
Construction of an ∼700-kb Transcript Map Around the Familial Mediterranean Fever Locus on Human Chromosome 16p13.3
- Michael Centola1,10,
- Xiaoguang Chen2,10,
- Raman Sood1,
- Zuoming Deng1,
- Ivona Aksentijevich1,
- Trevor Blake3,
- Darrell O. Ricke4,
- Xiang Chen1,
- Geryl Wood1,
- Nurit Zaks1,
- Neil Richards5,
- David Krizman6,
- Elizabeth Mansfield1,
- Sinoula Apostolou7,
- Jingmei Liu4,
- Neta Shafran1,
- Anil Vedula1,
- Melanie Hamon2,
- Andrea Cercek2,
- Tanaz Kahan2,
- Deborah Gumucio5,
- David F. Callen7,
- Robert I. Richards7,8,
- Robert K. Moyzis4,9,
- Norman A. Doggett4,
- Francis S. Collins3,
- P. Paul Liu3,
- Nathan Fischel-Ghodsian2, and
- Daniel L. Kastner1,11
- 1Arthritis and Rheumatism Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health (NIH), Bethesda, Maryland 20892-1820 USA; 2Departments of Pediatrics and Medical Genetics, Cedars-Sinai Medical Center, Los Angeles, California 90048-0750 USA; 3Genetics and Molecular Biology Branch, National Human Genome Research Institute, Bethesda, Maryland 20892 USA; 4Center for Human Genome Studies, Los Alamos National Laboratory, Los Alamos, New Mexico 87545 USA; 5Department of Anatomy and Cell Biology, University of Michigan, Ann Arbor, Michigan 48109-0616 USA; 6Laboratory of Pathology, National Cancer Institute, NIH, Bethesda, Maryland 20892 USA; 7Department of Cytogenetics and Molecular Genetics, Adelaide Women’s and Children’s Hospital, North Adelaide, South Australia 5006; 8Department of Genetics, The University of Adelaide, Adelaide, South Australia 5000
Abstract
We used a combination of cDNA selection, exon amplification, and computational prediction from genomic sequence to isolate transcribed sequences from genomic DNA surrounding the familial Mediterranean fever (FMF) locus. Eighty-seven kb of genomic DNA around D16S3370, a marker showing a high degree of linkage disequilibrium with FMF, was sequenced to completion, and the sequence annotated. A transcript map reflecting the minimal number of genes encoded within the ∼700 kb of genomic DNA surrounding the FMF locus was assembled. This map consists of 27 genes with discreet messages detectable on Northerns, in addition to three olfactory-receptor genes, a cluster of 18 tRNA genes, and two putative transcriptional units that have typical intron–exon splice junctions yet do not detect messages on Northerns. Four of the transcripts are identical to genes described previously, seven have been independently identified by the French FMF Consortium, and the others are novel. Six related zinc-finger genes, a cluster of tRNAs, and three olfactory receptors account for the majority of transcribed sequences isolated from a 315-kb FMF central region (betweenD16S468/D16S3070 and cosmid 377A12). Interspersed among them are several genes that may be important in inflammation. This transcript map not only has permitted the identification of the FMF gene (MEFV), but also has provided us an opportunity to probe the structural and functional features of this region of chromosome 16.
Saturating the human genome with mapped genes so that the structure and function of each can be determined is an ultimate goal of the human genome project (Collins and Galas 1993). In addition to random cDNA sequencing and subsequent mapping of expressed sequence tags (ESTs) (Milner and Sutcliffe 1983; Putney et al. 1983;Adams et al. 1995), another way of building transcript maps is by isolation of transcribed sequences from cloned chromosomal DNA. Such regional transcript maps not only facilitate the cloning of genes for Mendelian traits whose basic defects would otherwise remain elusive because of the lack of functional clues, but also provide us insights into the genomic structure of particular chromosomal regions. As part of our efforts to identify the genetic defect underlying familial Mediterranean fever (FMF), an autosomal recessive genetic disease characterized clinically by periodic attacks of fever and inflammation in the joints, peritoneum, and pleura (for review, see Kastner 1996), we physically mapped and cloned the genomic DNA surrounding this locus.
Assignment of the FMF locus to chromosome 16p13.3 by linkage (Pras et al. 1992) and regional mapping defined a candidate interval of 9 cM (Aksentijevich et al. 1993; Fischel-Ghodsian et al. 1993). We subsequently narrowed the interval to the region betweenD16S246 and D16S2622, and isolated a complete ∼1 Mb cosmid contig of the region (Sood et al. 1997). Later, our group and a competing consortium placed the gene for FMF in a region of <250 kb (The French FMF Consortium 1996; Balow et al. 1997).
In parallel with genetic and physical mapping studies, we wanted to construct a map of transcribed sequences from the FMF candidate interval. There are several methods available for such purposes. Exon amplification/trapping (Duyk et al. 1990; Buckler et al. 1991; Krizman and Berget 1993), cDNA selection (Wallace et al. 1990; Lovett et al. 1991; Parimoo et al. 1991), direct screening (Wallace et al. 1990), evolutionary conservation, transcript prediction by genomic sequencing (Oliver et al. 1992; Sulston et al. 1992), and CpG island-based methods (Patel et al. 1991; Valdes et al. 1994) have all been used to identify the transcribed portions of genomic DNA of this size. The advantages and disadvantages of these methods have been described (Collins 1992). In extensive comparative studies, no single method has been shown to be capable of isolating 100% of the transcripts encoded in a given stretch of genomic DNA (Brody et al. 1995; Harshman et al. 1995; Yaspo et al. 1995). For this reason, we used several transcript identification methods in parallel.
Exon amplification/trapping was chosen for its high success rate in prior positional cloning projects (Ambrose et al. 1992; Trofatter et al. 1993; Vulpe et al. 1993) and for its ability to recognize sequences as transcribed regardless of their tissue-distribution patterns. The latter is important because before we cloned the FMF gene we had no knowledge of its tissue-expression pattern, which turned out to be quite specific for granulocytes (International FMF Consortium 1997). Direct cDNA selection was also used because it is technically simple, and according to previous studies, has a satisfactory efficiency for transcript identification (Lovett et al. 1991; Parimoo et al. 1991). Moreover, the transition from selected cDNA fragments to full-length transcripts is usually easier than that from trapped exons, because the selected cDNA fragments are usually larger in size than the trapped exons. On the other hand it is a hybridization-based method, involving the use of a genomic probe and a pool of cDNA fragments. Successful identification of the transcribed sequences contained in the genomic probe depends heavily on whether such sequences are represented in the cDNA pool to be screened, making the choice of mRNAs from the right tissue or cell in the preparation of the cDNA pool a critical step for the success of the whole project. For unknown genes that may have tissue-specific expression, such a choice presents considerable challenge.
Transcript prediction by genomic sequencing was employed for the same reasons as exon amplification/trapping. Since its initial successful use in the positional cloning of the Werner’s syndrome gene and the gene for hemochromatosis (Feder et al. 1996; Yu et al. 1996), this expression-independent method is becoming increasingly popular because of the development of better prediction programs (Uberbacher and Mural 1991; Solovyev et al. 1994; Thomas and Skolnick 1994; Kulp et al. 1996), automation of sequencing, and, most importantly, accumulation of sequences in the database of expressed sequence tags (dbEST) (Schuler et al. 1996).
This report summarizes the transcript map we constructed during our FMF positional cloning effort. A broad interval of ∼700 kb was subjected to direct cDNA selection, with a more focused 285-kb segment scrutinized by multiple methods of transcript identification. During the review of this manuscript, Bernot and colleagues (1998) published their own partially overlapping transcript map of the FMF region. The two maps provide complementary data on an ∼225-kb region (D16S468/D16S3070–D16S3373) that was studied intensively by both groups. Each map shows a high gene density in this critical region, but, particularly in the telomeric part of this interval farthest from MEFV, there are transcripts unique to each map. Although several of these are characterized incompletely, the present report provides detailed sequence data on a novel caspase pseudogene not identified by Bernot et al. (1998) and also explores the genomic organization of a large tRNA cluster reported but not characterized in the other map. In their paper, Bernot et al. identified a novel metalloproteinase (MMP20) and zinc-finger gene (ZNF206) at the telomeric end of the ∼225-kb region that were not characterized in our transcript map. The two reports also provide complementary data on the transcript map centromeric toMEFV. Our map includes 17 transcripts in the ∼450 kb between D16S3373 and cosmid RT70, permitting a more complete analysis of the zinc-finger and olfactory-receptor clusters in the FMF region, whereas Bernot et al. (1998) present a number of new transcriptional units that are even more centromeric on 16p13.3. The combined data provide a valuable resource both for studying the genomic organization of this region, and for possible new positional cloning projects.
RESULTS
Transcript Identification
A combination of direct cDNA selection, exon amplification, and computational prediction from genomic sequence were used to screen for transcribed sequences in the 285 kb of genomic DNA betweenD16S468/D16S3070 and D16S3376, whereas only direct selection and sample sequencing of onefold (1×) clone coverage of the region were used for the 50-kb genomic DNA telomeric toD16S468/D16S3070 and the 400-kb genomic DNA centromeric toD16S3376. Figure 1 is a schematic representation of the transcripts identified in the overall ∼700-kb genomic region. Detailed information on each transcript is summarized in Tables 1 and 2. Representative Northerns showing the transcript size and tissue distribution pattern of four transcripts identified in this region are presented in Figure 2.

Schematic representation of the FMF transcript map. Positions of transcripts are shown relative to a composite genetic and physical map of the FMF region. Genetic markers are indicated by gray bars. All physical distances are indicated in kb. The map is drawn in three levels with resolution increasing on descending levels. Transcripts are not drawn to scale. (A) Transcripts identified by direct selection in the flanking intervals are indicated as are the physical map positions of large-insert genomic clones used for analysis. The position of cosmids subjected to partial DNA sequencing in the region telomeric of D16S468/D16S3070 and centromeric ofD16S3376 are also shown. (B) Transcripts identified using multiple methods are shown in the second level relative to a complete EcoRI restriction map of the interval. A contig of overlapping cosmids which were used for partial DNA sequence analysis are also shown, with those represented by blue lines sequenced at LANL and those in red sequenced at the NHGRI. Exon-trapping was performed on genomic DNA extending from cosmid 363D9 to PAC 273L24 (shown inA). (C) The 87-kb segment of genomic DNA sequenced to completion is indicated in purple. The organization of a tRNA cluster within this interval is indicated with the number, orientation, and map position of 18 tRNA genes. Nearby flanking Pol II transcripts are shown.
Transcripts Identified by Direct Selection Telomeric ofD16S468/D16S3070 and Centromeric ofD16S3376
Transcripts Located between D16S468/D16S3070 and Cosmid 377A12
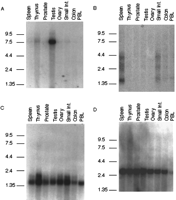
Northern blot analysis of four transcripts from the FMF interval. Autoradiograms of human multitissue Northern blots probed with [32P]dCTP-labeled cDNA are shown. Blots were probed with the following cDNAs: (A) clone CR17-26; (B) V9; (C) CR18-9; (D) ZNF174.
cDNA Selection
Five independent cDNA selection experiments were performed for the 700-kb genomic DNA extending from ∼50 kb telomeric toD16S468/D16S3070 to ∼50 kb centromeric to D16S2622(Fig. 1A). The cDNAs recovered were pooled to construct a regional genomic DNA-specific cDNA library of 1489 clones. Individual colonies were picked, and their inserts used as hybridization probes to determine their redundancy in the library. Forty different groups of cross-hybridizing clones were identified by Southern hybridization employing extensive blocking of repetititve sequences with both human Cot-1 DNA and sonicated denatured human placental DNA, and high stringency posthybridization washes. Each group included a minimum of two clones and a maximum of 54 cross-hybridizing clones from the original cDNA library. The average insert size was ∼600 bp, and thus they are more likely to be gene fragments than full-length cDNAs.
Partial or complete sequence (depending on the size of the insert) was determined for a representative clone from each group, and the sequence obtained was used to search the nonredundant GenBank database (nr), the database of expressed sequence tags (dbEST), and the combined finished/sample sequence of genomic DNA from the FMF candidate region to determine homology to known genes, presence of repetitive elements, and the likelihood that the cDNA was derived from the FMF genomic region. In addition, Southern hybridization of clone inserts toEcoRI digests of DNA from cosmids, chromosome 16-specific somatic cell breakpoint hybrids (Callen et al. 1992), and total human genomic DNA was performed on most of the clones to confirm their mapping to the FMF region. Assignment to the FMF region required either >90% sequence identity with the FMF region finished/sample sequence database or comparable hybridization patterns on total human genomic and somatic cell hybrid or cosmid DNA. For those clones that physically mapped to the region and had no appreciable repetitive elements, Northern analyses were performed to determine the size of the transcripts and to confirm that physically adjacent groups of cDNA clones really were fragments of the same gene.
Of the 40 groups of cDNA clones, 31 mapped back to the starting genomic DNA according to our mapping criteria. Nine different groups of cross-hybridizing clones were excluded from further study because they did not meet criteria in the FMF region genomic sequence database and gave inconclusive Southern patterns in mapping studies. Northern data were available for only 23 of the 31 groups that mapped to the region, because the other eight groups of clones contained low-copy-number repetitive elements such as MER9, MER11, and LINE repeats, and were not used as probes on Northern studies.
Table 1 summarizes transcripts identified by direct cDNA selection from genomic regions telomeric to D16S468/D16S3070 and centromeric to D16S3376. Together, 16 different transcripts were direct-selected from these two regions. Two of these are identical to, and two are similar to, genes described by others (Table 1). cDNA clones identical to genes that have been described previously are NC4-5 (ZNF174, Williams et al. 1995) and CR20-8 (DNASE1,Yasuda et al. 1995), both of which have been mapped to chromosome 16p13.3 already. Transcripts similar but not identical to previously described genes include NC15-25 and CR6-24. The former is homologous to two members of the human heat shock protein 90 (hsp90) family: human tumor necrosis factor type 1 receptor-associated protein (Song et al. 1995) and heat shock protein 75 (Chen et al. 1996), whereas the latter is homologous to ribosomal protein L18 (Puder et al. 1993).
Between D16S468/D16S3070 and D16S3376, direct selection identified transcripts Gene 1, NK4, part of PL59,ZNF213, ZNF200, FPM315, and Clone 7 (Table2). NK4 and FPM315 are genes that were described previously (Dahl et al. 1992; Yokoyama et al. 1997). In addition,ZNF213 (designated as CR53) and ZNF200 were found independently by a second group searching for the FMF gene (French FMF Consortium 1997; Bernot et al. 1998). Together, direct-selected clones account for seven of the 15 polymerase II (Pol II) directed transcripts identified from the D16S468/D16S3070–D16S3376 interval using a combination of several transcript identification methods (Fig. 1; Table 2). As expected, transcripts not present or present at very low levels in the cDNA pool used for direct selection, including V9,MEFV, olfactory receptors (OR1F1 and OR1F2), and the two V57 transcripts (V57c7 and V57c10), were not identified by this method.
Exon Amplification/Trapping
Internal exon trapping (Buckler et al. 1991) was done on a tiling path of cosmids from 363D9 to 23G10 and on PAC clone 273L24, and 3′ exon trapping (Krizman and Berget 1993) was done on cosmid 335H1 (Fig.1). Approximately 400 putative trapped exon clones were obtained and characterized in terms of their map locations and sequences. A total of 28 independent trapped exons with unique sequences were recovered and physically mapped to the interval. Both 5′ and 3′ RACE (Frohman et al. 1988; Loh et al. 1989) and solution hybridization (GeneTrapper, Life Technologies, Gaithersburg, MD) in several cDNA libraries were performed to extend these exon clones into full-length transcripts. Such efforts led to the identification of transcripts V9, PL59, V57c10, V57c7, and the gene responsible for FMF (MEFV) (International FMF Consortium 1997), with MEFV also cloned independently by another group (French FMF Consortium 1997). In total, trapped exons were present in eight of the 15 Pol II transcripts identified within the interval subjected to exon trapping (Table 2). Intronless genes, such as the olfactory receptors and the tRNA genes, were missed by this method.
Transcript Prediction by Partial Genomic Sequencing
An overlapping set of 16 cosmids were subjected to sample sequencing, using two different strategies at two different centers (Fig. 1B). A twofold (2×) coverage of these cosmids was achieved. In addition to BLASTN and BLASTX (Altschul et al. 1990) search of known genes and ESTs, sequences obtained were also subjected to exon prediction by GRAIL analysis (Uberbacher and Mural 1991). Although the depth of coverage within particular regions in the 363D9–377A12 tiling path was highly variable, sequences from all 16 Pol II transcripts identified from these cosmids were present in whole or in part within the combined 2× sample sequence database (Table 2). However, novel transcripts with weak homology or no homology to sequences identified previously, such as V57c7, and V57c10, though present in the 2× sample sequence, were not identified as Pol II transcripts by such analyses.
EST clones with greater than 85% homology to the partial DNA sequences were obtained from the IMAGE Consortium and physically mapped toEcoRI fragments within the interval. The validity of mapping was further evaluated by comparing the complete sequence of the IMAGE clone with the genomic sequence, where this was available. This led to the discovery of transcripts represented by EST clones R44239 andT99017 (ZNF210) (Table 2), both of which were also found byBernot et al. (1998) (and designated d5l2 and ZNF205) by a similar approach. In addition, EST clones highly homologous to Gene 1,NK4, ZNF200, and FPM315 also mapped back to the interval, confirming the presence of these transcripts within the FMF region (Fig. 1, Table 2). Transcripts ZNF213,OR1F1, OR1F2, OR2C1, and MEFV could also be identified from analysis of partial genomic DNA sequences. Significant homology to tRNA genes of multiple specificities from several species was identified in cosmids 360A8, 414G4, 347C2, and 399A10. Homology to several CpG island clones was also found in sequence from these cosmids.
Transcript Prediction by Complete DNA Sequencing
Relatively few cDNAs were identified in the region bordered by transcripts V9 and ZNF200 using the aforementioned combined approaches. To facilitate identification of transcribed sequences within this region, and to further delineate the genomic organization of ZNF213, V9, and ZNF200, an 87-kb DNA segment bordered by the 1.5-kb EcoRI fragment of cosmid 23G10 and the 0.5-kb EcoRI fragment of cosmid 442E2 was sequenced to completion (Fig. 1B; GenBank accession no. AF091512). The genomic sequences derived were analyzed for transcripts using exon prediction programs GRAIL-2 (Uberbacher and Mural 1991) and Gene Finder (Solovyev et al. 1994). A total of 16 exons, including four with Alu or LINE repetitive elements, were predicted by GRAIL-2, whereas 36 exons, including seven with Alu or LINE repetitive elements, were predicted by Gene Finder. The two exon-prediction programs identified relatively distinct DNA sequences as exons, with only one identical and four partially overlapping sequences considered as exons by both programs. Interestingly, the DNA sequence predicted as an exon by both programs corresponded precisely to that of V57c7 exon 2. Both 5′ and 3′ RACE were performed to extend the predicted nonrepetitive exons in cDNAs made from mRNAs of six distinct tissues and two myelogenous cell lines. Except for v57c7 exon 2, no bona fide extension products were identified, suggesting a relatively high rate of false positives for exon-prediction programs in this genomic region. Bernot et al. (1998) reported similar findings in their analysis of sequence from this region.
Transcript Descriptions
tRNA Cluster
Although no additional Pol II transcripts were found within the 87-kb region by complete DNA sequencing/exon prediction, DNA sequences likely to encode 18 tRNA genes (3 tRNAArg, 8 tRNAPro, 5 tRNALys, and 2 tRNA pseudogenes) clustered within a 46-kb genomic interval in the central portion of this region were identified by analysis of the sequence with tRNAscan-SE software (Lowe and Eddy 1997) (Fig. 1C). Previously characterized tRNA clusters are thought to have evolved by multiple duplication events (Santos and Zasloff 1981; Buckland 1989; Buckland et al. 1996). Consistent with a similar evolutionary heritage for this cluster, it had been noted that V57c7 exon 1 and V57c10 exon 1 share 73% sequence identity. In addition, DNA segments immediately adjacent to these exons hybridized to more than one location within the interval.
To characterize the repeat structure of this region more fully, DNA sequences between tRNA genes were compared and several fragments with DNA sequence similarity were identified. In addition, the location and orientation of tRNA genes, V57 exons, Alu repeats, and low complexity repeats within these regions was determined. This analysis revealed multiple copies of two major repeat motifs within the cluster, including seven copies of the repetitive segment designated A-repeat, and five copies of a second putative repetitive element designated B-repeat (Fig. 1 C).
The distinguishing structural elements of the A-repeat are, in order: tRNA, V57c7 gene exon 1-like sequence, tRNA, V57c7 exon 2-like sequence, adenine-rich simple-repeat, and V57c7 exon 3-like sequence. Two highly homologous nonadjacent copies of this repeat, present in an inverted orientation, were found in the DNA segments containing the V57 genes (Fig. 1C). It is interesting to note that although these two genes have homologous exon 1 sequences, splicing of the remaining exons is not identical. This differential splicing appears to be caused by mutations in the cis-acting splice regulatory elements of these two genes. This duplication event is therefore likely to be an example of nonreciprocal recombination. Sequence conservation among the remaining A-repeats is weaker, deviating because of apparent truncation of the full-length A-repeat sequence, introduction of Alu and low complexity repeats within the A-repeat sequence, and sequence drift. Although vestiges of the V57 exon sequences were identified in several of these additional A-repeat copies, no additional cDNAs were identified in the region.
The B-repeat has a relatively simple structure, with a tRNA followed by 6–8 Alu repeats. As with the A-repeat, copies of this repeat appear to diverge because of truncation, variation in the numbers ofAlu repeats, and sequence drift. The organization of the repeats within this cluster (Fig. 1C) are consistent with its having evolved from multiple nonreciprocal recombination events.
C2H2 Zinc-Finger Genes
At least six of the transcripts identified within the ∼700 kb genomic region encode proteins with C2H2 zinc-finger motifs. These zinc-finger encoding genes are, from telomere to centromere,ZNF210 (ZNF205), ZNF213, ZNF200,FPM315 (clone 10), clone 7, and ZNF174 (NC4-5) (Fig.1; Table 2). Clone 7 mapped between FPM315 and ZNF174and showed significant homology in both BLASTN and BLASTX searches toZNF75 (Table 2), the prototypic member of a subfamily of unique zinc-finger proteins (Villa et al. 1993,1996). Because it mapped outside of the minimal candidate interval for FMF (betweenD16S3405 and D16S3373), this particular zinc-finger gene was not characterized in detail. ZNF200 (Deng et al. 1998; French FMF Consortium 1997), FPM315 (Yokoyama et al. 1997), and ZNF174 (Williams et al. 1995) have been described. However, the map location of FPM315 was unknown previously.ZNF210 and ZNF213 were designated ZNF205 and CR53, respectively, by Bernot et al. (1998), but were only partially characterized. They map within 7 kb of one another and are transcribed in the same orientation (Fig. 1B). Detailed description of these two genes can be found elsewhere (Deng et al. 1998; X. Chen, M. Hamon, Z. Deng, M. Centola, R. Sood, K. Taylor, D.L. Kastner, and N. Fischel-Ghadsian in prep.). A seventh zinc-finger gene (ZNF206) ∼20 kb telomeric to ZNF210(ZNF205) was identified by EST hits, but has not been characterized fully (Bernot et al. 1998).
ZNF174 (Fig. 2D), ZNF213, and FPM315 are expressed in all of the eight tissues tested. Their predicted proteins all contain several zinc-finger motifs and a LeR domain/SCAN box (Fig.3A), a stretch of ∼70 amino acids rich in leucine that is tethered either alone or in association with a KRAB box to the C2H2 finger domains (Pengue et al. 1994; Williams et al. 1995). Such similarities suggest that they are members of the same subfamily of zinc-finger genes. In addition to the LeR/SCAN domain, a KRAB A domain (Bellefroid et al. 1991) is present in both ZNF213 andFPM315 (Fig. 3A). Although the number of zinc-finger motifs differ among these three genes, the zinc fingers themselves have a sequence identity as high as 77% at the amino acid level (Fig. 3B). However, no significant sequence homology was found among them in regions other than the functional domains mentioned above.
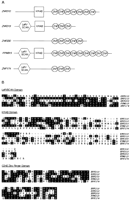
Conserved structural and sequence motifs in five zinc finger genes from the FMF region. (A) Spatial relationships of the zinc finger domains (ZNF), Krüppel-associated box (KRAB), and the LeR/SCAN domains for the five genes are shown. (B) Sequence alignment of the amino acid sequences of the five clones within the three conserved domains are shown. Residues shared among two or more clones are shaded.
ZNF210 (ZNF205), which also has a ubiquitous expression pattern, encodes a predicted protein with seven zinc-finger motifs and a KRAB box (both KRAB A and part of KRAB B) (Fig. 3A) (Deng et al. 1998). Its zinc fingers are strongly homologous to those ofZNF213 (Fig. 3B). ZNF200, however, has tissue-specific expression, with its message seen primarily in testis. Its predicted protein encodes only five zinc fingers and no other recognizable domains (Fig. 3A). Its zinc fingers are homologous to those found in ZNF210 (ZNF205), ZNF213,FPM315, and ZNF174, but the homology is not as strong as that found among the latter four zinc-finger proteins (Fig. 3B).
Alignment of the protein sequences of these five zinc-finger genes using Block Maker, a computer program that identifies conserved sequence blocks in multiple DNA/protein sequences (Henikoff et al. 1995), revealed blocks of amino acids in ZNF200 that are suggestive of vestigial LeR/SCAN and KRAB domains (Fig. 3B). However, significant sequence divergence has accumulated in these two domains to render them different from the others.
Genes of Immunological and Inflammatory Relevance
Transcript identification in the genomic region covered by cosmids 49B4 and 383E2 (Fig. 1B) by both direct cDNA selection and sequence-based transcript prediction recovered a transcript identical to NK4, a gene cloned previously from activated natural killer (NK) cells (Dahl et al. 1992). The ∼1-kb message of NK4 was recovered completely in cDNA form by cDNA selection, and was found to be encoded on seven exons interrupted by six introns (Fig.4A). Bernot et al. (1998) reported a similar genomic structure, but without the first 5′ untranslated exon. An in-frame alternative splicing event resulting in a short version of theNK4 protein was observed in which 138 nucleotides of the fourth exon were spliced out (Fig. 4A). In addition, a single nucleotide transition (G to A) at nucleotide 342 that changes the codon for aspartate to asparagine was also observed. Both the alternatively spliced form of NK4 and the missense change were observed as frequently in FMF patients as in controls, suggesting that they are common polymorphisms.
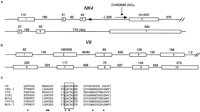
Genomic structure of NK4 and V9 and V9 caspase homology. Exon and intron sizes are shown in bp. (A) Genomic structure ofNK4 is shown with sizes of the two splice variants of exon 4 indicated. The location of marker D16S3082 within intron 3, and an Alu repeat within intron 6 are shown. (B) Genomic structure of V9 is shown with sizes of the two splice variants of exon 2 indicated. (C) Conserved residues of caspase family present in V9 are shown.(○) The P1 aspartate-binding residues; (•) catalytic residues in the amino acid sequence of 7 caspase family members and the putative protein sequence V9 encoding caspase homology. The conserved catalytic pentapeptide characteristic of the caspase family is boxed.
Two different exons were recovered by exon amplification/trapping using cosmid 23G10 as template, and mapped back to the 9.6-kb EcoRI fragment of this cosmid (Fig. 1B). Extension of these exons by solution hybridization recovered five cDNA clones that range in size from 1420 to 1435 bp. When sequenced, these clones had identical, overlapping DNA sequences and terminated with a consensus polyadenylation signal (AAUAAA) followed 12–18 bp downstream by a homoadenine stretch. The sequences of both exons were present in all five cDNA clones, suggesting that they are bona fide extension products of these exons. On a Northern blot made from mRNAs of multiple human tissues, at least four bands were discernible in mainly spleen and small intestine (Fig.2B). The band with the strongest intensity is ∼1.4 kb in size. The other bands have lower intensity and range in size from 2.4 to 4.4 kb (Fig. 2B). The 1.4-kb version of this gene, designated V9 after one of the initial trapped exons, is encoded by 10 exons (Fig. 4B).
To obtain cDNA for the other splice variants, 5′ RACE was done, and three additional cDNA clones were identified. Two of these had slightly longer 5′ ends, 3 and 5 bp, respectively, but were otherwise identical to cDNAs isolated previously. In the third 5′ RACE clone, the 5′ end of exon 1 was extended by 339 bp and the 3′ boundary of exon 2 was extended by 13 nucleotides relative to the splice form identified in the spleen and testis (Fig. 4B), consistent with multiple splice forms observed by Northern analysis. The 5′ most methionine codon (ATG) was found more than 600 bp downstream of the 5′ termini of the two splice variants identified. Preceding this ATG, in-frame stop codons were recognizable in both splice forms. However, in vitro transcription/translation failed to identify any translation products for either form, raising the possibility that these two splice variants might be nonfunctional.
No homology with V9 was demonstrated to any nucleotide sequences in either the nr or the EST databases. However, a search of the protein database by BLASTP (Altschul et al. 1990) using the putative translation product of V9 revealed significant homology to the caspase family of cysteine proteases (Alnemri et al. 1996; Henkart 1996). The catalytic pentapeptide QACRG, a hallmark of cysteine proteases (Walker et al. 1994; Wilson et al. 1994), as well as the conserved P1Asp binding and catalytic residues, was present in the putative translation product of V9 (Fig. 4C), suggesting that, if it is functional, this gene encodes a cysteine protease.
Olfactory Receptors
In the interval between D16S468/D16S3070 and cosmid 377A12, we identified by genomic sequencing three DNA sequences, OR1F1(OLFmf), OR1F2 (OLFmf2), and OR2C1(OLFmf3), with a high degree of sequence homology to olfactory receptor (OR) genes (Buck and Axel 1991). OR1F1 andOR1F2 were recovered independently in similar studies by another group (French FMF Consortium 1997; Bernot et al. 1998).OR2C1 maps to the centromeric boundary of the interval, and is strikingly homologous to the mouse olfactory receptor 3 gene (olf3–MUS) (Nef et al. 1992), displaying 81% sequence identity at the DNA level and 82% sequence identity at the amino acid sequence level (Fig. 5). Its putative open reading frame is colinear with the entire 310 amino acids of the mouse geneolf3–MUS, and thus is likely to encode an olfactory receptor of the 2C class, of which olf3–MUS is a prototypic member (Lancet and Ben-Arie 1993).

Olfactory receptor amino acid sequence alignment. The amino acid sequences of the open reading frame of the three FMF region olfactory receptor genes are shown. These sequences are aligned with those of prototypic members of the 2C (olf3–MUS) and 1F (or5–RAT) subfamilies. Amino acid residues present in more than one clone at a given position are shaded. Highly conserved residues, including two cysteines (•), a serine (○), and a SY and MAYDRFVAVC sequence motif (underlined), thought to be involved in OR structure or function are indicated. The seven hydrophobic putative transmembrane-spanning domains are demarcated and numbered with Roman numerals.
OR1F1 and OR1F2 map in the middle of the interval and are within <10 kb of one another. They have not only the same transcription orientation, but also 86% of their nucleotides are identical, suggestive of gene duplication. When searched against the nr database they are highly homologous to rat olfactory receptor 5 (or5–RAT), a prototypic member of the olfactory receptor 1F family (Lancet and Ben-Arie 1993). The predicted open reading frames of these two putative ORs bear 81% and 90% sequence identity at the nucleic acid level, and 76% and 88% sequence identity at the amino acid level to the rat sequence, respectively (Fig. 5). The putative translation start of OR1F1 coincides with the conserved translational start site for the 1F family of ORs, whereas the most 5′ ATG inOR1F2 is 99 bases downstream, resulting in a truncated protein lacking 33 amino acids that correspond to the extracellular amino-terminal domain and the proximal half of the first putative hydrophobic transmembrane region. Moreover, this ATG is not present in a sequence context that is conducive to in vivo translation (Kozak 1989), raising the possibility that OR1F2 is severely truncated or is a pseudogene, as has been suggested by others (French FMF Consortium 1997; Bernot et al. 1998).
Several conserved sequence motifs were recognizable in all three of the ORs identified in the FMF region (Fig. 5). They include two highly conserved cysteine residues at positions 97 and 169, a “MAYDRYVAIC” motif spanning residues 118–127, a SY motif at residues 217–218, and a serine residue (position 230) thought to be a site of phosphorylation (Ben-Arie et al. 1994). Given their strong sequence homologies to known ORs, these three OR-like sequences were not tested on Northerns, and therefore their tissue-expression profiles are unknown. However, one cDNA clone was identified from a human brain cDNA library, in which the majority of the OR1F1 sequences are spliced between the third and the fourth exons of V57c7 (Fig. 1B).
Other Genes
In addition to transcript identification, exon amplification/trapping and transcript prediction by genomic sequencing also helped to reveal the genomic structure of PL59, V57c7, and V57c10. By exon amplification, three independent exons were recovered that map to the 6.9-kb EcoRI fragment of cosmid 23G10 (Fig. 1B). Extension of these exons by RACE and solution hybridization in cDNA libraries identified six cDNA clones. They are nonidentical but related in that they all contain the three trapped exons. When sequenced and their sequences compared to each other and to the finished genomic DNA sequences in this region (Fig. 1B,C), these clones were found to be products of differential splicing from a single gene, designated PL59.
Similarly, extension of a trapped exon that maps to the adjacent 2.7- and 2.6-kb EcoRI fragments of cosmid 399A10 (Fig. 1B) recovered two cDNA clones (V57c7 and V57c10) (Table 2) from a human brain cDNA library. Comparison of their sequences to each other and to finished genomic sequences revealed that they encode two distinct genes transcribed in opposite orientations (Fig. 1B). cDNA V57c7 is 0.7 kb in size, has four exons and spans 40 kb of genomic DNA, whereas cDNA V57c10 is only 0.5 kb in length, has two exons and covers only 2.8 kb of genomic DNA. They are related through their 5′-most exons. The original trapped exon corresponds to the 5′-most exon in V57c7. However, the 5′-most exon of V57c10 has 78% sequence identity to the original trapped exon as well, suggesting possible inverted duplications in this genomic region.
DISCUSSION
In this report we present a transcript map encompassing ∼700 kb of genomic DNA on 16p13.3. This region is part of a 4-Mb contig stretching telomeric to the region containing the polycystic kidney disease locus (PKD1) (Dackowski et al. 1996) and centromeric into the contig surrounding the Rubinstein-Taybi syndrome locus (RSTS) (Giles et al. 1997). Thirty-two Pol II transcripts and 18 tRNA genes were identified from the 700 kb characterized in this report. Of these, 11 were described previously, including seven ( R44239, ZNF210/205, ZNF213, OR1F1,OR1F2, ZNF200, and MEFV) that were recovered independently by another group searching for the FMF gene (French FMF Consortium 1997; Bernot 1998), and four that were identified by others (NK4, FPM315, ZNF174, and DNASE1).
The transcript identification strategy used in this study has been dictated by our primary interest in the FMF gene. Such a strategy, in turn, has resulted in the construction of a transcript map with three levels of resolution. Telomeric to D16S468/D16S3070 and centromeric to D16S3376, the map is less comprehensive with only data obtained by direct selection and sample sequencing presented. Even if direct selection were 100% sensitive, the transcripts identified in these two regions still fall short of the coding potentials of the genomic DNA in these regions, because transcripts not present in the cDNA pool used for direct selection cannot be identified. In addition, transcripts identified from these areas were less well characterized than those recovered from the area betweenD16S468/D16S3070 and D16S3376, as refinement of the candidate interval excluded these flanking regions soon after the transcripts were identified. The only data available for each transcript identified in these two regions are its partial sequence, map location, size, and tissue distribution of expression. In contrast, the map between D16S468/D16S3070 and D16S3376 is more inclusive. The use of multiple transcript identification methods in this central region allowed a more comprehensive evaluation of its coding potential. In addition, most of the transcripts recovered from this region, especially those between D16S3405 andD16S3373, have been well characterized, including detailed information on their genomic structures. The portion of the map that contains the most detail is that between the 1.5-kb EcoRI fragment of 23G10 and the 0.5-kb EcoRI fragment of 442E2 (Fig.1B,C). In this area, four methods of transcript identification were utilized, including complete DNA sequencing of the interval. Exhaustive RACE analysis of all nonrepetitive putative exons identified in vivo by exon amplification/trapping or in silico by exon prediction was also completed. These studies, in conjunction with analysis of the DNA sequence by tRNAscan software, enabled the construction of a transcript map that is likely to be the most comprehensive of the region (Fig. 1B,C).
In agreement with previous studies (Brody et al. 1995; Harshman et al. 1995; Yaspo et al. 1995), we have found that individual methods of transcript identification were complementary and that no single method succeeded in recovering 100% of the transcribed sequences encoded in the genomic region studied. Of the 15 Pol II intron-containing transcripts identified in the ∼285 kb genomic region betweenD16S468/D16S3070 and D16S3376 subjected to exon amplification/trapping, eight were identified by this method. Nine of the 28 independent trapped exons were not assigned to specific transcripts. It is likely that most of these nine were false positives, given that each was subjected to RACE and solution hybridization with multiple cDNA libraries, though the possibility remains that one or more of these could be expressed in tissues that were not screened. Whereas intronless genes are unlikely to be identified by this method, its advantage of being expression-independent is demonstrated by its ability to recover V9 and MEFV, transcribed sequences not identified by direct selection presumably because of their lack of expression in the libraries used for cDNA selection. As expected, intronless genes, such as the three putative ORs and the tRNA genes, were not identified by exon amplification. Seven of the 15 Pol II transcripts found between D16S468/D16S3070 andD16S3376 were cloned by direct selection using mRNAs from fetal brain, fetal liver, and lymph node as sources for cDNAs. Genes not expressed in these tissues, including V9, MEFV, and the olfactory receptors, were not identified. In addition, ubiquitously expressed R44239 and ZNF210 (ZNF205) were also not identified by this method.
Transcript prediction by genomic sequencing proved to be a powerful adjunct to these analyses in that the 3 ORs and the tRNA genes, which were not identifiable by other methods, were found by this approach. In addition, sequences from all 16 Pol II transcripts from the ∼315-kb 363D9–377A12 tiling path were present in whole or in part within the combined finished/sample sequence database with at least one exon predicted for each of the exon-containing genes from the region (although predicted exon boundaries were not exact in all cases) (Table2). This should not, however, be taken as evidence that finished sequencing has a sensitivity and specificity of 100%. Actually, novel transcripts with weak homology or no homology to previously identified sequences, such as the V57 transcripts, were not recognizable by this method until their corresponding cDNA clones were recovered by other methods and sequenced. Even with definite transcribed sequences, such as ZNF213 and ZNF210 (ZNF205), only one or two of their exons were recognized as such by GRAIL analysis. Moreover, most of the exons predicted in the 87 kb of finished genomic sequences using either GRAIL or Gene Finder appear to have been false positives.
Several interesting properties of this genomic region have become clear in the construction of this transcript map. Our data indicate that the average Pol II transcript content in this region of chromosome 16p13.3 is at least one gene per 20 kilobases of genomic DNA, a content approximately twice of that estimated for the whole human genome (Antequera and Bird 1994; Fields et al. 1994). The actual gene content is likely to be higher than this estimate because in regions telomeric to D16S468/D16S3070 and centromeric to D16S3376, only a portion of the coding potential has been elucidated. The gene density of two other regions of the Giemsa-light 16p13.3 band have been published recently. Based only on exon-trapping, Burn et al. (1996)estimated the gene frequency recently in the 700-kb PKD1contig, which is ∼450 kb distal to the interval presented here, at a minimum of one gene every 40 kb. Flint et al. (1997) estimated the gene frequency recently in the distal region of 16p13.3 by analysis of ∼285 kb of sequence. Their estimate of one gene per 20 kb agrees well with the present report. It will, of course, be of great interest to determine the gene density for contigs in the Giemsa-dark, possibly gene-sparse, 16p13.2.
Structurally, the FMF genomic region is complex, with a high gene content and areas of possible duplications and inversions. Distribution of these transcribed sequences along this stretch of genomic DNA is uneven, with transcript-rich and -poor regions alternating. This is true especially in the 285-kb FMF central region (betweenD16S468/D16S3070 and D16S3376), where the transcripts identified more likely represent its full coding potential. As shown in Figure 1, the flanks of this region are rich in RNA polymerase II-directed transcripts, whereas the segment bordered by V9 andZNF200 contains few such sequences, and is comprised predominantly of RNA polymerase III-directed tRNA genes.
Three families of genes account for the majority of transcribed sequences encoded in the 285-kb central region. The largest gene family in this region, in terms of genomic coverage, is the zinc-finger gene family. The six members in this family identified in this report span almost the whole 285 kb between D16S468/D16S3070 andD16S3376 plus another ∼73 kb centromeric toD16S3376, and thus constitute the organizing structural feature for this region of genomic DNA. Bernot et al. (1998) described a seventh partially characterized zinc-finger gene (ZNF206), also in this interval. ZNF210/205, ZNF213, FPM315, and ZNF174 are closely related. They not only have the same ubiquitous tissue expression patterns but also encode proteins with similar structural features (Fig. 3A).
Clustered organization of related zinc-finger genes on both human and mouse chromosomes has been reported previously (Bellefroid et al. 1991;Crossley and Little 1991; Huebner et al. 1991; Hoovers et al. 1992;Lichter et al. 1992; Rousseau-Merck et al. 1992; Calabro et al. 1995;Shannon et al. 1996; Lee et al. 1997). Gene duplication during evolution has been suggested as the origin of such zinc finger clusters, although rarely have the evolutionary events leading to such duplications been defined (Bellefroid et al. 1991; Crossley and Little 1991; Shannon et al. 1996). The fact that ZNF213,FPM315, and ZNF174 all have significant sequence homologies to Zfp51 (or Zfp38), a member of a cluster of related zinc-finger genes on a portion of the mouse chromosome 17 that is deleted in the mouse embryonic lethal mutationtw18 (Crossley and Little 1991), suggests that the human and the mouse clusters might be homologs of one another. Adding further evidence to this contention is the observation that this mouse zinc finger cluster lies immediately centromeric to the mouse chromosomal area syntenic to human chromosome 16p13.3 (Himmelbauer et al. 1992;Olsson et al. 1995, 1996). If proven, this not only extends the human/mouse syntenic chromosomal region, but also gives us a clue to the origin of such a zinc-finger gene cluster in humans.
Likely, gene duplication in response to requirements posed by embryonic development provides the impetus for the formation of such a gene cluster in both mice and humans because of the association between the mouse zinc finger cluster and the mouse embryonic lethal mutationtw18 (Crossley and Little 1991). Even if this is the case, however, the duplication event must have happened long ago because individual zinc finger genes in both the human and the mouse clusters have accumulated significant sequence differences over regions free of functional constraints. Another line of evidence that these human zinc-finger genes are closely related and might be important in embryonic development and differentiation comes from the observations that their zinc fingers have an amino acid sequence identity as high as 77%. In lower organisms, it has been demonstrated that only those zinc finger genes that are important in the control of embryonic development and differentiation have their zinc fingers highly conserved (Zarkower and Hodgkin 1992; Garriga et al. 1993; Wimmer et al. 1993; Pieler and Bellefroid 1994).
Another gene family that constitutes a striking structural feature for this region of genomic DNA is the tRNA gene family. The human genome contains an estimated 1300 tRNA genes (Hatlen and Attardi 1971). Both individual genomic tRNA genes and tRNA gene clusters have been reported previously in humans (Zasloff and Santos 1980; Santos and Zasloff 1981;Roy et al. 1982; Buckland et al. 1983; Buckland 1989; van der Drift et al. 1994; Buckland et al. 1996). In none of these cases, however, is the complete sequence of a major human tRNA cluster fully delineated. The 46-kb genomic tRNA cluster identified in this work contains a total of 18 tRNA genes, including eight tRNAPro, 4 tRNALys, 4 tRNAArg, and two pseudo-tRNAs. Our analysis of this region has revealed two major repeat motifs the location and orientation of which are suggestive of extensive nonreciprocal recombination events having given rise to this cluster. The highest DNA sequence homology among the repeats is between the DNA segments containing V57c10 and the first three exons of V57c7. This repeat is easily visualized in the symmetrical arrangement of the tRNALys and tRNAArg genes around the two antiparallel transcripts (Fig. 1C). This structure is noteworthy in that the 5′ exons of these two genes are nearly identical, suggesting that the ancestral promoter of the original gene was maintained in the duplication event. In addition, these genes differentially splice, subsequent exons are not derived from homologous sequences within the repeat, and as such the divergence of these genes is likely to be an example of the early evolution of a novel human gene.
The last group of structurally related genes suggesting genomic duplication in this region is the OR family. ORs are G-protein coupled seven-transmembrane-domain receptor proteins involved primarily in the detection of odorant ligands, although their expression in testis suggests possible involvement in spermatogenisis or sperm function as well. Members of this superfamily share at least 35% amino acid homology, yet they are sufficiently different to have eight classes, with each class having several subclasses (Lancet and Ben-Arie 1993; Ben-Arie et al. 1994). Sharing 76% and 88% amino acid identity, respectively to Rat OLF-5, the prototypic member of the 1F subfamily, OR1F1 and OR1F2 likely encode additional members of this subfamily. On the other hand, OR2C1 bears strong sequence homology to olf3-MUS, the 2C subfamily prototype. Six additional human ORs of this subfamily were recently identified and mapped by FISH analysis to the interval 16p13.1–16p13.3 (Rouquier et al. 1998).
OR1F1 and OR1F2 are 86% identical at the nucleic acid level. They are tightly clustered, codirectional, and arranged in tandem with no cistrons identified in the intergenic region. Similar features have also been observed in a cluster of ORs identified on human chromosome 17p13.3, a cluster hypothesized to have arisen, in part, through a tandem-duplication event (Glusman et al. 1996), suggesting that a similar event may have also occurred in theOR1F1 and OR1F2 region.
Dispersed among members of the three gene families are three genes that are structurally different yet functionally similar in their probable involvement in host defense. Isolated from NK cells that have undergone stimulation by mitogens and interleukin 2 (IL-2), NK4 encodes a protein that constitutes a possible component in the activation pathway common to both NK and T lymphocytes (Dahl et al. 1992), and may have the potential to participate in both innate and specific immune responses. The second gene in this category is MEFV. With a granulocyte-specific message of 3.7 kb, this gene encodes a protein belonging to the RoRet gene family (International FMF Consortium 1997; French FMF Consortium 1997). Although its mechanism of action is unknown currently, mutations of MEFV in FMF patients assign this gene a role in regulating inflammation. Another gene in this region that may participate in host immune responses and inflammation control is the V9 gene. The conceptual translation product of this gene is homologous to caspases and contains the structural domains indispensable for caspase function. Caspases are regulators of programmed cell death, mediating both the transduction of the death signal and the degradation of cellular substrates. As such, they are required for proper embryonic development, cellular homeostasis, activated immune cell turnover, and regulated killing and removal of infected and metastatic cells by the immune system in complex multitissue organisms (Henkart 1996). Consistent with the functional properties suggested by its sequence homologies, V9 is expressed mainly in spleen and small intestine, organs rich in lymphoid tissues. Further study is needed, however, to confirm that this gene encodes a functional caspase because its two splice variants so far isolated by us are likely to be nonfunctional.
Another gene encoding a protein likely to have a role in inflammation was identified in the genomic region centromeric to D16S3376. NC15-25 is a direct-selected cDNA clone that may encode a heat shock protein that is homologous to the 3′ translated and untranslated regions of both human tumor necrosis factor receptor I (TNFR-1)-associated protein 1 (TRAP1) (Song et al. 1995) and human heat shock protein 75 (hsp75) (Chen et al. 1996) (Table 1). Both TRAP1 and hsp75 belong to the heat shock protein 90 family, but they have different functional profiles (Song et al. 1995; Chen et al. 1996). Whereas TRAP1 interacts with the intracellular portion of TNFR-1 and thus may regulate the many physiological/pathophysiological functions of TNF, one of which is regulation of inflammation (for review, seeEigler et al. 1997), hsp75 functions as a molecular chaperone for the retinoblastoma protein (Rb) that controls the progression of cell cycle and cellular response to external stimuli (Chen et al. 1996).
In conclusion, the transcript map described in this manuscript not only contributed to the identification of MEFV, the gene causing FMF, but also sheds light on the genomic organization and evolutionary history of this region of human chromosome 16. The establishment of a comprehensive transcript map provides a framework for understanding the genetic potential of a given segment of the human genome. Embedded in the DNA sequence is not only the coding potential of the various genes but also the physical relationship between the different members of multigene families. Clustered members of multigene families can undergo regulation in a locus specific manner by locus control regions (LCRs). The olfactory receptors and the members of the zinc-finger gene family constitute candidates for such coordinate control. Combined with the transcript maps of the neighboring PKD1 and RSTSregions, this map provides a rich resource for further defining the complex genomic organization of this region both in terms of full delineation of transcripts and characterization of functional relationships among related clusters of genes.
METHODS
Cosmids, BAC, and PAC Clones
Cosmids, BACs, and PACs from the FMF candidate interval have been described in detail elsewhere (Sood et al. 1997).
Direct cDNA Selection
Cosmids, BAC, and PAC clones in the FMF candidate region were biotinylated using BioPrime (Life Technologies, Gaithersburg, MD). cDNAs were prepared from combined mRNA from fetal brain, fetal liver, and human lymph node by reverse transcription and ligation of anEcoRI/NotI adapter, which also served as a PCR primer, to synthesize second-strand cDNAs. cDNAs were hybridized directly to biotinylated templates, which were recovered using streptavidin-coated magnetic beads. Conditions for blocking, hybridization, binding, and elution of cDNAs from magnetic beads (Dynal Inc., Lake Success, NY) were as described (Lovett et al. 1991; Parimoo et al. 1991). After two rounds of selection, eluted cDNAs were amplified with CUA-tailed EcoRI/NotI adapter primers and subcloned into the pAMP10 vector (Life Technologies) to yield libraries of selected cDNAs. Recombinant clones were arrayed on blots. Clones that hybridized to either repetitive or ribosomal sequences were excluded from further analysis. To confirm their origin, unique clones were hybridized individually to EcoRI digests of cosmid/BAC/PAC DNAs. Clones were then hybridized to each other and were binned into groups. Representative clones of each group were hybridized to multiple tissue Northern blots and sequenced.
Exon Amplification/Trapping
Internal exon amplification/trapping was done as described previously (Buckler et al. 1991) on a tiling path of cosmids from 363D9 to 23G10 and on PAC clone 273L24. Three-prime exon trapping was done on cosmid 335H1 as described (Krizman and Berget 1993). DNA from sublibraries of cosmid and PAC inserts was prepared, and partially digested with Sau3AI, size fractionated to select for fragments 2 kb and larger, and shotgun subcloned into theBamHI site of the exon trapping vector pSPL3 (Promega, Madison, WI). Such sublibraries were introduced into E. coliDH12B, and plasmid DNA was obtained by alkaline lysis of the transformed cells cultured en masse in LB with 200 mg/ml ampicillin for 16 hr at 37°C with shaking. Sublibrary plasmid DNA was then introduced, by transfection using lipofectACE reagent (Life Technologies), into COS-7 cells (ATCC30-2002) maintained in Dulbecco’s modified Eagle medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies), 2 mm l-glutamine (Life Technologies) and penicillin/streptomycin (Life Technologies). Cytoplasmic RNA was isolated from the COS-7 cells 24 hr post-transfection using Trizol (Life Technologies) extraction, followed by ethanol precipitation. The RNA was reverse-transcribed using a pSPL3 vector primer SA2. Trapped exons were then amplified using oligonucleotides SA2 and SD6, followed by digestion of the PCR products with BstXI. A second PCR reaction was performed on theBstXI digestion products using oligonucleotides dUSA4 and dUSD2. The resulting putative trapped exons were then cloned into the pAMP10 vector (Life Technologies), characterized, and sequenced. Three prime exon trapping was performed similarly using the vector pTAG4 (Krizman and Berget 1993).
Partial and Finished Sequencing of Cosmid Clones
Partial sequencing of cosmids was done at two sequencing centers using somewhat distinct strategies. At National Human Genome Research Institute (NHGRI) the cosmids were sheared by sonication and DNA fragments of ∼1 kb were shotgun cloned into an M13 sequencing vector and clones were subjected to one-pass sequencing using vector primers. A total coverage of ∼1.5× was achieved. The sequences derived were deposited in a common genomic sequence database. Partial sequencing at Los Alamos National Laboratory (LANL) was done using a similar strategy except that ∼3 kb DNA fragments were generated by partial Sau3A digestion, and these fragments were shotgun cloned into pBluescript and double-end sequenced to a 1× coverage.
Complete DNA sequencing of an 87-kb DNA fragment bounded by the 1.5-kbEcoRI fragment of cosmid 23G10 and the 0.5-kb EcoRI fragment of cosmid 442E2 was also done. DNA was subjected to partial digestion with Sau3A and 3-kb fragments were size selected and shotgun subcloned. Five hundred subclones were end sequenced (one end only) and the DNA sequences overlapped to generate sequence contigs. Gap closure was done in part by the identification and sequencing of subclones with unsequenced ends that fell within a gap and by PCR amplification and sequencing of DNA within a gap.
Northern Analysis
Northern blots with mRNA from multiple human tissues were purchased from Clontech (Clontech, Palo Alto, CA). For determination of transcript sizes and tissue-distribution patterns, cDNA fragments obtained by direct selection and trapped exons >200 bp in size were labeled with [32P]dCTP by random priming, and hybridized to Northern blots. Hybridization was done using Hybrisol I hybridization buffer (Oncor, Gaithersburg, MD). Autoradiography was done overnight at −80°C with intensifying screen.
cDNA Library Screening
cDNA library screening used both solution hybridization and plaque hybridization. Solution hybridization was carried out using the GeneTrapper cDNA Positive Selection System (Life Technologies). Briefly, oligos from trapped exons, selected cDNA fragments, and EST clones were biotinylated, and hybridized to single-stranded cDNA from adult and fetal brain, liver, spleen, leukocyte, kidney, and testis cDNA libraries. This was followed by hybrid capture using paramagnetic streptavidin beads and reconstruction of double-stranded cDNA using distinct nonbiotinylated gene-specific oligos. The resulting double-stranded recombinant clones were introduced into E. coli, and hybridization of colony lifts with [32P]dCTP end-labeled gene-specific oligos was used to identify positive colonies. Gel-purified inserts from positive clones were hybridized to cosmid contig blots to distinguish cDNA clones mapping to the FMF region from those that are false positives.
Library screening by conventional plaque hybridization was done primarily for cDNA fragments obtained by direct selection from theD16S468/D16S3070–D163376 interval. For each directly selected cDNA fragment representing a distinct transcript in this region, 1 × 106 plaques from a cDNA library of human peripheral blood leukocytes (Strategene, La Jolla, CA) were screened. Positive plaques were subjected to further plating and hybridization, and then converted to pBluescript plasmids by in vivo excision (Strategene).
Rapid Amplification of cDNA Ends
Rapid amplification of cDNA ends (RACE) was performed using RACE systems according to the manufacturer’s specifications (Life Technologies). Briefly, for 5′ RACE, antisense gene-specific oligos were designed from trapped exons, selected cDNA fragments, and mapped EST clones. First strand cDNA to mRNA from the tissue known to express the gene of interest was synthesized by reverse transcription with SuperScript RT II (Life Technologies) using the gene-specific oligo as primer. A poly(dC) stretch was then attached to the 5′ end of the first-strand cDNA using dCTP and TdT (Life Technologies). cDNA sequences upstream of the gene-specific oligo used for cDNA synthesis were then amplified by PCR using an anchor primer that anneals to the poly(dC) stretch (Life Technologies) and a nested gene-specific antisense oligo. For 3′ RACE, mRNA from the tissue known to express the gene of interest was reverse-transcribed using oligo(dT) tailed with an adapter primer. The 3′ unknown sequences of a gene were then retrieved by PCR amplification using a gene-specific primer and a universal adapter primer, followed by another round of PCR amplification using a nested gene-specific primer. The RACE products were cloned, mapped to the FMF interval, and sequenced.
DNA Sequence Determination and Computer Analysis
Selected cDNA fragments were sequenced using Sanger’s chain-termination method with gamma[33P]ATP end labeling of sequencing primers. Trapped exons, EST clones, and cDNA extension products were sequenced with flourescein-labeled dye primers or terminators (PE Applied Biosystems, Foster City, CA) according to manufacturer’s instructions and analyzed on an ABI 377 automated sequencer.
Determination of open reading frames within clones was done using the MapDraw program (DNAStar, Madison, WI). DNA and protein sequence comparisons were done with Megalign multiple sequence alignment program (DNAStar) using a clustal algorithim with a gap penalty and gap-length penalty of 10. Identification of transmembrane domains in the putative olfactory receptor open reading frame was done using Protean software (DNAStar). Sequence homology to previously characterized DNA sequences in the nr and dbEST was determined by the BLAST suite of software (Altschul et al. 1990). Exon prediction of partial DNA sequence data was done using the GRAIL2 program (Uberbacher and Mural 1991). Exon prediction of the 87-kb complete DNA sequence was done using both GRAIL2 and Gene Finder software (Solovyev et al. 1994).
Identification of DNA sequence similarities between the partial DNA sequences of cosmids sequenced at LANL and public database entries was done using an automated search program developed at Los Alamos National Labs, designated SCAN (Sequence ComparisonAnalysis). This program integrates search results from BLAST and FASTA analysis of the dbEST, GenBank, GenPept, PIR, SwissProt, and Repeats public databases, and in addition, summarizes suspected repeat, vector, and E. coli sequences in the query sequences while ignoring both short and low complexity homologies. Multiple homology hits to the same query region are displayed in a multiple alignment format and overlaps of query sequences are noted. In addition, the sequence analysis results from GRAIL 1, 1a, and 2 were included.
Identification of tRNA sequences was done using tRNAscan-SE software (Lowe and Eddy 1997). DNA sequences between tRNA molecules were compared and sequence similiarities identified using Sequencher software (Gene Codes). Repetitive DNA fragments within the cluster were determined using RepeatMasker2 software (A.F.A. Smit and P. Green, unpubl.). Identification of common structural elements within each DNA segment was done by visual inspection.
Acknowledgments
N.F.-G. and X. C. gratefully acknowledge the support of the Arthritis Foundation. D.O.R., R.K.M., and N.A.D. were supported by the U.S. Department of Energy under contract W-7405-ENG-36. D.L.G. is grateful for pilot grant support from the University of Michigan Multipurpose Arthritis Center, NIH-P60-AR20527.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵9 Present address: Department of Biological Chemistry, University of California at Irvine, Irvine, California 95616 USA.
-
↵10 These authors contributed equally to this work and are listed in alphabetical order.
-
↵11 Corresponding author.
-
E-MAIL kastnerd{at}arb.niams.nih.gov; FAX (301)402-0012.
-
- Received April 29, 1998.
- Accepted October 19, 1998.
- Cold Spring Harbor Laboratory Press








