
Microsatellite Data Support an Early Population Expansion in Africa
Abstract
We have developed a method for the analysis of microsatellite data that is useful in the elucidation of the demographic history of populations. This method, the PK distribution method of pairwise comparisons, is analogous to the mismatch distribution of sequence comparisons developed for the analysis of mitochondrial sequence data by Rodgers and Harpending and is defined as the distribution of the number of repeat unit differences between alleles when each allele in a sample is compared with every other allele in the sample. Using computer simulations of microsatellite loci, we show that the shape of the distribution of PK changes in a distinctive manner as a function either of time since population expansion or effective population size. Increases in both of these affect the PK distribution in a similar fashion leading to a change from a steep distribution with a P0 peak to one with a nonzero peak. Analysis of three data sets from surveys of microsatellite loci in ethnographically defined populations reveals that most (9/12) of the African populations analyzed, but none of the 30 non-African populations showed PK distributions with nonzero peaks. These PK distributions indicate either an earlier expansion or a larger effective population size for African populations. This observation is consistent with the hypothesized African origin of modern human.
Based on the topologies of mitochondrial and nuclear phylogenetic trees, it has been hypothesized that modern humans originated in Africa (Cann et al. 1987; Cavalli-Sforza et al. 1988; Nei and Roychoudhury 1993). If this were true, it would be expected that the genetic diversity of African populations would be greater than non-African populations as they would have expanded in size earlier. This increased genetic diversity of African populations, although clearly evident in the mitochondrial genome, has been more difficult to demonstrate for nuclear markers. Many classical polymorphic loci such as blood group, enzyme, and restriction fragment length polymorphic DNA markers demonstrate the highest levels of heterozygosity in European populations, reflecting the fact that these markers were first identified in European populations (Mountain and Cavalli-Sforza 1994;Rodgers and Jorde 1995). Because of their high degree of polymorphism, microsatellites are less affected by such ascertainment bias. Although large surveys of microsatellite loci in human populations have reported higher levels of heterozygosity in African populations (Bowcock et al. 1994; Deka et al. 1995a,b), most often these differences are not significant. We present a new method for analysis of microsatellite data that clearly shows an earlier expansion and/or a larger effective size of African populations.
Although the precise molecular mechanism of mutational change in repeat number of microsatellite loci has not been elucidated, it is evident from observed mutations and allele frequency distributions that they evolve via a forward–backward stepwise mutational process (Shriver et al. 1993; Weber and Wong 1993; DiRienzo et al. 1994). This observation implies that there will be evolutionary information in the allele size distribution, as alleles closer in size most likely share a more recent common ancestor than alleles with larger size differences. Several new measures of genetic distance have been developed that use the evolutionary information present in the size of microsatellite alleles (Goldstein et al. 1995a,b; Shriver et al. 1995; Slatkin 1995; Kimmel et al. 1996). We have developed a method for the analysis of microsatellite data that uses the evolutionary information inherent in microsatellite allele length to elucidate the magnitude of genetic diversity within populations. This method, the PKdistribution method of pairwise comparisons, is analogous to the mismatch distribution of sequence comparisons developed for the analysis of mitochondrial sequence data by Rodgers and Harpending (1992), and is defined as the distribution of the number of repeat unit differences between alleles when each allele in a sample is compared with every other allele in the sample.
RESULTS
We have applied the PK method to the interpretation of several sets of simulated microsatellite data. Genetic loci subject to forward–backward mutational events, when unconstrained, do not reach equilibrium with respect to the identity of alleles or their frequency profiles (Moran 1975). However, after a number of generations (∼4Ne, where Ne is the effective population size) a steady state is reached for certain summary statistics, including heterozygosity, number of alleles, and other measures of genetic diversity (Shriver et al. 1993). Similarly, the PK distribution also reaches a steady state acquiring a shape that does not change after 4Ne generations at a given mutation rate and effective population size (Moran 1975). Figure 1A shows the shape of the PK distribution at equilibrium for populations of different effective size. It is clear that the shape of the PKdistribution is related to Ne, with smaller populations having P0 peaks (most pairwise comparisons showing no difference in size), larger populations have P1peaks (most pairwise comparisons differing by one repeat), and intermediate sized populations (e.g.,Ne = 1000) having plateaus. When a population is not in equilibrium (e.g., a population that has recently expanded in size), the PK is also dependent on the demographic history (e.g., the magnitude of and time since population expansion). Figure 1B shows a series of PK distributions at different time points in generations since a population expansion event. For these computer simulations, parameter levels were set to reasonable estimates for human populations, namely μ = 0.001, finalNe = 5000, and magnitude of expansion = 1000 (see Methods for more detail). These simulations were performed as an instructive example, and we do not presume to accurately model human history with this simple design. Nonetheless, there is a clear relationship between time since expansion and the distribution of PK. After a population has expanded, the distribution has a peak at P0 from immediately following expansion to a point at which the peak shifts to P1. This distribution then flattens progressively until a steady-state distribution is reached at equilibrium, 4Ne generations after expansion.
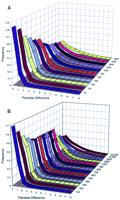
Computer simulations analyzed using PK distribution. The relationship between effective population size and PKdistribution shape is shown in A. Steady-state PKdistributions are shown for 11 populations ranging inNe from 50 to 5000. The relationship between the time since population expansion and the PK distribution is shown in B. PK distributions for 14 time points (in generations) after a 1000-fold increase in population size. Specifics for the simulations are given in Methods.
We have also compiled PK distributions for three independent sets of microsatellite data [our own (Deka et al. 1995a,b) and two from the literature (Bowcock et al. 1994; Jorde et al. 1995)]. These results are shown in Figure 2. Figure2A shows the results of data gathered in the laboratory of R.D., which includes 24 microsatellite loci in 15 human populations belonging to five major groups: three African, four Caucasian, three Asian Mongoloid, two Pacific Islander, and three Amerindian. Africans are the only populations with prominent nonzero peaks, whereas the Amerindian and Pacific Island populations show very steep PKdistributions, with the Caucasian and Asian populations having intermediate patterns. The steep slopes and high P0 peaks observed in the Amerindians and Pacific Island populations are consistent with their recent settlement and population history. Two populations, Brazilian whites and Bramin, show plateaus in the distribution of PK, where P0 ≅ P1. Figure 2B shows the PK distributions for a set of microsatellite frequency data on 30 loci in 13 populations (6 African, 2 European, and 5 Asian) reported by Jorde et al. (1995). Four of the six African populations again show distinct nonzero peaks, whereas for these loci the other two African, the two European, and one of the Asian populations show plateau PK distributions. The PK distributions of the other five Asian populations show steeper P0 peaks. Figure 2C shows the PKdistributions for 30 microsatellite loci reported by Bowcock et al. (1994). This group studied a total of 14 populations, including three African, two European, three Asian, three Amerindian, and three Austronesian. For these data, two of the three African populations show nonzero peaks and all other populations show peaks at P0. Overall, in three large microsatellite studies on ethnically and geographically well-defined populations, 9/12 African populations show PK peaks at P1, 2/12 show plateaus where P0 ≅ P1, and 1/12 shows a P0 peak; 0/8 Caucasian populations show a P1 peak, 4/8 show P0 peaks, and 4/8 show plateaus; 0/11 Asian populations show a P1 peak, 10/11 show a P0 peak, and 1/11 shows a plateau; 11/11 other populations have P0 peaks and none show peaks at P1 or plateaus.
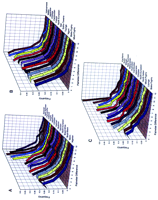
PK distributions for three sets of microsatellite data. Microsatellite data generated in the laboratory of R.D. on 24 loci in 15 populations were analyzed using PK and are shown inA. Data for 30 loci in 13 populations presented in Jorde et al. (1995) are shown in B. C shows PK distributions for data on 30 microsatellite loci on 14 populations by Bowcock et al. (1994).
DISCUSSION
There is good evidence from observed mutational events and the good fit of microsatellite loci to the stepwise mutation/drift model that the majority of di-, tri-, and tetranucleotide repeat loci evolve via a stepwise mutational mechanism, most likely replication slippage (Levinson and Gutman 1987; Shriver et al. 1993; Weber and Wong 1993;DiRienzo et al. 1994). It is also clear that there are differences in the mutational spectrum among specific microsatellite loci and classes of loci (Shriver et al. 1993; Weber and Wong 1993; DiRienzo et al. 1994; Chakraborty et al. 1997). Notwithstanding these interlocus differences, the PK distribution approach is applicable when the same set of loci are analyzed in all populations being considered. In addition, because the PK is the average distribution of pairwise differences at many loci, the effect of a rare locus that deviates from the stepwise mutational process or has an altered mutational spectrum in one or more populations will be diminished by the other loci in the survey.
The analysis of PK distributions among ethnically and geographically well-defined populations shows that African populations have PK distributions that are more similar in form to the simulated equilibrium distributions than the PK distributions of non-African populations. The PK distribution results can also be interpreted as African populations having a larger effective population size (Ne ) than non-African populations. Computer simulations of microsatellite loci show that both larger equilibrium Ne and longer time since population expansion can cause the P1 peaks that are characteristic of African populations. Because Ne is a function of the population size for many generations in the past, and the contemporary census size of African populations is not larger than European or Asian populations, we can conclude that the ancestral African population was larger than the ancestral populations of contemporary inhabitants of the other continents. This could be so if African population expansion preceded non-African population expansion. Alternatively, the smallerNe of contemporary non-African populations could be the result of a reduction in the population size, or population bottleneck, in the ancestral populations of non-Africans that Africans did not experience. Given other genetic and fossil evidence, it is likely that the actual course of history involved some combination of these two models. Population bottlenecks have been recognized to occur when migrating groups surmount geographical barriers as in the peopling of the Pacific and the Americas. It has been suggested that the movement of populations northward out of Africa would have resulted in a similar reduction in population size. We thus find that these PK distribution results are most consistent with an African origin of modern humans followed by migrations of smaller groups out of Africa. Although these data do not exclude admixture between populations moving out of Africa and local archaic populations, they are inconsistent with a European or Asian origin of humans unless likely scenarios for population bottlenecks related to population movement within Europe and Asia but not Africa can be advanced.
METHODS
Computer simulations of the one-step stepwise mutation model were performed to investigate the effects of population expansion and effective population size on the distribution of PK. The design of these simulations is based on previous software used to study the population dynamics of the stepwise mutation model (Shriver et al. 1993, 1995). Each generation consists of randomly drawing a number of alleles (2Ne alleles) from the previous generation. Once an allele is selected from the last generation, a second random number is used to determine whether this allele will mutate and if the allele does mutate (this random number is <0.001, the mutation rate), a third random number is used to determine in which direction the allele will mutate, one step larger or one step smaller. To reach a steady-state PK distribution, simulations were carried out for 4Ne generations. Previous simulation studies have shown that the stepwise mutation model reaches steady state by this point (Shriver et al. 1993). To simulate the effects of population expansion, we used equilibrium distributions for anNe = 5 and increased the effective population size to Ne = 5000 in one generation. This model for population expansion is similar to that used by researchers studying the dynamics of sequence mismatch distributions (Rodgers and Harpending 1992). In addition to this instantaneous increase model, we simulated exponential expansion rates and expansion rates of 1% per year [a conservative estimate of the rate of human population expansion (Rodgers and Jorde 1995)]. Within this range, the rate of increase in population size had negligible effect on the shape of the PK (data not shown). The simulated distributions show the results of sampling 50 individuals (100 chromosomes) for 100 independent microsatellite loci.
Acknowledgments
We thank Dr. R. Chakraborty for helpful discussion and encouragement in the course of this study. This work was supported in part by grants to M.S. from the National Institute of Justice (95-IJ-CX-0008) and the Keck Foundation for Advanced Training in Computational Biology, and from the National Institutes of Health (NIH) to R.D. (GM-45861), and an NIH Training Grant (T32-GS08404) to L.J.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
Present address: 2Allegheny University of the Health Sciences, Pittsburgh, Pennsylvania.
-
↵4 Corresponding author.
-
E-MAIL rdeka{at}helix.hgen.pitt.edu; FAX (412) 624-3020.
-
- Received January 31, 1997.
- Accepted April 25, 1997.
- Cold Spring Harbor Laboratory Press








