
The Unified Grass Genome: Synergy in Synteny
The grasses, members of the family Gramineae or Poaceae, are represented by over 10,000 species (Kellogg and Birchler 1993). Three of the domesticated grasses, rice, wheat, and maize, account for about half of total world food production. Although the oldest known grass fossils have been found in paleocene-eocene deposits that are ∼50–60 million years old (Crepet and Feldman 1991), morphological and molecular clock data suggest that the grasses had a monophyletic (single) origin over 70 million years ago (Linder 1986;Clark et al. 1995).
Grasses are morphologically distinct from other plant families but are also highly diverse in morphology and growth habit. Grass species differ greatly in chromosome number and genome size. The genome of rice, for instance, is >11-fold smaller than the genome of barley (Arumuganathan and Earle 1991), despite their equivalent diploid states and apparently similar morphological and physiological complexity.
Grass Genome Colinearity
The use of common sets of low-copy-number DNA markers, often coding sequences, in the mapping of grass genomes has indicated that the gene content of different grass species does not vary greatly (Hulbert et al. 1990; Ahn et al. 1993; Kurata et al. 1994). Rice and barley, for example, do not differ by even twofold in the average number of genomic restriction fragments that hybridize to the same low-copy-number probes (A.E. Van Deynze, M.E. Sorrells, and S.R. McCouch; http://greengenes.cit.cornell.edu:80/anchors/). Much of the difference in genome size is attributable to differences in amounts of repetitive DNA (Flavell et al. 1974); larger genomes like barley or wheat are composed of >75% repetitive DNA, whereas smaller genomes like rice contain less <50% repetitive DNA (Flavell et al. 1974;Deshpande and Ranjekar 1980). A recent study has shown that retrotransposons inserted between genes account for the majority of the repetitive DNA in some large genome grasses, like maize (SanMiguel et al. 1996).
The first genetic map generated for sorghum utilized DNA markers from its close relative maize (Hulbert et al. 1990). This study disclosed that these two species possessed largely conserved gene content and gene number and also exhibited extended regions of map colinearity. In contrast, the interspersed repetitive DNAs in these two species did not cross-hybridize detectably (Hulbert et al. 1990), in agreement with the general observation that most repetitive DNAs do not cross-hybridize between members of different plant genera.
The extensive conservation of gene content and gene order between maize and sorghum was not unexpected, because only ∼15–20 million years of independent descent differentiate these members of the tribe Andropogonae. However, subsequent comparative genetic maps of maize and rice (Ahn and Tanksley 1993; Ahn et al. 1993) and wheat and rice (Ahn et al. 1993; Kurata et al. 1994) using DNA probes also indicated long colinear segments. Rice and maize diverged ∼60–80 million years ago and thus represent a broad temporal range of grass genome separation (Clark et al. 1995). This observation of conserved gene content and order in the grasses gave rise to the model that individual grass species could be viewed best as manifestations of a single grass genome and that each of the strengths of studies in different grasses could be used to benefit all individual grass studies (Bennetzen and Freeling 1993; http://www-iggi.bio.purdue.edu).
Initial comparative studies of dicotyledonous genomes emphasized the chromosomal rearrangements that distinguish the genomes of closely related tomato and pepper (Tanksley et al. 1988). However, additional studies have indicated significant conservation of gene content and order in large segments of many dicot genomes (Bonierbale et al. 1988;Fatokun et al. 1992; Weeden et al. 1992; Prince et al. 1993; Kowalski et al. 1994), indicating that a parallel genome approach should also assist dicot research and development. Given that the monocot/dicot divergence is perhaps only threefold more ancient than the independent descent of the grasses is, it also seems likely that regions of synteny will be observed between monocots and dicots.
The numerous large rearrangements that do differentiate grass genomes are commonly inversions, translocations, or duplications that involve all or nearly all of a chromosome arm (Moore et al. 1995a). The extensive conservation of gene content and order among grass chromosome arms allowed the construction of a “Lego block” model of conserved chromosomal segments (Moore et al. 1995b) and assembly of a proposed progenitor genome structure from which all grass genomes have been derived. Figure 1 simplifies the model of Moore et al. (1995a) to indicate how three key grass genomes, maize, barley, and rice, are essentially reorganizations of a hypothetical ancestral grass genome.
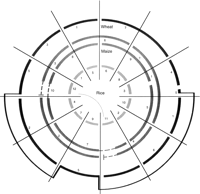
A simplified comparative map of rice, maize, and wheat. Chromosomes are represented by arcs, and the number below each arc indicates the chromosome number. The wheat genome used as reference is the D genome of bread wheat. Maize is represented by two concentric series of arcs because it is actually an ancient tetraploid. Inversions are not shown. The broken lines indicate regions duplicated in rice (on chromosomes 11 and 12; see connecting curved line) that are not duplicated in maize. (Modified, with permission, from Moore et al. 1995a. Copyright 1995 Current Biology, Ltd.)
However, the observed colinearity between grass genomes is laced with exceptions that are not explained simply by chromosomal rearrangements involving whole chromosome arms. Often, 20%–40% of the ordered/linked DNA markers in one grass species will not align in any predictably ordered/linked manner in another grass (J.L. Bennetzen, unpubl.). In addition, species-specific “adjacent” tandem duplications (Chen et al. 1997) and “distantly” tandem duplications (Sanz-Alferez et al. 1995) are often missed by, or confuse (Bennetzen 1996), comparative mapping studies. Finally, some low-copy-number DNA markers are missing completely from otherwise colinear chromosomes, as is exemplified best by studies in wheat that demonstrate single-copy markers found on only one of the homologous chromosome sets in this allohexaploid (Devos et al. 1993). Some of these uniquely present or placed low-copy-number markers may actually be single-copy retrotransposons (Camirand et al. 1990) or other types of mobile DNA, but many noncolinear DNA probes are characterized genes. Additional studies are warranted, and under way, to characterize the nature and mechanisms of these numerous small-scale rearrangements. Hence, genome colinearity needs to be used with caution when applied to individual and microsyntenous cases.
Out of Many, One: Grasses as a Single Genetic System
Despite its various and interesting exceptions, genome synteny in the grasses provides a powerful set of tools for understanding and manipulating grass biology. As a common theme, all of these tools utilize the synergy gained from applying similar or complementary studies in the whole array of grass species to each investigator’s own study species. A traditional disadvantage to both basic and applied research in plants has been the dilution of efforts across the entire range of species with agronomic significance. Using a comparative/parallel approach, information and materials from any plant species can now be utilized for the study and improvement of any other species.
The first, and most obvious, use of this approach is in understanding the nature of grass genome evolution. Already, data have accumulated to a point where the major chromosomal rearrangements punctuating the independent descent of various grasses could be arranged into a phylogenetic array. Such events as whole chromosome arm inversions or translocations would lead to lowered fertility of heterozygotes and, hence, should drive the speciation process. The duplications and deletions that differentiate species should also affect their evolutionary potential greatly to maintain old, or evolve new, functions for gene families.
The synergistic power of using grasses as a single genetic system can be applied whenever a functional allele—one that specifies a dominant or semidominant phenotype—is mapped onto the parallel grass genetic map. Microsynteny will allow the use of smaller grass genomes as a vehicle for the map-based cloning of genes in larger grass genomes. This technique has been utilized toward the isolation of a barley disease resistance gene by chromosome walking in rice (Kilian et al. 1995). In cases where microcolinearity holds up (Dunford et al. 1995;Avramova et al. 1996; Chen et al. 1997), this will be a relatively routine process. However, because of the many exceptions noted above, the investigator would be advised first to map carefully tightly linked DNA markers in both the small genome grass and the large genome grass to guarantee local colinearity prior to commencing such a study. Beyond gene isolation, syntenous genomes can also be used for identification of the genes on a large clone. Our recent studies (Avramova et al. 1996; Chen et al. 1997) have found that cross-hybridization between cloned colinear segments of different grass genomes efficiently identifies the conserved segments (e.g., genes) within a sea of unconserved repetitive and mobile DNAs.
Perhaps the most important use of parallel grass genomes will be in the genetic dissection of complex developmental and physiological processes (Bennetzen and Freeling 1993). Given the vagaries of mutational analysis, it is unlikely that any single species will ever have identified mutations affecting every step in a complex biological pathway. However, phenotype and map data from a full range of grass species might allow identification of the broadest scope of related loci in whatever species they have been uncovered. For instance, homologs of several maize genes involved in anthocyanin pigment production have not been detected by mutation in sorghum yet are present by gene hybridization and product profile criteria. We have mapped two pigmentation mutations in sorghum, using maize probes. One of them, Rs, maps in the comparable position to B1 of maize, whereas the other, P, maps in a position where no comparable maize gene has been identified yet (Bennetzen 1996). Interestingly, dominant alleles of the Rs gene lead to pigmentation only in the seedling of sorghum, whereas dominant alleles of B1 regulate pigmentation in the adult plant and seed. Hence, if Rs and B1 are trans-species alleles, then they have evolved to regulate pigment production at different times in development.
The Comparative Evolution of Developmental Design
Interspecies genetic homology determined by genome colinearity (i.e., orthology) can also be used to test the genetic basis of the evolution of key design principles. For instance, the morphology of the ligule, a fringe-like outgrowth of the leaf in grasses, is often used as a taxonomic trait because it is so polymorphic. Figure2 depicts a few grass ligule types. The maize ligule is a membranous epidermal extension of the sheath (Sylvester et al. 1990), whereas the rice ligule is a leaf-like organ with tissues, veins, and stomata (Chaffey 1983). If we could understand the genetic basis of the allelic variation that specified the variation in ligule designs, perhaps central principles of morphological design would emerge. The Lg1 and Lg2 genes have been cloned in maize, and because mutants for Lg1 and Lg2 exist and map to orthologous positions in both maize and rice (Paterson et al. 1995; M. Freeling, unpubl.), it is possible to swap either gene between these two species to determine the part of the morphological difference that is specified by a particular gene. We see experimental exchange of orthologous genes between grass species as an early step toward discovering the rules used by evolution and domestication to “engineer” the huge and potentially useful diversity evident in the grass family.
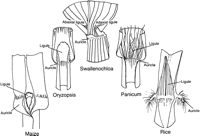
Diversity of structure at the sheath–blade boundary of leaves in the grass family. The ligule, or sheath extension, and the auricles are indicated. Both ligules and auricles of maize are missing in the absence of either LG1 or LG2 proteins. The ligule and auricles of maize and rice are very different. A species from each of three other grass genera is also represented. There is much variability of structure, presumably reflecting genetic polymorphism, for this region of leaf.
Comparative mapping of grass traits and identification of unexpected genetic relationships across species can now be accomplished even with grasses that lack detailed genetic maps, like Tripsacum dactyloides (Eastern gamagrass). Blakey and coworkers (1994) mapped in T. dactyloides a gynomonecious sex form variant allele, called Gsf1, onto a rudimentary genetic map using maize DNA probes. The recessive gsf1 allele conditions an alteration in maize floral structure that is broadly similar to the tassel seed mutants of maize. Gsf1 was found to be in a syntenous location with the ts2 gene of maize, which determines perfect flowers (and tassel seed) in the maize tassel. With this information, the investigators critically examined the phenotypes of gsf1 T. dactyloides and ts2 maize, and found similar phenotypic details. ts2 already had been cloned and sequenced in maize (DeLong et al. 1993) and is thus available for characterization of its possible contribution to the gynomonecious sex form trait in T. dactyloides.
Apomixis is the trait allowing reproduction by seed without sex. Apomictic variants must accomplish two different negative functions: blockage of the reductive division of meiosis and prevention of fertilization. Hence, it is likely that some inactivational mutations of some apomictic loci could lead to a failure of meiosis. PerhapsApo, the dominant allele that confers diplosporic apomixis to some Tripsacums (Savidan et al. 1995), or the dominant aposporic apomictic chromosomal region of the millet Pennisetum squanulatum (Ozias-Akins et al. 1993), might map to regions carrying known meiotic mutants in other grass species, including some that have been or soon will be cloned.
Cross-species identification of the full set of genes involved in a given process, using the full array of grasses, will not only allow more comprehensive studies, but once the orthology of genes from different species has been identified, then information gained in the study of the gene in one species can be applied to the study of this gene in other species. If genes are orthologous between species, then the scientist or genetic engineer can choose the best allele, regardless of the species of origin, for study or modification of any grass. This pan-species use of information and genes can be applied both to simple traits and to quantitative traits influencing such important plant characteristics as drought tolerance or growth habit. For instance, Paterson and coworkers (1995) have shown that most of the mutations selected by early farmers in the domestication of grasses from weedy relatives were the same genes in such different species as rice, maize, and wheat.
Several potentially useful genetic traits exist only in exotic grass species or ecotypes. For example, apomixis has been documented in 145 different species of exotic grasses (Carman 1995). A first step toward learning more about these useful traits would be to map them on a comparable grass genetic map and to see whether known mutations in any species appear to have orthologous location and similar function. Standard sets of grass DNA markers could be used for this mapping (A.E. Van Deynze, M.E. Sorrells, and S.R. McCouch;http://greengenes.cit.cornell.edu:80/anchors/), and very wide crosses could be performed (Laurie et al. 1990; O. Reira-Lizarazu, H.W. Rubes, R.L. Phillips, and D.W. Davis, pers. comm.) to introgress important traits into genetically tractable relatives of exotic plants.
Gene Redundancy and Differentiation
Even the apparent disadvantages of some genetic systems can be converted to novel strengths with a comparative approach. Polyploidy is common in the grass family and has been viewed traditionally as an unfortunate complication to mapping and other genetic characterizations. An evolutionarily recent polyploid would have multiple copies of each gene, thereby making the identification of recessive mutations more difficult. Hence, many of the traits of interest in polyploids are specified by apparent gain-of-function mutations as dominant alleles. If polyploidy is more ancient, as is the case in maize (Helentjaris et al. 1988), then there may be little polyploidy-generated functional redundancy of the sort that masks recessive mutations. Rather than a problem, the presence of an enlarged gene family in a polyploid allows for the evolution of functional or tissue-specific separation between members of the family. Many genes in a true diploid, if fully inactivated, would confer the relatively uninformative phenotype of lethality. However, if the essential function is duplicated and diverged, then mutational inactivation of one or the other gene might give informative phenotypes. An example of this phenomenon is shown in maize, where the essential chalcone synthase gene family can have some of its members inactivated, in one case (c2) giving rise to a seed (and plant) with reduced anthocyanin pigments, and in a second case (whp) yielding no morphological phenotype unless in a double c2/c2 whp/whpmutant where white pollen and self-sterility result (Coe et al. 1981).
Synteny and Synergy
In short, the synteny of grass genomes will allow their synergistic study, reengineering, and improvement. With common gene content and similar physiological processes, information and genes can be applied across species boundaries. Small genome relatives will facilitate map-based gene cloning, including access to genes from large genome grasses. Alleles of genes can be transformed into amenable grasses with a reasonable likelihood that the other genes with which they need to interact will be in place. The creative genius of evolution, where minor changes in gene content, function, or regulation give rise to major new organismal properties, can now be studied across a range of species and times that will inform us not only of what is there and what it does, but also why such outcomes have arisen.
Acknowledgments
We thank Sheri Frank for producing Figure 1 and Mark Mooney for the hand drawings of Figure 2. This work was supported by grants from the U.S. Department of Agriculture (94-37300-0299 and 94-37310-0661) to J.L.B. and the Department of Energy (91ER20028) to M.F.
Footnotes
-
↵2 Corresponding author.
-
E-MAIL maize{at}bilbo.bio.purdue.edu; FAX (317) 496-1496.
- Cold Spring Harbor Laboratory Press








