
High-Resolution Mapping and Transcript Identification at the Progressive Epilepsy with Mental Retardation Locus on Chromosome 8p
- Susanna Ranta1,
- Anna-Elina Lehesjoki2,
- Maria de Fatima Bonaldo1,
- James A. Knowles1,
- Aune Hirvasniemi3,
- Barbara Ross1,
- Pieter J. de Jong4,
- Marcelo Bento Soares1,
- Albert de la Chapelle2, and
- T. Conrad Gilliam1,5
- 1Departments of Psychiatry, Genetics and Development, and Columbia Genome Center, College of Physicians and Surgeons at Columbia University and New York State Psychiatric Institute, New York, New York 10032; 2Department of Medical Genetics, University of Helsinki, and the Department of Molecular Genetics, the Folkhälsan Institute of Genetics, Helsinki, Finland; 3Department of Pediatrics, Kainuu Central Hospital, Finland; 4Department of Human Genetics, Roswell Park Cancer Institute, Buffalo, New York 14263
Abstract
Progressive epilepsy with mental retardation (EPMR) is an autosomal recessive central nervous system disorder characterized by childhood onset epilepsy and subsequent mental retardation. The locus for EPMR has been mapped to human chromosome 8p23. We recently reported the construction of a YAC contig across the 4 centimorgan minimum genetic region that harbors the disease locus. We now report further delineation of the critical region to <700 kb. Our mapping strategy relied on the identification of nine novel microsatellite markers and the construction of a complete BAC contig across the critical region. Several partial gene sequences have been identified from the region and are being analyzed as candidate genes for EPMR.
[The sequence data described in this paper have been submitted to the GenBank data library under accession nos. AF009188–AF009214.]
Epilepsy is a relatively common symptom of a diverse group of disorders that, together, affect ∼2%–3% of the population (Scheuer and Pedley 1990; Hauser et al. 1993). Although clearly a heterogeneous disorder, genetic factors are known to contribute to the etiology of at least a subset of epilepsy cases. To date, seven independent loci have been genetically mapped for monogenic forms of epilepsy with clear Mendelian patterns of inheritance (Leppert et al. 1989; Lehesjoki et al. 1991; Lewis et al. 1993; Tahvanainen et al. 1994; Ottman et al. 1995; Phillips et al. 1995; Guipponi et al. 1997). A subset of families for one of the multifactorial forms of epilepsy, juvenile myoclonic epilepsy, may be linked to chromosome 6p (Greenberg et al. 1988; Durner et al. 1991; Weissbecker et al. 1991;Liu et al. 1995). Moreover, evidence for involvement of a locus on chromosome 8q in idiopathic generalized epilepsy has been reported (Zara et al. 1995).
One strategy to better understand the pathophysiology underlying epileptic seizures has been to positionally clone and characterize the genes whose mutations cause monogenic forms of epilepsy. This approach has led to the identification of a missense mutation and, recently, a 3-nucleotide insertion, in the coding region of the α4 subunit of the neuronal nicotinic acetylcholine receptor that cosegregates with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) (Steinlein et al. 1995, 1997). The gene responsible for a form of progressive myoclonus epilepsy (EPM1) was identified as a protease inhibitor, cystatin B, by Pennacchio et al. (1996).
Progressive epilepsy with mental retardation (EPMR), also known as Northern epilepsy, is an autosomal recessive disorder so far reported only from a small rural region in Northern Finland. To date, 25 patients with the disorder have been identified. Given their descendance from a small number of founders and their subsequent relative genetic isolation, Finnish families carrying a rare Mendelian disorder are likely to harbor clonal copies of the identical defective allele (Norio et al. 1973). Thus, the EPMR families are ideal for a positional cloning study.
The main clinical features of EPMR are normal early development followed by childhood onset epilepsy and subsequent mental retardation. Epilepsy begins between the ages of 5 and 10 and is characterized by generalized tonic–clonic seizures that increase in frequency until puberty. In addition, one-third of all patients suffer complex partial seizures. All epileptic activity decreases during early adulthood. Mental retardation begins 2–5 years after the onset of seizures and continues through adulthood. All of the adult patients need help in their daily lives. Of the antiepileptic drugs used, clonazepam has been the most effective (Hirvasniemi et al. 1994, 1995).
The EPMR gene has been localized to the telomeric region of chromosome 8p (Tahvanainen et al. 1994). We recently reported the construction of a yeast artificial chromosome (YAC) contig across theEPMR disease locus (Ranta et al. 1996). Using the contig to physically map densely spaced microsatellite markers, we have continued to narrow the disease gene region by precisely mapping meiotic breakpoints that refine the boundaries of the locus and by identifying regions homozygous by descent in affected individuals. The combination of these mapping strategies allowed us to identify a region of <700 kb that harbors the EPMR locus. A contig consisting of 17 overlapping bacterial artificial chromosome (BAC) clones was constructed across the region. The BAC clones were used as targets for exon trapping, whereas two midi-YAC clones were immobilized to nylon filters and hybridized to brain cDNA libraries to identify protein-coding DNA sequences. A partial transcript map of the region is presented. These candidate gene sequences are currently being used to scan for disease-causing mutations.
RESULTS
Physical Mapping
To generate additional microsatellite markers across theEPMR disease locus, we set out to construct a BAC contig across the minimum genetic region identified from our previous study (Ranta et al. 1996). Because all affected individuals, with the exeption of the one haplotype we considered different (see Haplotype Analysis, below), were homozygous for microsatellite markers spanning a 3-centimorgan (cM) region between D8S504 andAFMa054td9, we limited the contig construction to this region. Randomly selected sequence tag sites (STSs) mapping to the previously reported EPMR–YAC contig (Ranta et al. 1996), together with probes obtained from the Whitehead Institute database, were used to screen the commercially available Research Genetics Human BAC library. BAC clones that were positive for STSs in the region were screened for the presence of microsatellite repeat sequences (see below), and nine new markers were characterized as described in Table 1. Because recombination breakpoints were identified with these markers (see below and Fig. 1), we were able to concentrate our efforts on a much smaller region defined by the new flanking markers, 140CA and 159CA. Figure 2 depicts the physical map of the new minimal genetic region. A total of 17 BAC clones were identified, each of which shared at least two STSs with its overlapping neighbors. To secure the overlap between BAC clones, we directly sequenced most BAC ends to develop PCR primers. When this method failed, we used ligation-mediated PCR (Kere et al. 1992) to obtain the BAC end sequences (Table 2). Because the T7 end of BAC clone 233K17 was chimeric, we rescreened the library with another STS derived from the end sequences of an overlapping P1 clone (91o6) (data not shown) and found two additional BAC clones, 104G24 and 369H9. Two sets of BAC end sequences were identical; 108T7/239SP6 and 93T7/293SP6. BAC 44D1 may have undergone a deletion event, as it failed to amplify STS WI-11694.
Microsatellite Markers Spanning the Minimum Genetic Region

Refinement of the EPMR region by meiotic breakpoint mapping. DNA marker genotypes across the EPMR locus in two segments of the extended pedigree from Northern Finland (Hirvasniemi et al. 1994). The haplotypes reveal key recombination events that reduce theEPMR minimum genetic region to <1 cM. Both recombinations occur in healthy siblings (arrows). Disease homologs are in boldface type. The boxed regions contain the segment of recombinant chromosome marking the sites of chromosomal recombination. (O) Untyped alleles.
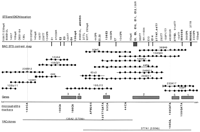
Physical map of the EPMR minimum genetic region. Seventeen BAC clones with their corresponding STS, exon, or microsatellite positions are shown in the middle. The BAC contig spans the region between the telomeric flanking marker 140CA and the centromeric flanking marker 159CA. (•) The BAC location of an STS, exon, or microsatellite marker, with its corresponding name at the top. Names in boldface type indicate exons or STSs from cDNA clones. (X) A chimeric BAC-end sequence; (▪) a repeat element. Exon sequences obtained from exon trapping are indicated as vertical bars protruding down from the top border. New microsatellite loci are indicated below the bottom border. Two midi-YAC clones are indicated at the bottom. Six putative gene locations are indicated by shaded boxes along the bottom border.
Primer Sequences for the BAC-End STSs Listed by the BAC Address
A total of nine microsatellite markers were identified by systematically screening all BAC clones for hybridization to poly [d(C-A)n] ⋅ [d(G-T)n] and oligonucleotide (GATA)5, probes. Clone 159F7 contained three markers (159CA, 159GATA, 159CA-TA), one of which (159CA) proved to be a new centromeric flanking marker for EPMR. Five additional microsatellite markers were identified by this method along with one marker (125 GATA) that was revealed while sequencing the T7 end of clone 125H11.
Finally, we used marker AFM265wb5 to screen the Centre d’Etude du Polymorphisme Humain (CEPH) midi-YAC library (Albertsen et al. 1990; Green and Olson 1990) to obtain a small set of clones that span the entire disease gene region but only a limited amount of the surrounding region. Two additional YAC clones were identified (136A2 and 377A1) that span the entire region. Characterization of the YAC-end sequences revealed no chimerism (data not shown). We estimate the maximum size of the critical genetic region to be the sum of these two partly overlapping YACs, ∼700 kb (370 + 330, respectively).
Haplotype Analysis
The EPMR locus previously had been mapped to a 4-cM region between the flanking markers AFM185xb2 andAFMa054td9. Nine new microsatellite markers (Table 1) generated from the BAC contig clones described above, together with one new marker from the Whitehead Institute for Biomedical Research database, were used to genotype the two EPMR families known to harbor recombination breakpoints between the disease locus and flanking markers. Haplotype analysis shows that markers 140CA and159CA recombine in healthy individuals, both clearly beyond the age of risk for disease (Fig. 1). Marker 156CA is uninformative in these families. The recombination has therefore occurred between 140CA and 186CA on the telomeric side and between 44CA and 159CA on the centromeric side. In family 2, individual II-3 is homozygous 2/2 for marker93CA. This could result from a double recombination event, which we consider very unlikely given the small physical and genetic distances between the markers, or from a new mutation within the dinucleotide repeat sequences of the marker. In support of the latter, alleles 1 and 2 differ by a single dinucleotide repeat. The two recombination events shown in Figure 1 refine the minimum genetic region to an area of <1 cM between markers 140CA and159CA.
Our previous genetic analysis identified two distinct haplotypes associated with the EPMR disease allele, thereby suggesting two independent disease mutations that segregate through the Northern Finnish pedigrees (Tahvanainen et al. 1994; Ranta et al. 1996). We therefore genotyped the single EPMR family segregating this putative distinct disease haplotype with the new set of densely spaced markers. Our data show that the affected individuals who appeared to be compound heterozygotes at the EPMR locus are clearly homozygous for markers closer to the disease locus and identical to those affected individuals shown in Figure 1. Thus, homozygosity mapping places the disease gene between markersD8S504 and 159CA, consistent with the meiotic breakpoint predictions (Table 3) and suggests only a single ancestral mutation.
Chromosomal Region Identical By Descent at the EPMR Locus
Transcript Map
A combination of exon trapping (Buckler et al. 1991; Church et al. 1994) and cDNA library screening (Bonaldo et al. 1994) was used to identify putative protein-coding DNA sequences within the minimum genetic region. A total of 15 BAC clones were used for exon trapping, and the two midi-YAC clones were used to screen a normalized infant brain cDNA library.
A total of 22 exons was selected, which map unambiguously to the EPMR BAC contig. Of these, three map beyond the telomeric border of the minimal genetic region and another five appear to identify a single gene that spans the centromeric border of the region. Thus, a total of 14 exons map clearly within the minimal genetic region as described in Table 4 and as depicted as vertical bars at the top of Figure 2. The sizes of the exons are given in Table 4. The five exons spanning the centromeric border show complete sequence homology with the 165-kD skeletal muscle protein gene (GenBank accession no.X69089) (Fig. 2, gene 1). We have mapped several of these exons centromeric to the flanking marker 159CA on BAC 159F7. This gene lies at least partially outside the region of interest, although it is possible that one or more exons and/or regulatory sequences lie within the critical region where they would be targets for a disease-causing mutation.
Exonic DNA Sequences from the Minimum Genetic Region
Of the 14 exons from the critical interval, 5 map to BACs 104G24, 125H11, 44D1, 246K11, and 293D11 (Fig. 2, E8-12). We used oligonucleotide primers from these exons to prime reverse transcriptase PCR (RT–PCR) amplification of poly(A)+ RNA from total human brain. Three of the exons (E10–12) amplify 410 bp of a contiguous cDNA transcript and presumably encode a single gene (Fig. 2, gene 2). Several of these exons were used to screen a Clontech human cerebral brain cDNA library. Exons E2 and E3 both identify a single cDNA clone. Further library screening for this gene revealed four overlapping clones spanning 3412 bp of cDNA sequence that includes a third exon from the trapping experiments (E4) and a BAC-end STS (156SP6) within the 3′-untranslated region (UTR). Homology searches with the entire cDNA sequence revealed strong homologies with the rat PSD-95/SAP90 (postsynaptic density95/synapse-associatedprotein 90)–associated protein 2 mRNA (GenBank accession no. U67138) and the human GKAP (guanylate kinase-associatedprotein) mRNA (GenBank accession no. U67988). This novel gene (Fig. 2, gene 3, GenBank accession no. AF009204) detects RNA transcripts of 1.7 and 2.5 kb in all tissues tested (heart, brain, placenta, lung, liver, skeletal muscle, kidney, and pancreas), along with a 10-kb transcript detected only in brain. Exons E10–12 identified a single cDNA clone. We used 3′ rapid amplification of cDNA ends (RACE) to extend this clone. These two overlapping clones span 4353 bp and include exons E9 and E14, an additional exon not originally placed on the contig, and BAC end-STS 44T7 (GenBank accession no. AF009205). This gene detects a 6-kb RNA transcript in all tissues tested (see above).
A normalized brain cDNA library was screened using the inserts of YACs 136A2 and 377A1 according to a protocol described previously (Bonaldo et al. 1994; Soares et al. 1994). Clone 136A2 detected one new cDNA clone (136E3, GenBank accession no. AF009202) that mapped back to the EPMR contig and clone 377A1 detected two novel cDNA clones (377H7 and 377E3). No significant sequence homologies were detected with these clones. 377H7 was identical to the 3′ end of gene 2. These gene segments are shown as genes 4, 2, and 5 in Figure 2. We also searched the National Council for Biotechnology Information Gene Map web site (http://www.ncbi.nlm/nih.gov/SCIENCE96/) corresponding to the Genome issue of Science (Schuler et al. 1996) for expressed sequence tags (ESTs) and were able to map four new sequences within the BAC contig; a004f35, a004a42, cda0eh6, and bcd1479. Of these, we were not able to link a004f35 to any of our exons or cDNAs. This EST is therefore listed as gene 6 in Figure 2. The remaining three ESTs are component parts of cDNA clone 377E3 (GenBank accession no. AF009203). An EST from the 3′ UTR of 377E3 spans the centromeric boundary of the minimal genetic region.
DISCUSSION
The minimum genetic region harboring the EPMR locus has been refined to a region of <700 kb. Previously, we reported the construction of a YAC contig across a 4-cM region that genetically defined the EPMR locus (Ranta et al. 1996). A high-resolution BAC contig was constructed across the region of homozygosity, and nine new microsatellite markers were identified and physically ordered across this region. This dense set of DNA markers resolved a more defined, 1-cM region of homozygosity common to all affected individuals, notably including one disease haplotype previously assumed to carry an independent disease allele based on its unique signature in regions more distant to the disease locus (Ranta et al. 1996). Two recombinant breakpoints were identified in unaffected members of the EPMR pedigrees, which further narrowed the disease locus region to <700 kb. Having exhausted the search for microsatellite markers in the region, we have now begun a DNA sequencing effort to pinpoint more precisely the remaining two recombination breakpoints and thereby the minimum genetic region harboring the EPMR locus.
Various neurotransmitter receptors have long been considered targets for epilepsy mutations (Elmslie and Gardiner 1995). In the past few years several genetic linkage studies and mutation reports have produced data in support of this hypothesis. For example, mutations in the α4 subunit of the neuronal nicotinic acetylcholine receptor gene has been demonstrated in ANDFLE in two pedigrees (Steinlein et al. 1995, 1997). Mutations in a chromosome 12p, Shaker-related voltage gated K+ channel gene, KCNA1, are associated with an autosomal dominant episodic ataxia–myokymia disorder in several families (Browne et al 1994, 1995; Comu et al. 1996) and mutations in a gene encoding the α1 subunit of the glycine receptor (GLRP1) cosegregate in families affected with familial hereditary hyperekplexia (Shiang et al. 1993, 1995; Rees et al. 1994). The fact that the gene responsible for a form of progressive myoclonus epilepsy (EPM1) is a protease inhibitor (Pennacchio et al. 1996), however, cautions one against limiting the epilepsy candidate gene pool.
The BAC contig provides valuable reagents for isolating candidate genes for EPMR, as well as sequencing this entire region of the human genome. The collection of trapped exons from BAC clones, together with homologous cDNAs selected with the two YAC clones (Fig. 2) was used to identify six putative independent genes from the region. The most interesting candidate at this point is gene 3 (Fig. 2), a human GKAP homolog. GKAP is a novel synaptic protein that appears to play a role in the organization of ion channels and signaling molecules at the synaptic junction (Kim et al. 1997). GKAP interacts directly with a carboxy-terminal domain of the PSD-95 family of proteins, which in turn bind to Shaker-type K+ channel proteins via their amino-terminal domain. In Drosophilia, mutation of the PSD-95 homolog (Discs Large, DLG) disrupts the normal synaptic clustering of Shaker proteins and alters synaptic morphology (Lahey et al. 1994; Tejedor et al. 1997). As noted above, mutations in the human KCNA1 Shaker-related K+ channel gene produce epilepsy-like symptoms. One of the exons (E4) comprising the GKAP homolog maps to the same bin as the microsatellite marker 186CA, which is therefore an intragenic polymorphic marker. Because there are no recombinations between186CA and the disease gene, this cDNA is an excellent candidate gene for EPMR. Therefore, for the GKAP homolog and other genes in the minimal genetic region, we will continue our efforts to clone the full-length cDNAs, identify intron–exon junctions, and systematically screen patient and control DNA (or RNA when the gene is expressed in lymphoblasts) for disease mutations.
Positional cloning studies targeted at epilepsies with monogenic etiologies are clearly in a very productive phase. The limitation of the EPMR locus to a region of <700 kb and the construction of a BAC contig spanning this minimum genetic region will greatly facilitate the cloning of the defective gene causing this particular type of epilepsy. The imminent discovery of genes that mutate to cause monogenic forms of epilepsy will provide new molecular tools to study the normal and pathological functions of these key genes and their protein products. It is hoped that through discovery of allelic variants of these same genes, or through identification of new genes from common molecular pathways, these discoveries will provide new insights to the investigation of more common forms of epilepsy.
METHODS
BAC and YAC Library Screening
BAC clones were isolated from the Research Genetics Human BAC library by PCR screening. For BACs 6K9, 60o3, 93H7, 104G24, 108o11, 124A4, 140H1, 156G21, 159F7, 186J15, 233K17, and 369H9, only the superpools and plate pools were screened, followed by the individual BAC plate. Eight of the BAC plates were contaminated, giving more than one address for the same BAC clone. In these cases the BAC address was determined by colony plating. For BACs 44D1, 125H11, 239B12, 246K11, and 293D11, BAC library pools for rows and columns were available and afforded definite BAC addresses for each clone.
The YAC clones were isolated by PCR screening of the CEPH midi-YAC library (Albertsen et al. 1990; Green and Olson 1990).
BAC-End Isolation
BAC-end sequences were determined by direct sequencing with an automated ABI 373A DNA sequencer except in cases where direct sequencing failed (124SP6, 239T7, 239SP6) or did not provide sufficient sequence to read through an Alu repeat (156T7). In these four cases, the ends were isolated by ligation-mediated PCR amplification with minor modifications of published protocols (Kere at al. 1992). One-hundred nanograms of BAC DNA was digested in separate 15-μl reactions with RsaI, EcoRV, and PvuII for the SP6 end isolation, and PvuII, BglII, andDdeI for the T7 end isolation. The ends for fragments digested with BglII and DdeI were filled using Klenow (Boehringer Mannheim). A short linker sequence (2 pmoles), T4 DNA ligase, and buffer recommended by the enzyme supplier were added to the reactions and incubated for 1 hr at 37°C. One microliter of the reaction mixture was amplified by PCR in 20-μl reactions using the linker primer (3 pmoles) and a BAC vector arm-specific primer (30 pmoles). One microliter of reaction mixture was reamplified in 50 μl of PCR reaction using the linker primer and an internal BAC arm-specific primer. The primer sequences are as follows: T7 I: 5′-AATTGTAATACGACTCACTATAGGGCG-3′; T7 II, 5′-ATCCTCTAGAGTCGACCTGC-3′; SP6I, 5′-GTTGTGTGGAATGTGAGCGG-3′; SP6II, 5′-AGCTATTTAGGTGACACTATAGAATAC-3′.
Identification of Microsatellite Markers
With the exception of clone 239B12, all clones from the BAC contig (Fig. 2) were subcloned into pUC18. The colonies were screened with nick-translated poly[d(C-A)n] ⋅ [d(G-T)n] and kinase-labeled (GATA)5. Positive clones were sequenced and oligonucleotide primers flanking the repeat sequences were designed. To estimate the number and frequencies of alleles we typed at least 20 unrelated individuals. All microsatellite genotyping was performed by kinase labeling of one of the oligonucleotide primer pairs with [γ-32P]ATP using standard PCR amplification conditions.
Exon Trapping
Exon trapping was performed using GIBCO’s exon trapping kit (cat. no. 18449-017) according to manufacturer’s and published protocols (Buckler et al. 1991; Church et al. 1994) with minor modifications; we used vector pSPL3b instead of pSPL3. Also, to minimize the number of false positives in the final PCR we added an additional step after the second subcloning with vector pAMP: colony hybridization with kinase-labeled oligonucleotide (5′-GTAAGTAATACATGTAATGCAACCTATAC-3′) derived from the HIV portion of vector PSPL3b.
cDNA Library Screening and 3′ RACE
Clontech’s cerebral brain cDNA library (cat. no. HL3002a) was screened using standard protocols with exons E1–6 and E10–12. Only one positive clone containing exons E2 and E3 was identified in the initial screening with exons E1–6. The library was then rescreened with a 735-bp PCR product from this cDNA clone. Four additional clones were identified in the second screening. One clone was identified after screening with exons E10–12. In preparation for the cDNA library selection, YAC inserts were separated from yeast chromosomes by pulsed-field electrophoresis using a Pharmacia LKB apparatus with a hexagonal electrode. Approximately 10 ng of YAC DNA was denatured with 0.5 m NaOH for 10 min at room temperature. The DNA (2–5 ng) was then spotted onto a GeneScreen plus (DuPont) membrane (0.5 cm2), neutralized with 1 m Tris-HCL (pH 7.5), washed with 2× SSC, and UV cross-linked. The conditions for cDNA selection have been described by Bonaldo et al. (1994). Selected cDNAs were recovered by alkaline denaturation, eluted from the membrane, and ethanol precipitated. The cDNAs were then converted to the double-stranded forms by primer extension using M13 universal primer and T1 polymerase (Sequenase, v. II, U.S. Biochemical), electroporated into bacteria (DH10B), and grown in 2.5 ml of 2× YT (Bio 101) at 37°C for 1 hr. The entire culture was plated on 2× YT ampicillin agar. The colonies were amplified by PCR and hybridized with Cot I DNA (GIBCO BRL) and YAC DNA. (20× SSC = 3 m NaCl, 0.3m Na3citrate H2O at pH 7.0).
To clone the 3′ end of gene 2, 3′ RACE was performed using the Marathon Human Brain cDNA Library (Clontech, 7400-1). The following oligonucleotide primers were used: 5′-CCGTGGTTGCTAACGCGAAACC-3′ and the adaptor primer AP1. PCR was carried out using the following cycling conditions: 94°C for 1 min, 94°C for 10 sec, 68°C for 5 min for 30 cycles, and 72°C for 4 min using Perkin Elmer 9600. The PCR product was gel-purified and cloned for sequence analysis.
Northern Blotting
Northern blot MTN7760 (Clontech) was hybridized with a 735-bp PCR product from the first cDNA clone with exons E2 and E3 and with a 110-bp product from the 3′ RACE clone from gene 2 according to the manufactors protocols.
Acknowledgments
We thank Trecia Henriques for her assistance with building the BAC contig. This work was supported by the Ulla Hjelt Foundation (S.R.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵5 Corresponding author.
-
E-MAIL TCG1{at}columbia.edu; FAX (212) 543-6002.
-
- Received June 9, 1997.
- Accepted July 25, 1997.
- Cold Spring Harbor Laboratory Press








