as an alternative tool for sequence display and analysis. SeqHelp emphasizes integrated, sequence-level information presentation and provides color display of alignments from local and public databases, allowing for easier analysis of the sequence at the base level. It maintains hypertext links to database search results before linking to the remote database entries, allowing for more user involvement in decision-making to select results for further study. SeqHelp allows for incorporation of information on repeat elements, predicted exons and CpG islands, as well as allowance for miscellaneous features. Moreover, SeqHelp generates a hypertext-linked report for all sequences in a sequencing project to allow for fast examination of results. Because SeqHelp generates hypertext reports, genomic data can be analyzed on any computer, even remotely, via a web server. Taken together, SeqHelp is more flexible in organizing relevant information for analysis.
The alignment of multiple sequences is another highly important and well-studied process in molecular genetics. Rigorous algorithms (for review, see Waterman 1989) have been studied, and various computer programs such as GCG (GCG 1994) and CLUSTAL (Higgins and Sharp 1988) were developed for this purpose. As a by-product of the display of database search results in general, SeqHelp provides a less rigorous, but quick, answer to the examination of relationships among multiple sequences displayed with each other, borrowing the local alignments of BLAST, with the added advantage that results from public database searches can be studied simultaneously with these sequences. Insertions/deletions (indels) in alignments in gene identification projects are less critical but are more important in population biology context. These alignments will be improved as indels are properly handled (the current version of SeqHelp is not suitable for detecting indels properly but is being modified with a simple dynamic programming algorithm to handle this). Sequence variations and, alternatively, identical sequences, can be identified from multiply aligned sequences. Experimental application of this method to search for identical sequences is being conducted in our research.
Conclusions
SeqHelp enables us to accomplish several tasks relatively efficiently for genome sequencing and other sequence analysis projects. The investigator can quickly study the summary report to identify a sequence of interest. It allows minimal effort for the experimental biologist to visualize database search results by displaying them along with the data sequence. The possible genomic structure of a data sequence can be studied because the genomic or amino acid sequences of known genes are displayed where they align with each other. Further information for any genetic entity of interest identified from the database search can be readily obtained following the hypertext links to more complete records. For each contig, visual analysis of the alignment of constituent sequences allows the investigator to explore the reliability of the sequence data. In principle, a DNA sequence of any length can be studied with this approach.
The ability to study genomic structure, identify candidate genes, extract genetic information from a novel sequence, and evaluate relationships among similar sequences are fundamental needs for scientists in the Human Genome Project and other laboratories involved in molecular genetic research. Sophisticated computational tools are required for these analyses. Given the various levels of computer knowledge among experimental biologists, easy-to-use, readily available computational tools are very helpful. In addition, as different computers have different operating systems, the ability to analyze the same data on different computer platforms with minimal software requirements will be beneficial. SeqHelp was designed to identify candidate genes, study genomic structures, organize data, and compare multiple sequences to aid positional cloning efforts. It has successfully met our objectives and can also serve to meet the more general needs mentioned above in genomic research.
METHODS
SeqHelp is written in the C programming language, currently running on the UNIX platform. Its availability, user’s manual, auxiliary programs, future upgrades (including the version for managing indels), and examples are announced athttp://polaris.mbt.washington.edu.
Program Components
Identification of Repeat Elements
The program RepeatMasker [http://ftp.genome.washington.edu/RM/RepeatMasker.html (A. Smit, unpubl.)] is used to identify repeat elements in the DNA sequences against the latest database of known repeats, from which regions containing repeat elements are masked before database searches.
Database Search
The programs BLASTN and BLASTX (Altschul et al. 1990) are used to search for sequences (in nonredundant public nucleic, EST, and amino acid databases) similar to each data sequence. All individual sequences generated from the underlying sequencing project (and any other sequence of interest) are built into a local database suitable for search with BLAST to identify sequences similar to the data sequence, in a format consistent with other database search results.
Exon Prediction
Exons are predicted with the computer program Genefinder (Wilson and P. Green, unpubl.) and are indicated by color in the corresponding ORFs.
SeqHelp collects results from the above programs and performs the following steps for each data sequence to generate information for visual analysis.
Collection of Database Search Results
Database search results for ESTs, genomic or cDNA, and local sequence matches with a given level of identity below a specific probability of being random matches as calculated by BLASTN are included in the report. For amino acid sequences, matches with a given level of similarity are included, but matching subsequences with low complexity are filtered out using local complexity statistics (Wootton and Federhen 1993), where thresholds for inclusion are derived from the distribution of complexity statistics of simulated amino acid sequences, using amino acid frequencies taken from 100 independent, complete human genes in GenBank, version 95.
CpG Island Prediction
CpG islands are predicted based on the CG contents in a genomic region. Using a counting method similar to the Window module of GCG (GCG 1994) the number of CG pairs are counted within a 100-base window of a base (in 3-base increments) for 100 independent human genes chosen from GenBank, version 95. These CG-pair counts are pooled to obtain an average (m) and standard deviation (s.d.). For the data sequence, the CG frequencies are calculated at 3-base increments for windows of 100 bases. A region at least 300 bases long with CG frequencies greater than m + s.d. is indicated as a possible CpG island.
Information Presentation
For each data sequence, SeqHelp organizes its ORFs, database search results, predicted exons and CpG islands, and identified repeat elements into an HTML file of multiply aligned sequences. Hypertext links point to database search results and their relevant records in the remote databases. A summary report with hypertext links to all data sequences in the same sequencing project and to entries in their respective database search results is also generated. The hypertext files can then be studied as web pages using any computer program capable of browsing hypertext files.
Acknowledgments
We thank P. Green, C. Wilson, A. Smit, B. Ewing, and D. Gordon for providing software. This work was supported by National Institutes of Health grants R01-CA27632 and R01-DC01076, and the Markey Molecular Medicine Center, University of Washington.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL mlee{at}u.washington.edu; FAX (206) 616-4295.
- Received September 10, 1997.
- Accepted February 2, 1998.
- Cold Spring Harbor Laboratory Press








